Introduction
Leukemia is one of the most common malignancies in
China and the United States of America (1,2). Leukemia
was estimated to have effected ~54,270 individuals in 2015,
according to the cancer statistics of the USA, accounting for 4 and
3% of new cancer cases in males and females, respectively (2). With the advances in new chemotherapeutic
drugs and the application of hematopoietic stem cell
transplantation (HSCT), the overall survival rates of patients with
leukemia have markedly increased in the past several decades
(3,4).
However, patients treated with chemotherapy and HSCT have various
complications, including hematological, hepatic, renal and
gastrointestinal toxicity, as well as diarrhea (5,6). Diarrhea
is one of the most common complications, and the incidence of acute
diarrhea has been reported to be 43% in patients receiving bone
marrow transplantation (7).
Currently, the antidiarrheal agent loperamide is effectively and
frequently used to treat these patients (8). However, the effects of loperamide on
leukemia cells have not been studied. Loperamide was reported to
have antitumor effects in solid tumors, including liver, breast,
lung, drug-resistant ovarian, gastric, osteosarcoma, renal and
canine cancer cell lines (9,10). However, whether loperamide exerts
cytotoxic effects on leukemia cells remains unknown.
Cell apoptosis, one of the cell death pathways,
serves a vital role in antitumor effects (11). Apoptosis induced by DNA damage is an
important mechanism by which a variety of antitumor drugs exhibit
their effects, including mitoxantrone, etoposide and bendamustine
(12,13). Once DNA damage is triggered by these
agents, the DNA damage response (DDR) is activated, including the
damage sensor rH2ax and the subsequent signal transduction pathways
(14–16). These pathways, including ATM-Chk2
pathway activation, phosphorylate a panel of substrates involved in
cell cycle control, transcription, DNA repair and apoptosis
(17,18). If the lesion is mild, DNA damage is
repaired through DDR, otherwise, it will lead to mutation of genes,
chromosomal rearrangement or apoptosis (19). In addition, during the process of
apoptosis triggered by DNA damage, elimination of the
anti-apoptotic protein myeloid cell leukemia (Mcl-1) is required,
which performs an important role in apoptosis initiation (20). Loperamide has been reported to induce
apoptosis in the aforementioned cancer cell lines (9,10), but the
mechanisms are unclear.
To the best of our knowledge, the present study
evaluated the effect of loperamide on the growth of leukemia cells
for the first time. To better represent the biological properties
of leukemia, primary leukemia cells from patients and leukemia cell
lines were included in the present study. It was found that
loperamide potently inhibited the growth of leukemia cells in a
dose-dependent manner in leukemia cell lines, as well as primary
leukemia cells from patients. Additionally, cell apoptosis was
dose-dependently induced by loperamide. Of note, the present study
demonstrated that loperamide increased rH2ax, activated the ataxia
telangiectasia mutated serine/threonine kinase (ATM)-checkpoint
kinase 2 (Chk2) signaling pathway, and induced DNA damage in
leukemia cells. Therefore, the present study provides new insights
into the therapeutic potential of loperamide in leukemia and
unravels a novel mechanism through which loperamide inhibits
leukemia cells.
Materials and methods
Chemicals and reagents
Loperamide hydrochloride was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and dissolved in
dimethyl sulfoxide (DMSO) at a stock concentration of 40 mmol/l.
MTT was also purchased from Sigma-Aldrich (Merck KGaA).
Ficoll-Hypaque solution and Wright-Giemsa stain solution were
purchased from Solarbio Life Sciences (Beijing, China). The FITC
Annexin V Apoptosis Detection kit II was purchased from BD
Biosciences (San Jose, CA, USA). The CometAssay® kit
(25×2 well slides) was purchased from Trevigen, Inc. (Gaithersburg,
MD, USA). SYBR-Green was purchased from BioTeke Corporation
(Beijing, China). Mouse anti-phospho (p)-ATM (Ser1981) antibody
(cat. no. AA866; 1:1,000) was purchased from Beyotime Institute of
Biotechnology (Haimen, China). Rabbit anti-caspase-3 (cat. no.
9662; 1:1,000), cleaved caspase-3 (cat. no. 9661; 1:1,000), rabbit
anti-B-cell lymphoma-2 (Bcl-2; cat. no. 2872; 1:1,000), rabbit
anti-Bcl-2-associated X protein (Bax; cat. no. 2774; 1:1,000),
rabbit anti-AKT (cat. no. 4691; 1:1,000), rabbit anti-p-AKT
(Ser473; cat. no. 9018; 1:1,000), rabbit anti-signal transducer and
activator of transcription 3 (STAT3; cat. no. 12640; 1:1,000),
rabbit anti-p-STAT3 (Ser727; cat. no. 94994; 1:1,000), rabbit
anti-p-histone H2A.X (Ser139; cat. no. 9718; 1:1,000 for western
blotting, 1:100 for Immunofluorescence) rabbit anti-ATM (cat. no.
2873; 1:1,000) and rabbit anti-p-Chk2 (Thr68; cat. no. 2197;
1:1,000) antibodies were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Rabbit anti-p65 (cat. no. sc-372; 1:100),
rabbit anti-poly(ADP-ribose) polymerase (PARP; cat. no. sc-7150;
1:1,000), mouse anti-Mcl-1 (cat. no. sc-377487; 1:1,000) and rabbit
anti-Chk2 (cat. no. sc-377487; 1:1,000) antibodies, and all of the
secondary antibodies including mouse anti-rabbit IgG-horseradish
peroxidase (HRP; cat. no. sc-2357; 1:5,000) and rabbit anti-mouse
IgG-HRP (cat. no. sc-358914; 1:5,000) were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cell lines and culture conditions
A total of 10 cell lines, including 5 acute
myelocytic leukemia (AML) and 5 acute lymphocytic leukemia (ALL)
cell lines, were used in the present study. The 5 AML cell lines,
consisting of Thp1, U937, Kg-1, Kasumi-1 and Kg-1a, and 5 ALL
lines, consisting of Nalm6, Molt4, CCRF-CEM, Jurkat and SUP-B15,
were obtained from the American Type Culture Collection repository
(Manassas, VA, USA). The SUP-B15 cell line was cultivated in
Iscove's modified Dulbecco's medium with 10% fetal bovine serum
(FBS) (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
while the other cell lines were maintained in RPMI-1640 medium
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.).
All of the cell lines were cultivated in a humidified incubator at
37°C in a 5% CO2 atmosphere.
Primary cells from patients with acute
leukemia
Bone marrow samples were obtained by bone marrow
aspiration from 15 patients with acute leukemia, and the primary
cells were isolated using Ficoll-Paque PREMIUM (GE Healthcare Life
Sciences, Little Chalfont, UK) subsequent to obtaining written
informed consent from all patients included in the present study.
