Introduction
Cancer immunotherapy exerts beneficial effects
through mediating tumor cell regression (1), which relies on activating antitumor T
cells. Co-stimulatory molecules, such as CD80 (B7-1) and CD86
(B7-2), expressed on the surface of dendritic cells (DCs) or other
antigen presenting cells (APCs), regulate T cell activation and
proliferation by interacting with CD28 (2,3).
Conversely, upon activation, cytotoxic T lymphocyte-associated
antigen 4 (CTLA-4) is expressed on the T cell surface, and also
interacts with CD80 and CD86. However, contrary to CD28, CTLA-4
serves an inhibitory co-stimulatory role in regulating T cell
activation and proliferation (4,5). It was
reported that CTLA-4 downregulates the activation of T cells by
inhibiting IL-2 production and cell cycle progression (6). It has been verified that CTLA-4 competes
with CD28 and acts as an inhibitory receptor to influence T cell
function (7).
Adoptive cell therapy (ACT) is a promising
anticancer therapy option (8).
Cytokine-induced killer (CIK) cells, a heterogeneous subset of
expanded lymphocytes, exhibit notable cytotoxic activity against
tumor cells (9–11). CIK cells are generated from peripheral
blood mononuclear cells (PBMCs), with a cytokine cocktail including
interferon (IFN)-γ, anti-CD3 monoclonal antibody, interleukin
(IL)-2 and IL-1α (12). Clinically, a
limited expansion rate and low in vivo toxicity are
significant shortcomings of CIK cell therapy. The present study
hypothesized that CTLA-4 expression increases when PBMCs are
induced into CIK cells during in vitro culture. In the
present study, lentivirus-mediated RNA interference was employed to
knock down CTLA-4 expression in CIK cells and explore the effects
of CTLA-4 inhibition on the proliferation and toxicity of CIK
cells.
Materials and methods
Cell culture
The A549 human lung carcinoma cell line (no.
59239596; American Type Culture Collection, Manassas, VA, USA) was
cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Thermo Fisher Scientific, Inc.). PBMCs from healthy adults were
collected by cell apheresis (Fresenius Kabi Asia-Pacific, Ltd.,
Wanchai, China). CIK cells were generated by culturing PBMCs in
RPMI-1640 supplemented with 10% FBS and containing 1,000 IU/ml
recombinant IFN-γ (Shanghai Chemo Wanbang Biopharma Co., Ltd.,
Shanghai, China). At 24 h, 100 ng/ml anti-CD3 antibody (Wuhan
Institute of Biological Products Co., Ltd., Wuhan, China), 1,000
IU/ml IL-2 (Four Rings Biotechnology, Beijing, China) and 1 ng/ml
IL-1α (Invitrogen; Thermo Fisher Scientific, Inc.) were added. From
day 5, the cells were replenished every 3 days with fresh medium
containing 1,000 IU/ml IL-2. All cells were culturedat 37°Cin 5%
CO2.
Cell transduction of CTLA-4 shRNA
(shCTLA-4) lentiviral particles
shCTLA-4 lentiviral particles (Hanbio Biotechnology
Co., Ltd., Shanghai, China), containing a 29-mer shRNA sequence
5′-GGAATGAGTTGACCTTCCTAGATGA-3′, were used to knockdown the
expression of CTLA-4 in CIK cells. On day 10, CIK cells were
transduced with shCTLA-4 lentiviral particles at multiplicity of
infection of 10. A total of 96 h later, the transduction efficiency
was estimated by detecting CIK cells expressing green fluorescence
protein under a fluorescence microscope.
Polymerase chain reaction (PCR)
Total RNA was isolated using an RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany), and reverse transcribed as
single-stranded cDNA. cDNA was then used as the template to amplify
CTLA-4 via PCR with the following primers: Forward,
5′-GACCTGGCCCTGCACTCTCCTGTTT-3′ and reverse,
5′-ACTGTCACCCGGACCTCAGTGGCTT-3′. GAPDH was employed as the internal
control. Primers for the amplification of GAPDH are as follows:
Forward, 5′-TGCCTCCTGCACCACCAACT-3′ and reverse,
5′-CCCGTTCAGCTCAGGGATGA-3′. PCR was carried out at 94°C for 30 sec,
55°C for 30 sec and then 72°C for 30 sec.