All of the patients with leukemia were diagnosed according to the
standard diagnostic criteria of World Health Organization (21). A total of 7 male patients and 8 female
patients were included in the present study. The age range of these
patients was 18–76 years and the primary cell percentage as
determined by flow cytometry ranged between 86 and 95%. All of the
patients who were admitted to the Second Affiliated Hospital
(Hangzhou, China) between February 2011 and November 2013 were
newly diagnosed patients without any prior treatment. The study was
approved by the ethics committee of The Second Affiliated Hospital,
School of Medicine, Zhejiang University (Zhejiang, China) and
followed the Declaration of Helsinki principles. The clinical
characteristics of the 15 patients, including 9 AML and 6 ALL
patients, are listed in Table I.
 | Table I.Clinical characteristics of patients
with leukemia. |
Table I.
Clinical characteristics of patients
with leukemia.
| Patients | Subtype | Age | Sex | WBC
(×109/l) | Primary cell
percentage, % | Gene mutation | Chromosome
karyotype |
|---|
| AML1 | M2 | 18 | Male | 66.8 | 94 | AML/ETO | 46, XY,
der(3)t(3;8) (p26;q13), t(8;21) |
| AML2 | M2 | 30 | Female | 39.5 | 87 | NO |
47,XY,+mar[16]/46,XX[4] |
| AML3 | M5 | 51 | Female | 7.0 | 90 | NO | 46,XX,t(8;19)
(q13;p13.1), del(9) (q22q34;del(11)(q23)[5] |
| AML4 | M1 | 56 | Female | 6.0 | 95 | CEBPA |
47,X,i(X)(q10),+10[5]/46,X, -X,+10[4] |
| AML5 | M4 | 45 | Male | 2.5 | 88 | FLT3-ITD |
46,X,-Y,+8,t(8;21)(q22;q22)
[18]/46,XY[2] |
| AML6 | M5 | 60 | Male | 244.7 | 93 | FLT3-ITD,
dupMLL | 46,XY[8] |
| AML7 | M4 | 46 | Male | 35.5 | 92 | CEBPA/MYH11 | 46, XY,
inv(16)(p13,1q22)[20] |
| AML8 | CML-BC/M | 32 | Female | 23.9 | 90 | BCR/ABL(P210),
AML1/MDS1, EVI1 |
46,XX,t(9,22)(q34;q11.2)
[18]/46,idem,t(3;21) (q26;q22)[2] |
| AML9 | M5 | 29 | Male | 58.0 | 95 | EVI1 |
46,XY,t(11,19)(q23,p13.1) |
| ALL1 | B-ALL | 39 | Female | 4.5 | 94 | NO | 46,XX[2] |
| ALL2 | B-ALL | 76 | Female | 229.0 | 89 | BCR/ABL(P210) |
46,XX,t(9,22)(q34;q11.2)[14] |
| ALL3 | B-ALL | 68 | Female | 127.3 | 86 | BCR/ABL(P190) |
46,XX,t(9,22)(q34;q11.2)
[13]/49,idem,+1,+5,+6[2]/46, XX[2] |
| ALL4 | T-ALL | 18 | Male | 107.8 | 93 | SIL/TAL1 | 46,XY[8] |
| ALL5 | B-ALL | 53 | Male | 1.5 | 88 | NA | NA |
| ALL6 | B-ALL | 21 | Female | 2.8 | 93 | NO | 46,XX[2] |
Cell viability assay
Cell viability was measured by MTT assay. Briefly, 5
AML (Thp1, U937, Kg-1, Kasumi-1 and Kg-1a) and 5 ALL (Nalm6, Molt4,
CCRF-CEM, Jurkat and SUP-B15) cell lines and primary cells from the
aforementioned 9 AML and 6 ALL patients were seeded onto 96-well
plates at ~70% confluence and a total volume of 200 µl. Following
exposure to 0, 2.5, 5, 10, 20 and 40 µM loperamide for 24, 48 or 72
h, the cells were incubated with MTT (20 µl, 5 mg/ml) solution at
37°C for 4 h. Supernatant (100 µl) was then pipetted gently and 100
µl of the joint fluid (10% SDS, 5% isobutanol and 1% hydrochloric
acid) was added to each well to dissolve the formazan crystals. The
absorbance value of each well was determined at a test wavelength
of 570 nm and the half-maximal inhibitory concentration
(IC50) of drugs was calculated.
Morphological observation
Molt4 and Thp1 cells were cultivated on 6-well
plates at 37°C overnight and treated with 0 µM (DMSO-treated) or 20
µM loperamide at 37°C for 24 h. The cells were then collected and
transferred to glass slides. The morphological changes were
observed under light microscopy (magnification, ×400) and images
were captured using a camera.
Flow cytometric cell apoptosis
analysis
The assay was performed according to the
manufacturer's protocol (BD Biosciences, San Jose, CA, USA).
Briefly, A total of 1×106 Molt4, Thp1, ALL-P1 and AML-P1
cells were seeded on 6-well plates and treated with 0
(DMSO-treated), 5, 10 or 20 µM loperamide. The cells were collected
after incubation for 24 h and washed with PBS twice. For apoptosis
analysis, the cells were re-suspended in 1X Binding Buffer (BD
Biosciences) at a concentration of 1×106 cells/ml. Then,
100 µl cell resuspension solution was incubated with 5 µl of
fluorescein isothiocyanate (FITC) Annexin V and 5 µl of
7-aminoactinomycin D+ (7AAD+) at room
temperature for 15 min in the dark. Another 400 µl of 1X Binding
Buffer was added and the cells were analyzed by flow cytometry
within 1 h.
Western blot analysis
Cells treated with loperamide were collected and the
total proteins were extracted by radioimmunoprecipitation assay
buffer (Sigma-Aldrich; Merck KGaA) containing protease and
phosphatase inhibitors (Sigma-Aldrich; Merck KGaA). The proteins
were determined colorimetrically, and then 30 µg/lane protein was
loaded and separated by 10% SDS-PAGE. The separated proteins were
then blotted to polyvinylidene fluoride membranes (BD Biosciences,
San Jose, CA, USA). The membrane was incubated in TBS containing 5%
skimmed milk powder at room temperature for 1 h and then probed
with primary antibodies specific to anti-caspase-3 (cat. no. 9662;
1:1,000), cleaved caspase 3 (cat. no. 9661; 1:1,000), PARP (cat.
no. sc-7150; 1:1,000), Bcl-2 (cat. no. 2872; 1:1,000), Bax (cat.
no. 2774; 1:1,000), Mcl-1 (cat. no. sc-377487; 1:1,000), Akt (cat.
no. 4691; 1:1,000), p-Akt (Ser473; cat. no. 9018; 1:1,000), ATM
(cat. no. 2873; 1:1,000), p-ATM (cat. no. AA866; Ser1981; 1:1,000),
Chk2 (cat. no. sc-377487; 1:1,000), p-Chk2 (Thr68; cat. no. 2197;
1:1,000), p-histone H2A.X (Ser139; cat. no. 9718; 1:1,000), STAT3
(cat. no. 12640; 1:1,000) or p-STAT3 (Ser727; cat. no. 94994;
1:1,000) at 4°C overnight. Then, the membrane was washed with TBST
3 times and incubated with the secondary antibodies, horseradish
peroxidase-conjugated rabbit anti-mouse (cat. no. sc-358914;
1:5,000) and mouse anti-rabbit (cat. no. sc-2357; 1:5,000), at room
temperature for 2 h. The membrane was washed again with TBST 3
times. Then, the protein bands were visualized using an enhanced
chemiluminescence western blotting detection system (BD
Biosciences, San Jose, CA, USA) and analyzed using Quantity
One® software (version 4.62; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Immunofluorescence staining
Cells treated with loperamide were transferred to
the slides at 80–90% confluence by a slide centrifuge at 500 × g
for 5 mins at room temperature. The cells were then fixed with
freshly prepared 4% paraformaldehyde in PBS at 37°C for 15 min and
permeabilized with 0.2% Triton X-100 in PBS for 15 min at room
temperature. Subsequent to being blocked with PBS containing 3%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) for 30 min at room
temperature, the samples were incubated with primary rabbit
anti-phospho-histone H2A.X (Ser139) antibody (cat. no. 9718; 1:100)
or rabbit anti-p65 antibody (cat. no. sc-372; 1:100) at 4°C
overnight followed by the anti-rabbit-FITC secondary antibody (cat.
no. sc-2359; 1:200) for 1 h at room temperature in the dark. The
cells were mounted in mounting medium (Sigma-Aldrich; Merck KGaA)
with DAPI for 15 min at room temperature in the dark and sealed.
Immunofluorescence was observed and recorded immediately on a Zeiss
Confocal Laser Scanning Microscope (magnification, ×400; Carl Zeiss
AG, Oberkochen, Germany).
Comet assay
To detect smaller amounts of damage, including
single and double-stranded breaks, the alkaline comet assay was
used. The assay was performed following instructions from Trevigen,
Inc. Briefly, cells treated with loperamide were harvested and
combined at 1×105/ml with molten LMAgarose (at 37°C) at
a ratio of 1:10 (v/v), and then transferred to comet slides. The
slides were immersed in 4°C Lysis Solution (Trevigen, Inc.
Gaithersburg, MD, USA) for 1 h and later in freshly prepared
alkaline unwinding solution (pH>13.0) for 20 min at room
temperature. The slides were then placed in an electrophoresis
slide tray with the alkaline electrophoresis solution (200 mM NaOH,
1 mM EDTA) and electrophoresis was performed under the conditions
of power up to 21 V for 30 min. The cells in the circle of dried
agarose were stained with diluted SYBR-Green (BioTeke Corporation,
Beijing, China; cat. no. 9718; EP1601-1) for 30 min at room
temperature and then viewed on a Zeiss Confocal Laser Scanning
Microscope (Carl Zeiss AG; magnification, ×400). The percentage of
DNA in the tail and tail length are two common descriptors of DNA
damage for the alkaline comet assay, which were analyzed by the
Comet Assay Software Project (CaspLab-Comet Assay Software, CASPLab
1.0.1, http://casplab.com/) (22). At least 50 randomly selected cells
were analyzed per sample.
Statistical analysis
Data were confirmed through three independent
experiments. The results are expressed as the mean ± standard
deviation. The differences between groups were assessed by unpaired
t-test, or one-way ANOVA with a Least Significant Difference test,
depending on the number of groups compared. The statistical
analysis was performed by SPSS 16.0 software (SPSS, Inc., Chicago,
IL, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
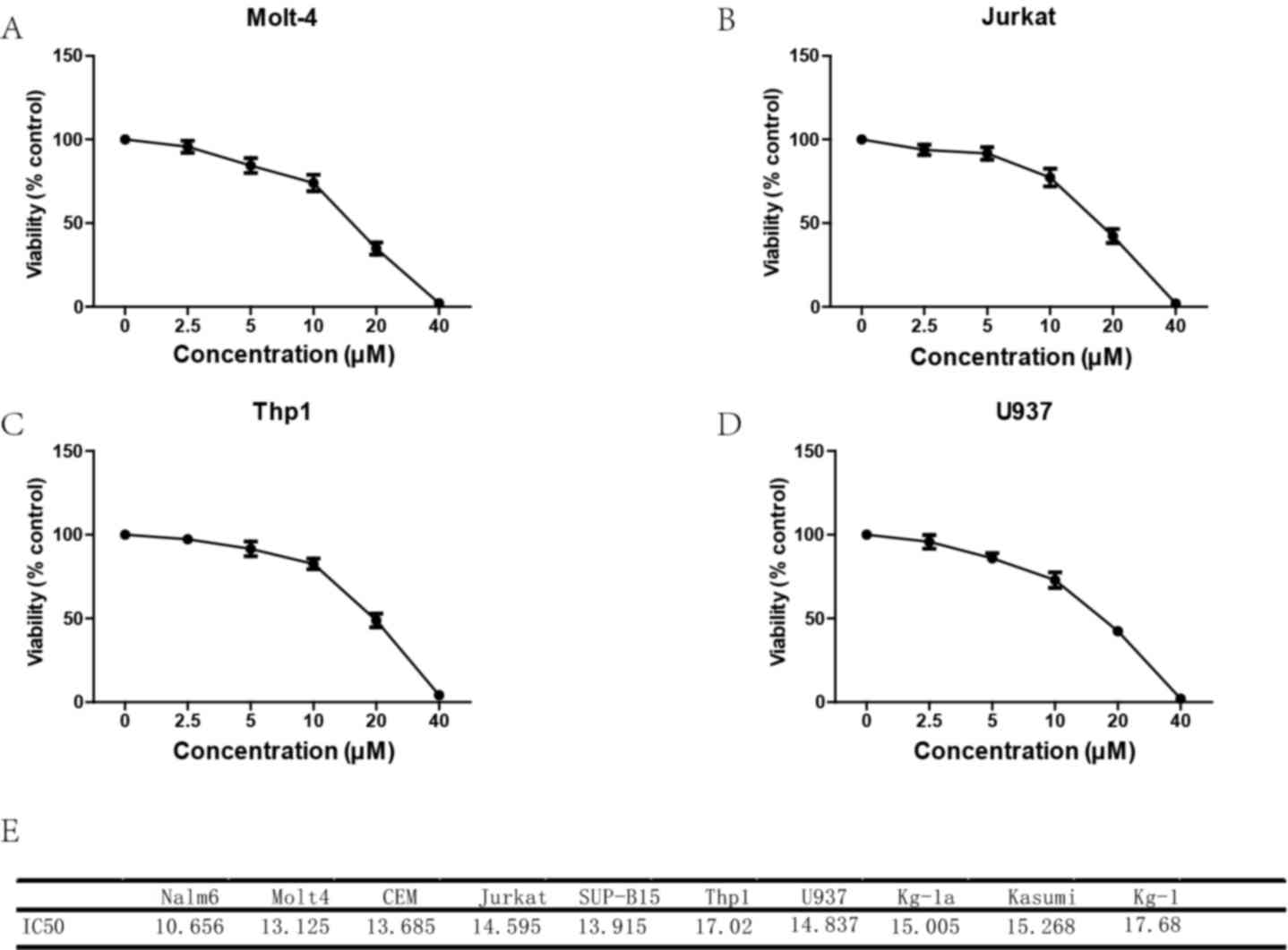
Loperamide inhibits growth of leukemia
cell lines
To evaluate the effect of loperamide on leukemia
cells, the MTT assay was performed in 5 AML cell lines and 5 ALL
cell lines with different concentrations of loperamide. Loperamide
significantly inhibited the growth of leukemia cell lines in a
dose-dependent manner between 2.5 and 40 µM (Fig. 1). Loperamide (40 µM) inhibited growth
of almost all the leukemia cells, to a cell viability of <1%.