Western blotting
For protein analysis, total protein was extracted
from 1×107 CIK cells using RIPA lysis buffer (BestBio,
Shanghai, China) and then quantified using a BCA kit (Pierce;
Thermo Fisher Scientific, Inc.). Equal amount of whole cell lysates
(20 µg per lane) were separated using SDS-PAGE on a 10% gel and
transferred to a polyvinylidene difluoride membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The membrane was blocked
using 2.5% non-fat milk for 1 h at room temperature and then
incubated with rabbit monoclonal antibody against human CTLA-4
(cat. no. SC-9094; 1:200; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) or rabbit polyclonal antibody against human GAPDH (cat.
no. SC-25778; 1:1,000; Santa Cruz Biotechnology, Inc.) for 2 h at
25°C followed by incubation with a horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G antibody
(cat. no. SC-2004; 1:2,000; Santa Cruz Biotechnology, Inc.) for 1 h
at 25°C prior to detection with chemiluminescence
(FluorChem™ HD2 system; ProteinSimple; Bio-Techne,
Minneapolis, MN, USA). The expression of CTLA-4 was normalized to
that of GAPDH.
Cytotoxicity assay
CIK cell-mediated cytotoxicity was assessed using
the CytoTox 96® Non-Radioactive Cytotoxicity Assay
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol. Briefly, shCTLA-4 lentiviral
particle-transduced CIK cells, control shRNA (shControl) lentiviral
particle-transduced CIK cells and the CIK cells without lentivirus
transduction were suspended in RPMI-1640 medium, supplemented with
5% FBS, at a density of 2×106 cells/ml. The control and
experimental wells were set up using a round-bottom 96-well culture
plate. The wells which only contained CIK cells served as the
control for the spontaneous LDH release effector cells; the
experimental wells contained CIK cells and A549 cells at a ratio of
20:1. Cells were centrifuged at 250 × g for 4 min at 20°C after
incubation at 37°C for 4 h. For target cell maximum LDH release