The IC50 values of the AML cell lines at 24 h were
calculated, ranging between 14.83 and 17.68 µM, while for ALL cell
lines, the IC50 values ranged between 10.66 and 14.60 µM
(Fig. 1). It was then determined
whether loperamide inhibits the growth of these leukemia cell lines
in a time-dependent manner. No significant difference in cytotoxic
effect was apparent following treatment for 24, 48 or 72 h in AML
and ALL cell lines, which indicated that loperamide exerted its
inhibitory effect on leukemia cell lines to the maximal extent at
24 h (data not shown).
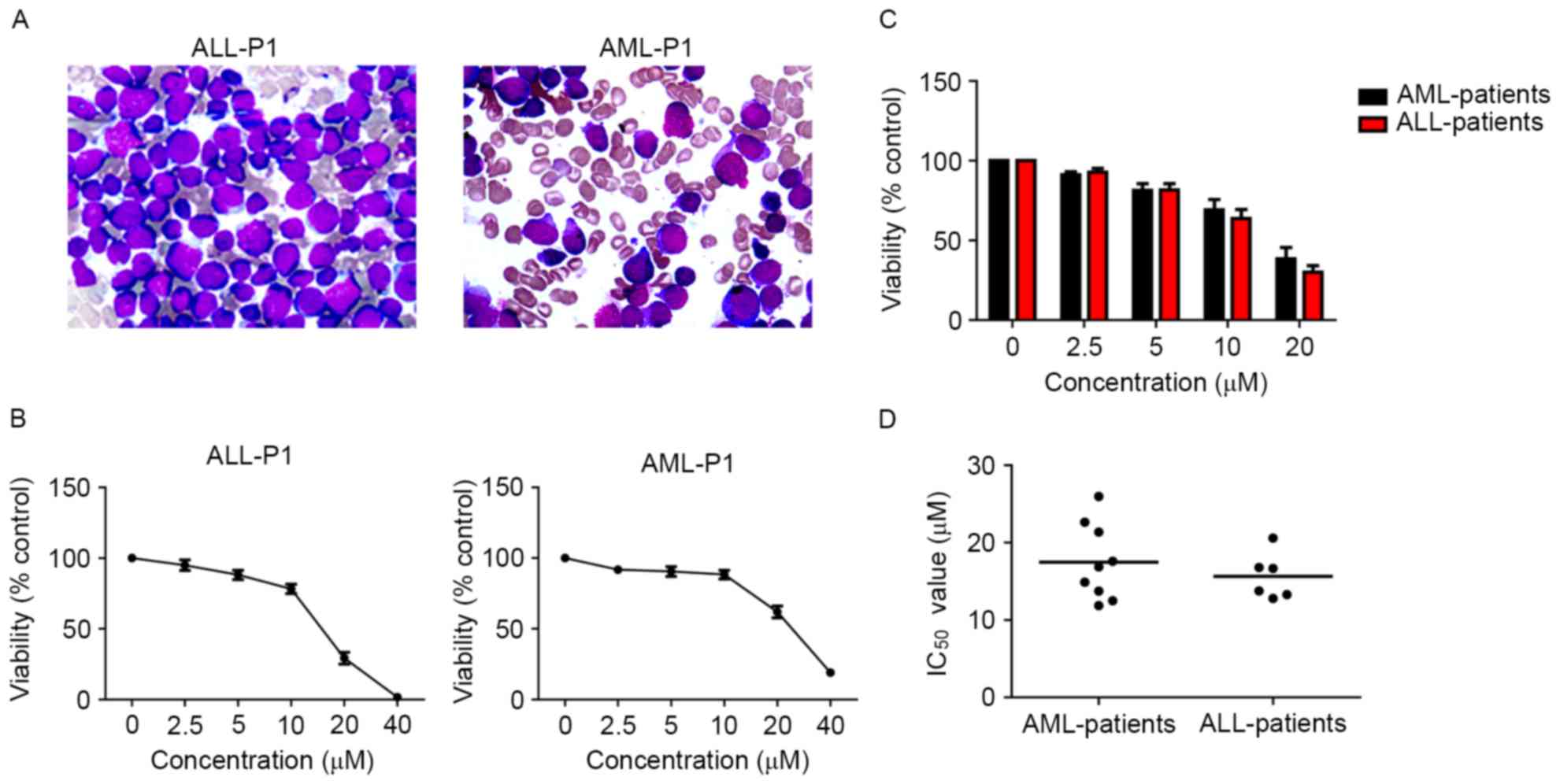
Loperamide inhibits the growth of
primary cells from patients with acute leukemia
Primary cells more accurately reflect the biological
properties of tumors from patients. Primary leukemia cells were
isolated from patients with ALL and AML. The subtypes of patients
with AML were: 1 with M1; 2 with M2; 2 with M4; 3 with M5; and 1
with chronic myelogenous leukemia-blast crisis/myeloid (M), while
there were four B-ALL and one T-ALL patients. Gene mutations and
chromosome karyotype abnormalities were present in 10 patients. The
detailed clinical characteristics of the patients are described in
Table I. The inhibitory effect of
loperamide on primary cells was then assessed and it was found that
loperamide could reduce the survival of the primary cells from AML
and ALL primary cells, even for those with complex chromosomal
abnormalities and adverse genetic mutations, in a dose-dependent
manner (Fig. 2). The IC50
values of AML primary cells at 24 h were 11.87–25.96 µM, while for
ALL primary cells, the IC50 values were 12.78–20.58 µM
(Fig. 2). Furthermore, no significant
difference was observed for the inhibitory ratio of loperamide on
primary cells in the time studied (data not shown).