control wells, the lysis solution was added 45 min prior to
supernatant harvest. A total of 50 µl supernatant from each well of
the assay plate was transferred to a flat-bottom 96-well plate that
was pre-loaded with 50 µl/well reconstituted substrate mix.
Following incubation at room temperature while protected from light
for 30 min, 50 µl stop solution was added to each well followed by
reading the absorbance at 490 nm. The cytotoxicity of CIK cells was
calculated as follows: [(Experimental-effector spontaneous-target
spontaneous)/(target maximal-target spontaneous)]x100.
To further confirm the cytotoxicity of CIK cells, a
more convenient and intuitive cell counting assay was used to
detect the cytotoxicity of the shCTLA-4 lentiviral
particle-transduced CIK cells. As aforementioned, the CIK cells
transduced with shCTLA-4 lentiviral particles or shControl
lentiviral particles, and the CIK cellswithout lentiviral
transduction were co-cultured with A549 cells at a ratio of 20:1
(effector cells:target cells). For the negative group, only A549
cells were seeded and cultured for 24 h. According to the
morphology of adhesive A549 cells and suspended CIK cells, the
residual A549 cells from all groups were observed under a
microscope and counted following trypan blue dye exclusion. The
cytotoxicity of effector cells could be deduced from the numeration
of A549 cells that survived the CIK cells.
Statistical analysis
All statistical analyses were performed using SPSS
software (version 19.0; IBM Corp, Armonk, NY, USA). The band
signals were analyzed using Quantity One software (version 4.6.2;
Bio-Rad Laboratories, Inc.). The data are presented as the mean ±
standard error of the mean. Two or multiple group comparisons were
performed using a Student's t-test or one-way analysis of variance
with Fisher's least significant difference post hoc test,
respectively. P<0.05 was considered to indicate a statistically
significant difference.