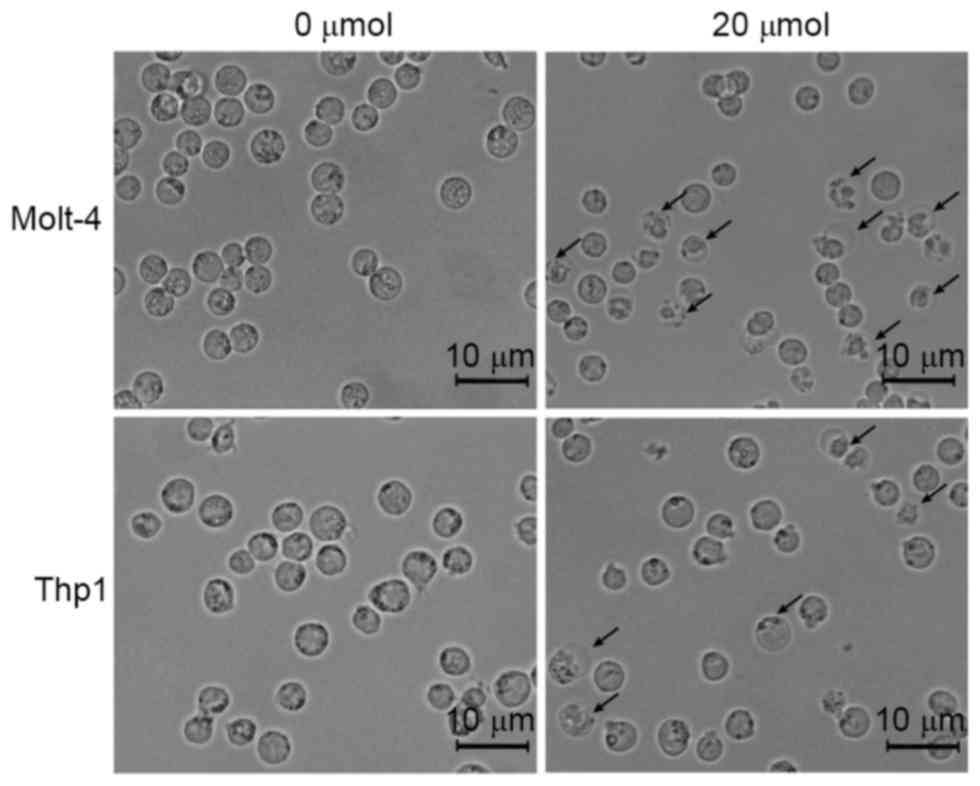
Loperamide induces apoptosis in
leukemia cell lines and primary cells
To investigate the potential mechanism of the
inhibitory effects of loperamide on leukemia cells, the
morphological changes in cells treated with 20 µM loperamide were
observed. As shown in Fig. 3, nuclear
shrinkage and membrane vacuolization were observed in Molt-4 and
Thp1 cells following 20 µM loperamide treatment, which are features
of cell apoptosis. These results indicated that the inhibitory
effects of loperamide on leukemia cells may be exerted via cell
apoptosis.
To confirm that the antitumor role of loperamide is
mediated by cell apoptosis, cells treated with loperamide were
stained with Annexin V and 7AAD+ and analyzed by flow
cytometry. The results showed a dose-dependent increase in cell
apoptosis for cells treated with various concentrations of
loperamide. In the absence of loperamide treatment, cell apoptosis
was <10%. At a concentration of 20 µM, the apoptotic cell
percentage in Thp1 was 62.3±5.7%, while it was 78.6±3.7% in Molt4
cells (Fig. 4A and B). The apoptotic
percentage was 73.9±4.7% for ALL-P1 cells and 45.8±4.15% for AML-P1
cells (Fig. 4A and B). To determine
the mechanisms of apoptosis induced by loperamide, western blot
analysis was performed to assess caspase-3 activity and PARP,
Bcl-2, Bax and Mcl-1, which are important in inducing apoptosis
(23,24). As shown in Fig. 4C and D, the expression of
cleaved-caspase-3 and cleaved-PARP increased in a dose-dependent
manner with loperamide treatment. To identify the upstream
mechanism of apoptosis induced by loperamide in leukemia cells, the
activation of the nuclear factor (NF)-κB, c-Jun N-terminal kinase
(JAK)-STAT3 and phosphoinositide 3-kinase (PI3K)/Akt pathways was
measured, which perform important roles in apoptosis (25–27).
Western blot analysis was performed to detect the expression of
STAT3, p-STAT3, Akt and p-Akt, and immunofluorescence staining was
performed to test the cellular localization of p65. The expression
of STAT3, p-STAT3, Akt and p-Akt and the cellular localization of
p65 was not changed following loperamide treatment (data not
shown). These data indicated that loperamide did not trigger the
activation of the NF-κB, JAK-STAT or PI3K/Akt pathways. Although
the expression of Bcl-2 and Bax was not significantly changed,
Mcl-1, an important anti-apoptosis protein (28), was downregulated following loperamide
treatment. These results indicated that loperamide inhibited the
proliferation of leukemia cells and induced cell apoptosis.
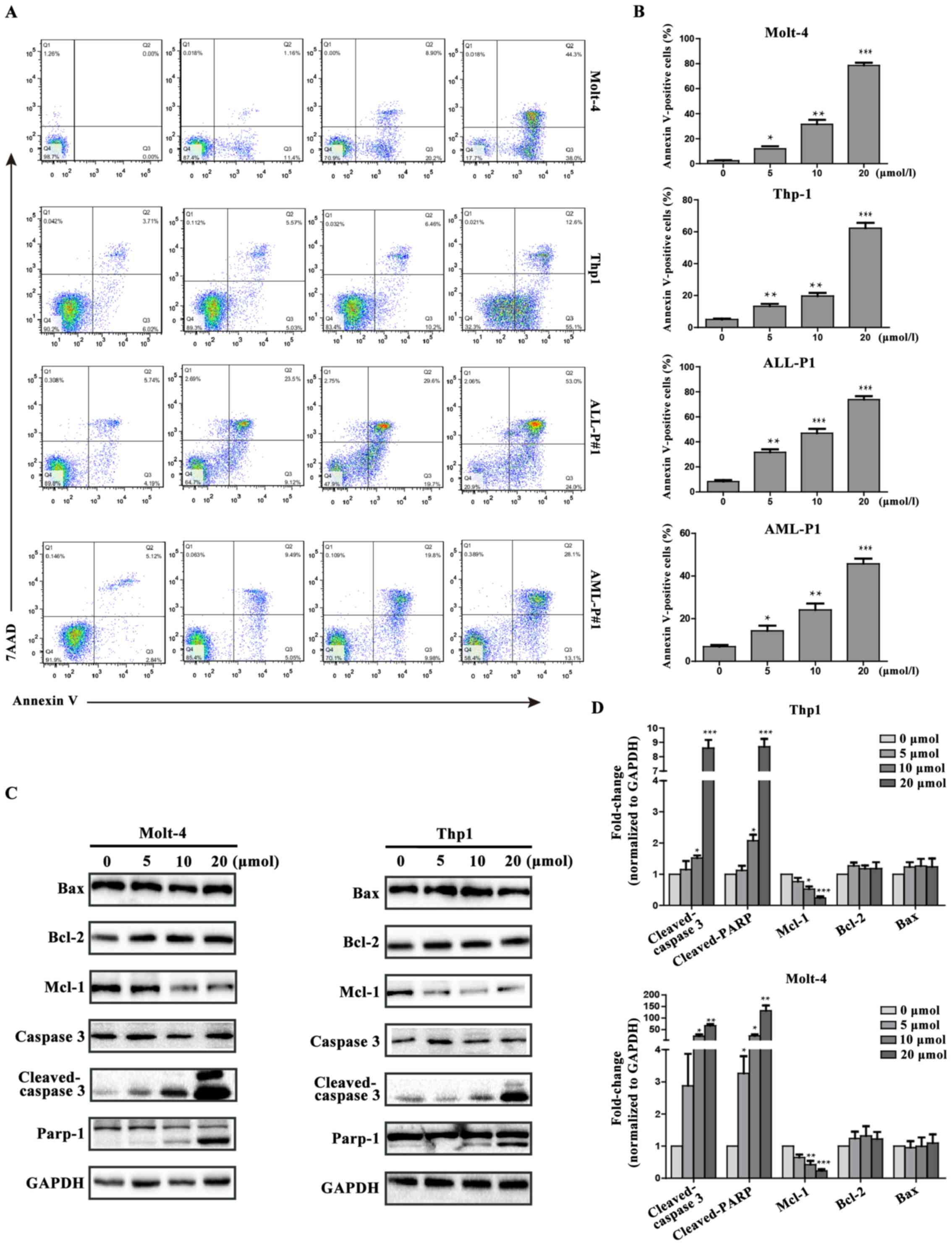 | Figure 4.Loperamide induces apoptosis in
leukemia cell lines and primary leukemia cells from patients in a
concentration-dependent manner. (A) Molt-4, Thp1, ALL-P1 and AML-P1
cells were treated with various concentrations of loperamide for 24
h and apoptosis was determined by flow cytometry. (B) The
percentage of cell apoptosis following treatment with loperamide.
(C) Western blot analysis of caspase-3, cleaved-caspase-3, PARP,
Bcl-2, Bax and Mcl-1 in Molt-4 and Thp1 cells treated with
loperamide for 24 h. (D) Band quantification is shown as the fold
change from control. The results are presented as the mean ±
standard deviation of three independent experiments. *P<0.05;
**P<0.01; ***P<0.001 vs. control group. PARP,
poly(ADP-ribose) polymerase; Bcl-2, B-cell lymphoma-2; Bax,
Bcl-2-associated X protein; Mcl-1, myeloid cell leukemia 1; acute
lymphocytic leukemia-patient 1; AML-P1, acute myeloid
leukemia-patient 1. |
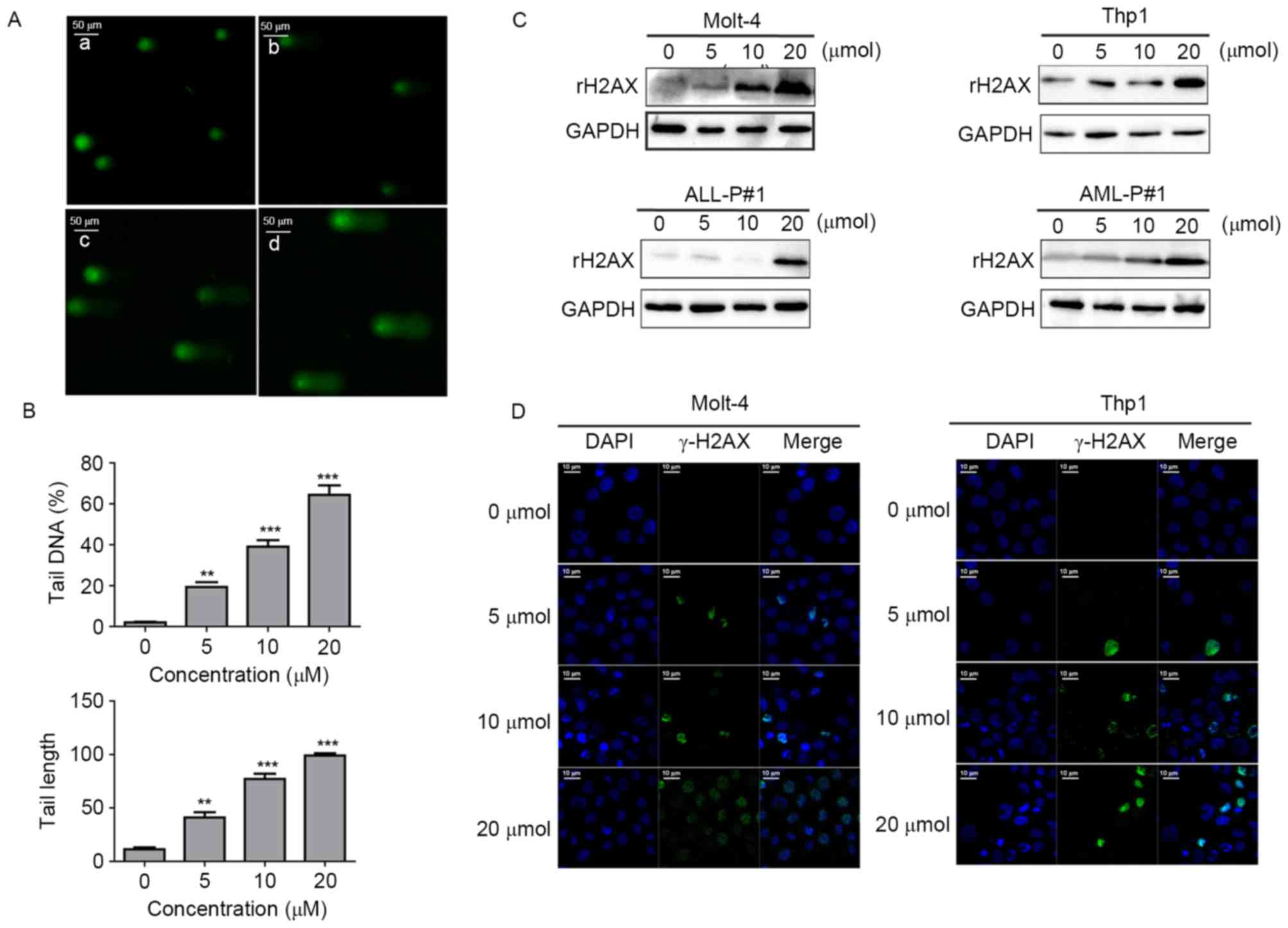
Loperamide induces DNA damage and
activation of the ATM-Chk2 pathway in leukemia cells
Considering the specific changes in Mcl-1 and
cleaved PARP expression and the significant role of Mcl-1 and
cleaved PARP in DNA damage repair, it was examined whether
loperamide induced DNA damage (29,30). The
comet assay detects DNA damage in vitro (31). It was revealed that the length of the
tail and the percentage of cells with a long tail increased as the
concentration increased, which demonstrated the DNA damage-inducing
effect of loperamide in a concentration-dependent manner (Fig. 5A). As demonstrated by histogram
statistics, at a concentration of 20 µM, the percentage DNA in the
tail (measured as the percentage of total DNA in the tail) reached
64.3±8.1%, while the tail length was 99±3.6 µm (Fig. 5B). The phosphorylation level of H2Ax,
a sensitive DNA damage response marker, was then determined by
western blotting and immunofluorescence staining, and the rH2Ax
levels were found to be markedly elevated following treatment with
20 µM loperamide for 24 h (Fig. 5C).