Results
The expression of CTLA-4 increases
with the induction of CIK cells
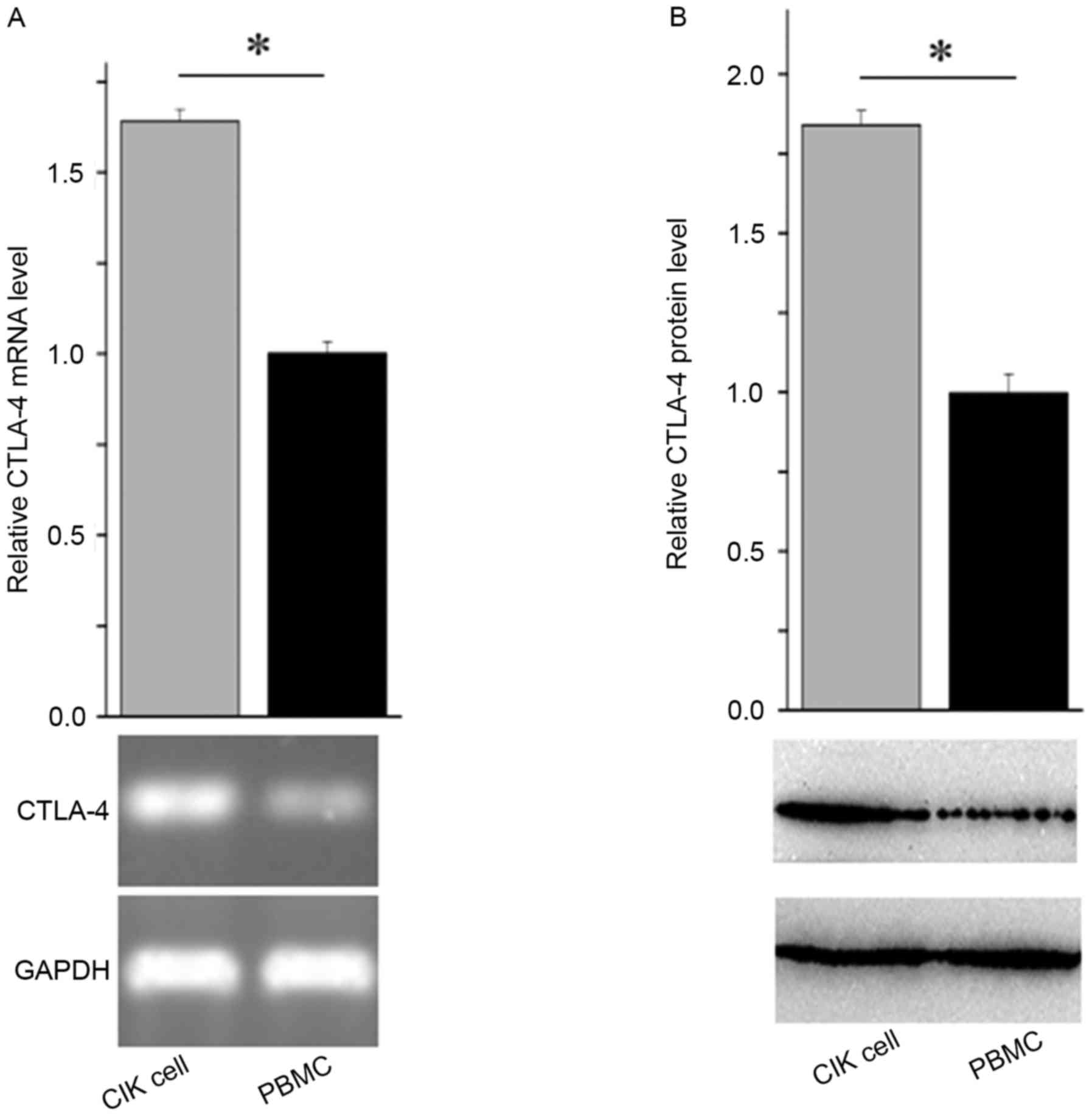
To compare the expression levels of CTLA-4 between
CIK cells and the corresponding PBMCs, CIK cells and PBMCs were
used to prepare total RNA and protein. As evaluated using RT-PCR
and western blot analysis, the mRNA and protein expression levels
were much higher in CIK cells than those in PBMCs (Fig. 1A and B). These observations suggested
that CTLA-4 expression increases during the induction of CIK
cells.
shCTLA-4 lentivirus efficienctly
inhibits CTLA-4 expression
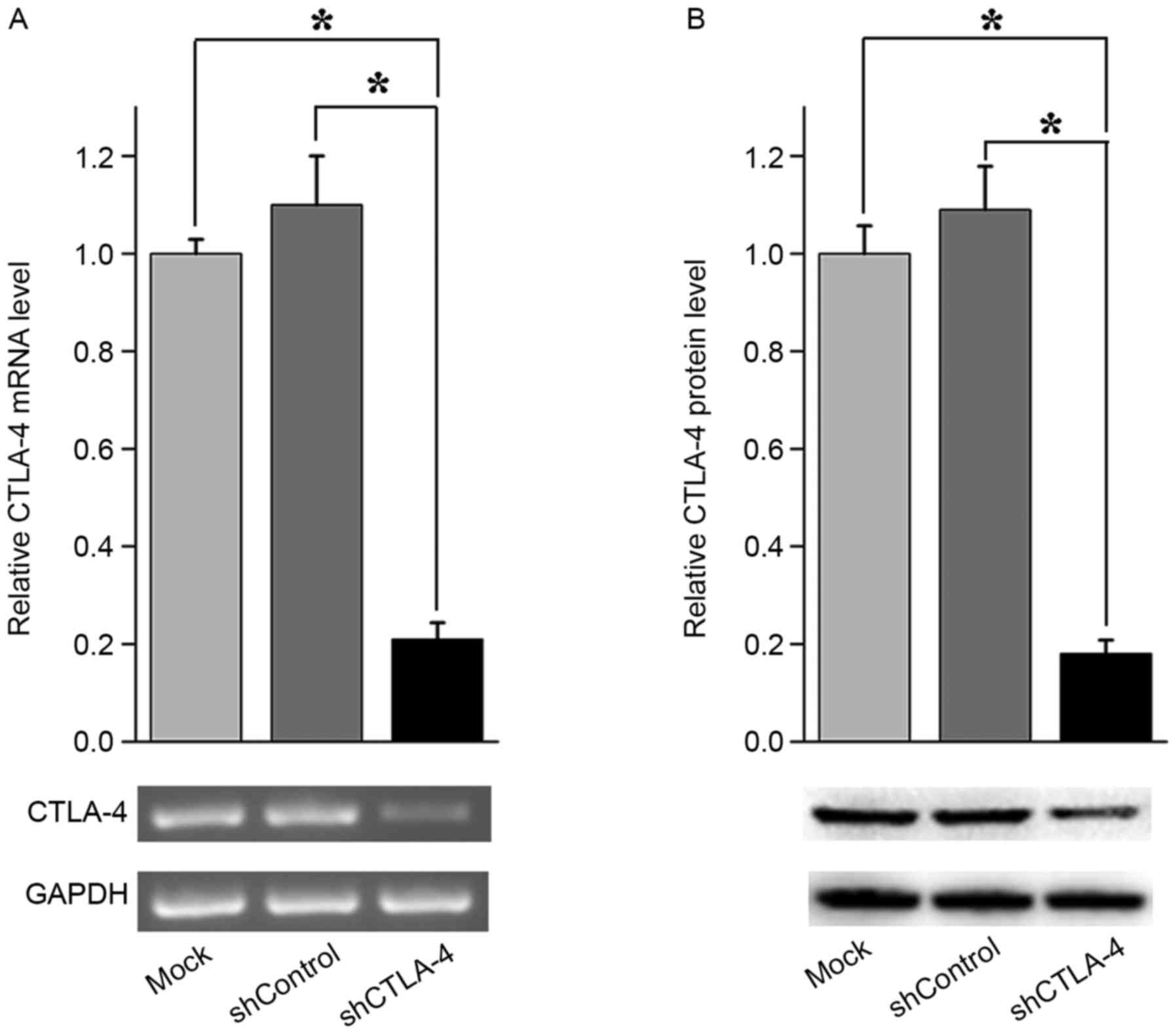
The shCTLA-4 lentiviral particles were employed to
knock down CTLA-4 expression in CIK cells. As determined by PCR,
shCTLA-4 lentiviral particles significantly decreased CTLA-4 mRNA
levels in CIK cells when compared with the shControl lentiviral
particles (Fig. 2A). As analyzed by
western blotting, shCTLA-4 lentiviral particles significantly
reduced the expression levels of CTLA-4 protein in CIK cells when
compared with the shControl lentiviral particles (Fig. 2B). These data indicate that shCTLA-4
lentiviral particles can successfully silence the expression of
CTLA-4 in CIK cells.
Silencing CTLA-4 promotes the
proliferation of CIK cells
To examine whether silencing CTLA-4 with shCTLA-4
lentivirus affects the proliferation rate of CIK cells, CIK cells
were transduced with shCTLA-4 lentivirus or shControl lentivirus. A
total of 96 h after viral transduction, cell counting was
performed. Morphologically, the shCTLA-4 lentivirus-transduced CIK
cells presented with more agglomeration than the shControl
lentivirus-transduced CIK cells did (Fig.
3A). Compared with control transduction of shControl
lentivirus, the transduction of shCTLA-4 lentivirus promoted the
ex vivo expansion of CIK cells (Fig. 3B). These findings indicated that
transduction of shCTLA-4 lentivirus enhances the proliferation of
CIK cells.