The results of immunofluorescence staining showed that the
intensity of rH2Ax was enhanced significantly (Fig. 5D). These results suggested that
loperamide increased the expression of phosphorylated H2Ax and the
length of the tail, causing DNA damage in leukemia.
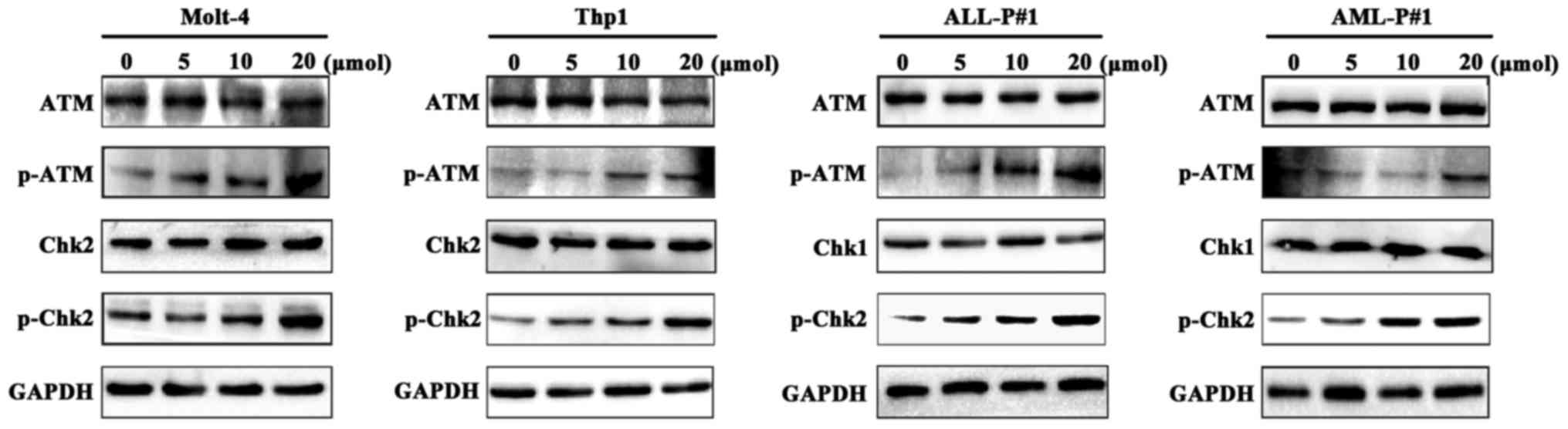
The ATM-Chk2 pathway performs an important role in
the DNA damage response (32). It was
investigated whether loperamide induced DNA damage by activating
the ATM-Chk2 pathway in leukemia cells. The results showed that the
phosphorylation level of ATM (Ser1981) in the Molt-4 and Thp1 cell
lines and primary cells treated with 5–20 µM loperamide increased
compared with cells treated with DMSO (Fig. 6). Chk2, the downstream effector of ATM
(32), was also examined by western
blotting. The phosphorylation level of Chk2 (Thr68) was also
increased in a dose-dependent manner (Fig. 6). These data indicated that loperamide
induced DNA damage and activated the ATM-Chk2 pathway in leukemia
cells.
Discussion
Loperamide, first synthesized in 1973, is widely
prescribed and used as an antidiarrheal agent around the world
(33). Blocking the µ-opioid receptor
of the gastrointestinal tract and antagonizing calmodulin are the
two main molecular mechanisms for the antidiarrheal effect of
loperamide (34–37). In previous years, loperamide was also
found to possess anti-neoplastic activity in a variety of tumors
in vitro, including liver, breast, lung and canine cancers
(9,10). However, the specific mechanism of its
antitumor effect remains unclear. To the best of our knowledge, the
present study investigated the effects of loperamide treatment on
the growth of leukemia cells for the first time, and demonstrated
that loperamide effectively inhibited the growth of leukemia cell
lines and primary cells from patients with leukemia in a
dose-dependent manner by inducing cell apoptosis. DNA damage
triggered by loperamide treatment was involved in the molecular
mechanism of its anti-leukemia effects. Furthermore, the ATM-Chk2
signaling pathway in leukemia cells was activated and associated
with the DNA damage following loperamide treatment. These findings
uncover the anti-leukemia role and mechanism of loperamide. Thus,
loperamide, used widely to treat leukemia, possesses therapeutic
potential as an anti-leukemic agent.
The cancer cell lines used in preclinical studies
have a vital role in the study of biological mechanisms of cancer
and high throughput screening of effective antitumor drugs.
However, gene aberrations in cancer cell lines occurring with
increasing passage number has led to misrepresentative clinical
scenarios, and thus limits their clinical correlation (38,39). In a
previous study, Gillet et al (38) investigated the multidrug resistance
transcriptome of six cancer types in established cancer cell lines
and clinical samples, and found no association between established
cancer cell lines and clinical samples. Furthermore, considering
that there are no two genetically identical samples even from the
same patient, it is difficult for a small number of cancer cell
lines to represent the genetic and epigenetic variation of millions
of patients with cancer (40).
Therefore, driven by personalized medicine, it is necessary to use
primary tumor models to test the effectiveness of antitumor drugs
to ensure the authenticity of patient-dependent tumor variability.
In the present study, primary leukemia cells from 9 patients with
AML and 6 patients with ALL were isolated. Notably, gene mutation
and chromosome karyotype abnormalities existed in 10 patients,
representing the patient-dependent tumor variability. Therefore,
the result that the cytotoxic effect of loperamide was also
observed in primary cells from patients with leukemia supported the
anti-leukemia potential of loperamide.
Cell apoptosis, also termed programmed cell death,
is a process occurring primarily through an evolutionarily
conserved form of cell suicide and performs a significant role in
animal development (41,42). The dysregulation of this process is
involved in the pathogenesis of a panel of human diseases,
including cancer (41). Therefore,
inducing tumor cell apoptosis is the pathway by which the majority
of antitumor agents take effect (43). To investigate the cytotoxic mechanism
of loperamide on leukemia cells, leukemia cells were treated with
loperamide for 24 h, and morphological changes were observed and
flow cytometric cell apoptosis analyses were performed. The results
supported the hypothesis that loperamide exerts a cytotoxic effect
via inducing cell apoptosis. Caspase-3 activation followed by
caspase-3-mediated cleavage of PARP has a critical role in
apoptosis (44,45). The results determined by western
blotting of a dose-dependent generation of cleaved caspase-3 and
cleaved-PARP validated the assumption of the apoptosis-inducing
effect of loperamide.