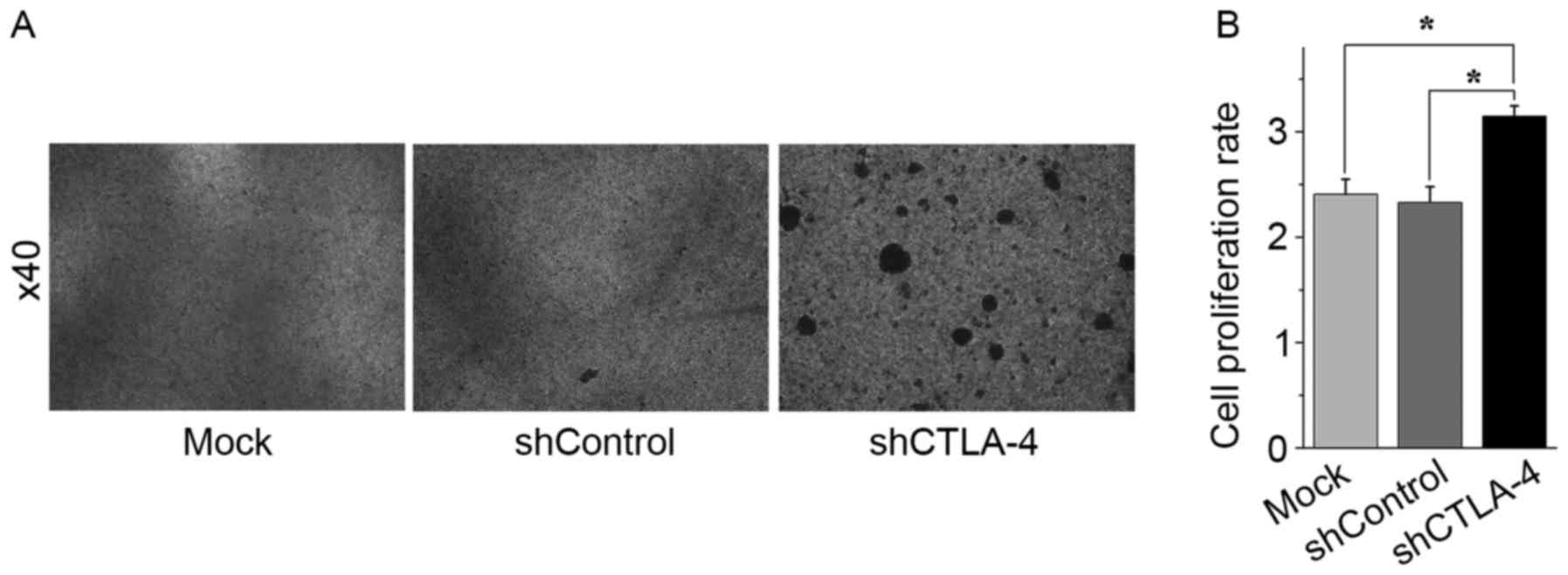 | Figure 3.Inhibition of CTLA-4 expression
promoted CIK cell expansion in vitro. CIK cells on day 10
were transduced with shCTLA-4 lentiviral particles, shControl
lentiviral particles, or mock transduced. A total of 96 h after
cell transduction, CIK cells were harvested and counted to evaluate
cell expansion efficiency. (A) CIK cells mock transduced (left
panel), transduced with shControl lentiviral particles (middle
panel), or transduced with shCTLA-4 lentiviral particles (right
panel). Magnification, ×40. (B) Compared with mock transduced CIK
cells or shControl lentiviral particles transduced CIK cells,
shCTLA-4 lentiviral particles transduced CIK cells exhibited a
higher expansion rate. *P<0.01. Each experiment was performed
three times. The data are plotted as the mean ± standard error of
the mean. CTLA-4, cytotoxic T lymphocyte-associated antigen 4; CIK,
cytokine-induced killer; PMBC, peripheral blood mononuclear cells;
qPCR, quantitative polymerase chain reaction; sh, short
hairpin. |
Silencing CTLA-4 improves the
cytotoxicity of CIK cells
As determined by a CytoTox 96 Non-Radioactive
Cytotoxicity Assay, the shCTLA-4 lentivirus-transduced CIK cells
caused more LDH release against A549 cells compared with the
shControl lentivirus-transduced CIK cells did (Fig. 4A). A further cell counting assay
indicated that, compared with the shControl lentivirus-transduced
CIK cells, a decreased number of A549 cells survived under the
cytotoxic activity of shCTLA-4 lentivirus-transduced CIK cells
(Fig. 4B and C). These findings
indicate that the cytotoxicity of CIK cells against A549 cells can
be enhanced through the inhibition of CTLA-4 expression in CIK
cells.
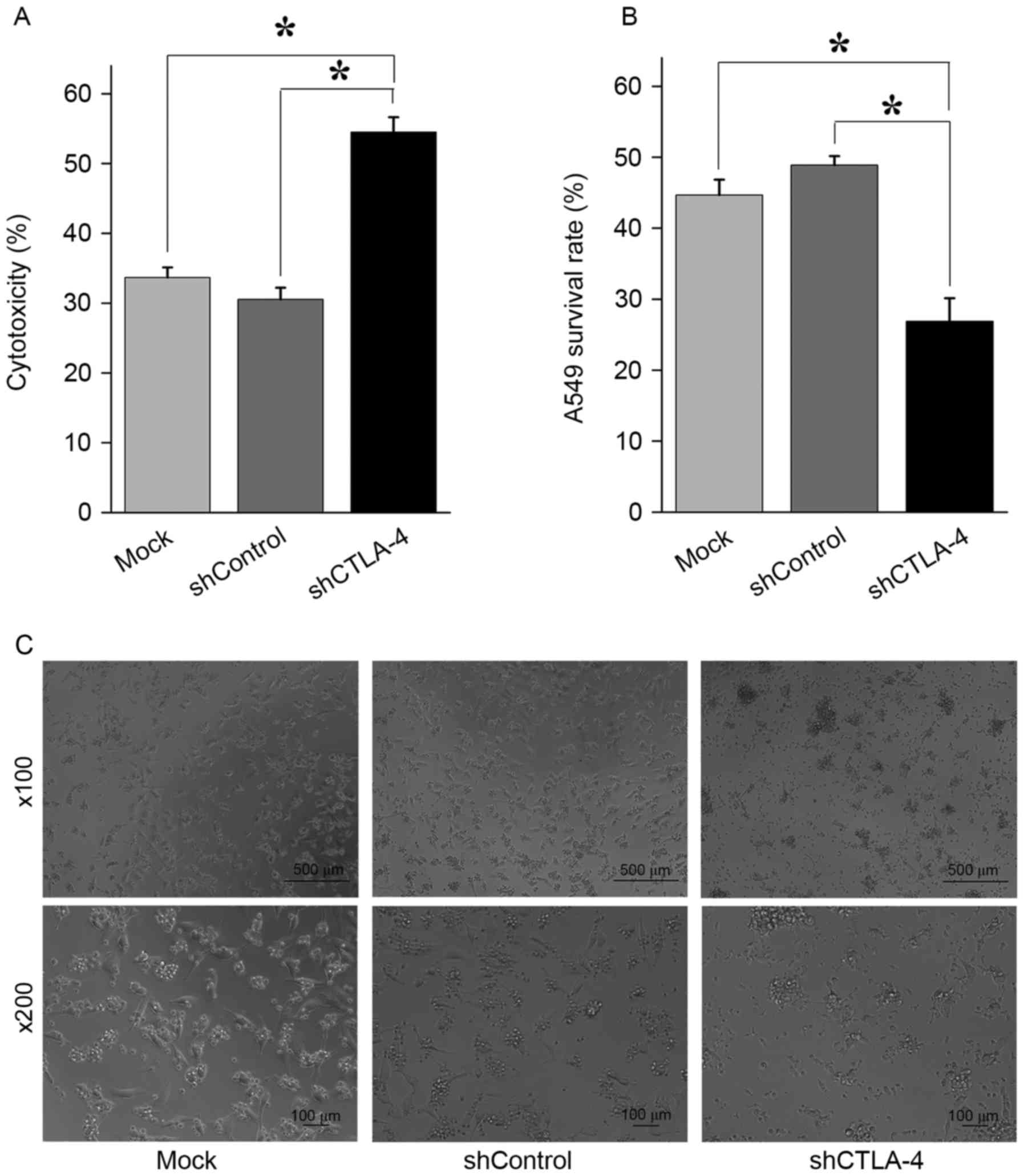 | Figure 4.The inhibition of CTLA-4 expression
increased cytotoxicity of CIK cells against A549 cells. (A)
Determined by CytoTox 96 Non-Radioactive Cytotoxicity Assay,
compared with CIK cells mock transduced or transduced with
shControl lentiviral particles, CIK cells transduced with shCTLA-4
lentiviral particles exhibited significantly higher cytotoxicity
against A549 cells. *P<0.001. (B) A549 cells were co-cultured
with CIK cells for 24 h before cell counting. Compared with the
number of viable A549 cells co-cultured with CIK cells mock
transduced or transduced with shControl lentiviral particles, the
number of viable A549 cells co-cultured with CIK cells transduced
with shCTLA-4 lentiviral particles were significantly lower.
*P<0.01. (C) Representative figures of A549 cells co-cultured
with CIK cells mock transduced (left panel), transduced with
shControl lentiviral particles (middle panel), and transduced with
shCTLA-4 lentiviral particles (right panel) for 24 h. Each
experiment was performed three times. The data were plotted as the
mean ± standard error of the mean. CTLA-4, cytotoxic T
lymphocyte-associated antigen 4; CIK, cytokine-induced killer;
PMBC, peripheral blood mononuclear cells; qPCR, quantitative
polymerase chain reaction; sh, short hairpin. |
Discussion
As a potential anti-tumor ACT, CIK cells have been
extensively tested in clinical practice (13–16). The
ex vivo expansion rate and cytotoxicity are two key factors
that determine the therapeutic efficacy of CIK cells. CIK cells
specifically kill tumor cells via the secretion of IFN-γ, tumor
necrosis factor-α or perforin (16).
However, the activation of immune suppression pathways induces T
cell anergy (17). By competing with
CD28, CTLA-4 serves anegative role in the regulationof T-cell
activation. CTLA-4 prohibits T cell activation through the
inhibition of signal transduction of the T cell receptor and
transmitting the inhibitory signal to T cells (17–19). These
maybe associated with the regulation of CTLA-4: CD28/CD80 axis. It
was reported that CD80 serves a pivotal role in immune
cell-mediated cytotoxicity. Chambers et al (20) indicates that NK cell-mediated
cytotoxicity is a result of interactions between triggering signals
and the inhibitory receptors and ligands, during which CD80 isthe
triggering signal for tumor cell lysis.
In the present study, we demonstrated that the
expression of CTLA-4 on CIK cells is upregulated compared with that
on the corresponding PBMCs. This is concordant with the results of
previous reports, in that the responsiveness of IL-2 led to the
induction of CLTA-4, particularly for the IL-2-expanded immune
effector cells (21,22). It was hypothesized that PBMCs, when
induced with a CIK cell cytokine cocktail, are activated and this
trigged the activation of inhibitory signal to balance the immune
response of CIK cells represented by the concurrent upregulation of
CTLA-4 and CD28 (23,24).
It was further demonstrated that the surpression of
CTLA-4 expression by lentiviral vector-mediated RNA interference
(RNAi) on CIK cells promoted proliferation and the killing
efficiency of CIK cells, which confirmed CTLA-4's inhibitory effect
on CIK cells. Since regulatory T cells (Tregs) are
crucial for the maintenance of immune tolerance, especially for
autoimmunity and the tumor microenvironment (25–27), it
was hypothesized that the effect of inhibiting CTLA-4 expression on
CIK cells may alter the balance of Tregs.
Tregs express a high level of CD25 (the IL-2 receptor α
chain) and compete with other effector cells to bind IL-2,
resulting in cytokine-mediated immune suppression (6,28).
Furthermore, it has been reported that the interaction of CTLA-4 on
Tregs with CD80 and CD86 on APCs can block and even
downregulates the expression of CD80 and CD86 (29). It was also reported that blocking
CTLA-4 has an impact on the balance of Tregs and
downregulates Tregs-mediated suppression, but the
mechanisms are still unclear (23,30).
Although the results have revealed the immune
suppression of CIK cells mediated by CTLA-4, they were unable to
determine the exact mechanisms by which CTLA-4 regulates the
proliferation and activation of CIK cells, which is the distinct
limitation of this study.
To conclude, the present study demonstrated that
CTLA-4 expression increased when PBMCs were induced into CIK cells
by cytokine cocktail, RNAi mediated by lentiviral particles
efficiently inhibited the expression of CTLA-4 on CIK cells,
suppression of CTLA-4 expression significantly promoted the
proliferation of CIK cells in vitro and enhanced the
cytotoxicity of CIK cells against lung cancer cells. These findings
indicate that the blockade of CTLA-4 signaling has potential
therapeutic significance for CIK cell therapy and further clinical
study is warranted to verify these results.
References
|
1
|
Rosenberg SA: Progress in human tumour
immunology and immunotherapy. Nature. 411:380–384. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bour-Jordan H and Blueston JA: CD28
function: A balance of costimulatory and regulatory signals. J Clin
Immunol. 22:1–7. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Gool SW, Barcy S, Devos S,
Vandenberghe P, Ceuppens JL, Thielemans K and de Boer M: CD80
(B7-1) and CD86 (B7-2): Potential targets for immunotherapy. Res
Immunol. 146:183–196. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Slavik JM, Hutchcroft JE and Bierer BE:
CD28/CTLA-4 and CD80/CD86 families: Signaling and function. Immunol
Res. 19:1–24. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakajima A and Azuma M: Costimulatory
molecules in autoimmunity: Role of CD28/CTLA4-CD80/CD86. Nihon
Rinsho. 55:1419–1424. 1997.(In Japanese). PubMed/NCBI
|
|
6
|
Krummel MF and Allison JP: CTLA-4
engagement inhibits IL-2 accumulation and cell cycle progression
upon activation of resting T cells. J Exp Med. 183:2533–2540. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soskic B, Qureshi OS, Hou T and Sansom DM:
A transendocytosis perspective on the CD28/CTLA-4 pathway. Adv
Immunol. 124:95–136. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darcy PK, Neeson P, Yong CS and Kershaw
MH: Manipulating immune cells for adoptive immunotherapy of cancer.
Curr Opin Immunol. 27:46–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Margolin KA, Negrin RS, Wong KK,
Chatterjee S, Wright C and Forman SJ: Cellular immunotherapy and
autologous transplantation for hematologic malignancy. Immunol Rev.
157:231–240. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schmeel LC, Schmeel FC, Coch C and
Schmidt-Wolf IG: Cytokine-induced killer (CIK) cells in cancer
immunotherapy: Report of the international registry on CIK cells
(IRCC). J Cancer Res Clin Oncol. 141:839–849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume
KG and Weissman IL: Use of a SCID mouse/human lymphoma model to
evaluate cytokine-induced killer cells with potent antitumor cell
activity. J Exp Med. 174:139–149. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mesiano G, Todorovic M, Gammaitoni L,
Leuci V, Giraudo Diego L, Carnevale-Schianca F, Fagioli F,
Piacibello W, Aglietta M and Sangiolo D: Cytokine-induced killer
(CIK) cells as feasible and effective adoptive immunotherapy for
the treatment of solid tumors. Expert Opin Biol Ther. 12:673–684.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chung MJ, Park JY, Bang S, Park SW and
Song SY: Phase II clinical trial of ex vivo-expanded
cytokine-induced killer cells therapy in advanced pancreatic
cancer. Cancer Immunol Immunother. 63:939–946. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang XF, Ma DC, Ding ZY, Liu ZZ, Guo F,
Liu L, Yu HY, Han YL and Xie XD: Autologous cytokine-induced killer
cells therapy on the quality of life of patients with breast cancer
after adjuvant chemotherapy: A prospective study. Zhonghua Zhong
Liu Za Zhi. 35:764–768. 2013.PubMed/NCBI
|
|
15
|
Yu X, Zhao H, Liu L, Cao S, Ren B, Zhang
N, An X, Yu J, Li H and Ren X: A randomized phase II study of
autologous cytokine-induced killer cells in treatment of
hepatocellular carcinoma. J Clin Immunol. 34:194–203. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhong R, Han B and Zhong H: A prospective
study of the efficacy of a combination of autologous dendritic
cells, cytokine-induced killer cells, and chemotherapy in advanced
non-small cell lung cancer patients. Tumour Biol. 35:987–994. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Robert L, Harview C, Emerson R, Wang X,
Mok S, Homet B, Comin-Anduix B, Koya RC, Robins H, Tumeh PC and
Ribas A: Distinct immunological mechanisms of CTLA-4 and PD-1
blockade revealed by analyzing TCR usage in blood lymphocytes.
Oncoimmunology. 3:e292442014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qureshi OS, Zheng Y, Nakamura K, Attridge
K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z,
et al: Trans-endocytosis of CD80 and CD86: A molecular basis for
the cell-extrinsic function of CTLA-4. Science. 332:600–603. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Catalfamo M, Tai X, Karpova T, McNally J
and Henkart PA: TcR-induced regulated secretion leads to surface
expression of CTLA-4 in CD4+CD25+ T cells.
Immunology. 125:70–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chambers BJ, Salcedo M and Ljunggren HG:
Triggering of natural killer cells by the costimulatory molecule
CD80 (B7-1). Immunity. 5:311–317. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Geldhof AB, Moser M, Lespagnard L,
Thielemans K and De Baetselier P: Interleukin-12-activated natural
killer cells recognize B7 costimulatory molecules on tumor cells
and autologous dendritic cells. Blood. 91:196–206. 1998.PubMed/NCBI
|
|
22
|
Stojanovic A, Fiegler N, Brunner-Weinzierl
M and Cerwenka A: CTLA-4 is expressed by activated mouse NK cells
and inhibits NK Cell IFN-γ production in response to mature
dendritic cells. J Immunol. 192:4184–4191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Quezada SA, Peggs KS, Curran MA and
Allison JP: CTLA4 blockade and GM-CSF combination immunotherapy
alters the intratumor balance of effector and regulatory T cells. J
Clin Invest. 116:1935–1945. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Egen JG, Kuhns MS and Allison JP: CTLA-4:
New insights into its biological function and use in tumor
immunotherapy. Nat Immunol. 3:611–618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamaguchi T and Sakaguchi S: Regulatory T
cells in immune surveillance and treatment of cancer. Semin Cancer
Biol. 16:115–123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sakaguchi S: Naturally arising
Foxp3-expressing CD25+CD4+ regulatory T cells
in immunological tolerance to self and non-self. Nat Immunol.
6:345–352. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Friedline RH, Brown DS, Nguyen H, Kornfeld
H, Lee J, Zhang Y, Appleby M, Der SD, Kang J and Chambers CA: CD4+
regulatory T cells require CTLA-4 for the maintenance of systemic
tolerance. J Exp Med. 206:421–434. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Josefowicz SZ, Lu LF and Rudensky AY:
Regulatory T cells: Mechanisms of differentiation and function.
Annu Rev Immunol. 30:531–564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Onishi Y, Fehervari Z, Yamaguchi T and
Sakaguchi S: Foxp3+ natural regulatory T cells
preferentially form aggregates on dendritic cells in vitro and
actively inhibit their maturation. Proc Natl Acad Sci USA. 105:pp.
10113–10118. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Read S, Greenwald R, Izcue A, Robinson N,
Mandelbrot D, Francisco L, Sharpe AH and Powrie F: Blockade of
CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in
vivo. J Immunol. 177:4376–4383. 2006. View Article : Google Scholar : PubMed/NCBI
|