In addition to engaging in apoptosis, PARP is also
involved in and has a vital effect in DNA damage repair by
recognizing DNA strand breaks and acting as a critical regulatory
component (29). Furthermore,
downregulation of Mcl-1 expression following loperamide treatment,
together with evidence that Mcl-1 regulates the DNA damage response
and overexpression promotes resistance to DNA damage, suggested the
possible DNA damage-inducing effect of loperamide (30,46). In
the present study, the comet assay, which is a sensitive method of
evaluating DNA damage in cells, showed a long tail length at the
concentration of 20 µM loperamide, evidence of DNA damage induced
by loperamide treatment. The phosphorylation of histone H2A acts as
a sensitive marker of DNA damage, which forms nuclear foci at sites
of DNA damage and behaves as a signal to recruit other repair
proteins (14). The results of
western blotting and immunofluorescence staining indicated that the
molecular sensor of DNA damage, rH2ax, increased in a
dose-dependent manner following treatment with loperamide. To
investigate the signaling pathway activated by DNA damage, the
ATM-Chk2 pathway was investigated by western blotting. ATM is a
kinase belonging to the PI3K signaling family and is commonly
activated by double-strand DNA breaks (DSBs) (47). Once triggered by DSBs, ATM is
activated and phosphorylates its downstream signaling proteins,
including Chk2, which serves as a signal transducer and effector
that is involved in the process of apoptosis initiation (32). Western blotting assays indicated that
ATM was activated via its phosphorylation at Ser1981 subsequent to
DNA damage being induced by loperamide treatment and subsequently
initiating the downstream molecular Chk2. In addition, the
alteration of numerous signaling pathways that perform crucial
roles in tumor survival and apoptosis was also determined,
including the NF-κB, JAK-STAT and PI3K/Akt pathways (25–27). The
results revealed that these pathways were not activated (Fig. 3).
To conclude, the present study demonstrated for the
first time that loperamide, an old drug used as an antidiarrheal
agent, has therapeutic potential as an anti-acute leukemia agent.
Loperamide effectively inhibits the growth of leukemia cell lines
and primary leukemia cells through inducing cell apoptosis. In
addition, a new antitumor mechanism of loperamide was identified in
the present study: DNA damage. Furthermore, the ATM-Chk2 pathway
activated by DNA damage in response to loperamide treatment is
found in leukemia cell lines and primary leukemia cells. Therefore,
the present study provides new insights into the therapeutic
potential and antitumor mechanism of loperamide in leukemia.
Acknowledgements
The present study was supported by the Leukemia
Research Innovative Team of Zhejiang Province (grant no.
2011R50015), Medical and Health Research Foundation of Zhejiang
Province (grant no. 2017KY369).
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eryilmaz E and Canpolat C: Novel agents
for the treatment of childhood leukemia: An update. OncoTargets
Ther. 10:3299–3306. 2017. View Article : Google Scholar
|
|
4
|
Showel MM and Levis M: Advances in
treating acute myeloid leukemia. F1000prime Rep. 6:962014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turpin F, Tubiana-Hulin M, Meeus L, Goupil
A, Berlie J and Clavel B: Complications of antitumor and
antileukemic chemotherapy. 1. Sem Hop. 58:2047–2057. 1982.(In
French).
|
|
6
|
Hatzimichael E and Tuthill M:
Hematopoietic stem cell transplantation. Stem Cells Cloning.
3:105–117. 2010.PubMed/NCBI
|
|
7
|
Cox GJ, Matsui SM, Lo RS, Hinds M, Bowden
RA, Hackman RC, Meyer WG, Mori M, Tarr PI, Oshiro LS, et al:
Etiology and outcome of diarrhea after marrow transplantation: A
prospective study. Gastroenterology. 107:1398–1407. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Geller RB, Gilmore CE, Dix SP, Lin LS,
Topping DL, Davidson TG, Holland HK and Wingard JR: Randomized
trial of loperamide versus dose escalation of octreotide acetate
for chemotherapy-induced diarrhea in bone marrow transplant and
leukemia patients. Am J Hematol. 50:167–172. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong XW, Xu YH, Chen XL and Wang YX:
Loperamide, an antidiarrhea drug, has antitumor activity by
inducing cell apoptosis. Pharmacol Res. 65:372–378. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Regan RC, Gogal RM Jr, Barber JP,
Tuckfield RC, Howerth EW and Lawrence JA: Cytotoxic effects of
loperamide hydrochloride on canine cancer cells. J Vet Med Sci.
76:1563–1568. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smart DJ, Halicka HD, Schmuck G, Traganos
F, Darzynkiewicz Z and Williams GM: Assessment of DNA double-strand
breaks and gammaH2AX induced by the topoisomerase II poisons
etoposide and mitoxantrone. Mutat Res. 641:43–47. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leoni LM, Bailey B, Reifert J, Bendall HH,
Zeller RW, Corbeil J, Elliott G and Niemeyer CC: Bendamustine
(Treanda) displays a distinct pattern of cytotoxicity and unique
mechanistic features compared with other alkylating agents. Clin
Cancer Res. 14:309–317. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Attikum H and Gasser SM: The histone
code at DNA breaks: A guide to repair? Nat Rev Mol Cell Biol.
6:757–765. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou BB, Chaturvedi P, Spring K, Scott SP,
Johanson RA, Mishra R, Mattern MR, Winkler JD and Khanna KK:
Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by
inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol
Chem. 275:10342–10348. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gatei M, Sloper K, Sorensen C, Syljuäsen
R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J and
Khanna KK: Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent
phosphorylation of Chk1 on Ser-317 in response to ionizing
radiation. J Biol Chem. 278:14806–14811. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lavin MF, Birrell G, Chen P, Kozlov S,
Scott S and Gueven N: ATM signaling and genomic stability in
response to DNA damage. Mutat Res. 569:123–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Niida H and Nakanishi M: DNA damage
checkpoints in mammals. Mutagenesis. 21:3–9. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nijhawan D, Fang M, Traer E, Zhong Q, Gao
W, Du F and Wang X: Elimination of Mcl-1 is required for the
initiation of apoptosis following ultraviolet irradiation. Genes
Dev. 17:1475–1486. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sabattini E, Bacci F, Sagramoso C and
Pileri SA: WHO classification of tumours of haematopoietic and
lymphoid tissues in 2008: An overview. Pathologica. 102:83–87.
2010.PubMed/NCBI
|
|
22
|
Końca K, Lankoff A, Banasik A, Lisowska H,
Kuszewski T, Góźdź S, Koza Z and Wojcik A: A cross-platform public
domain PC image-analysis program for the comet assay. Mutat Res.
534:15–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Catlett-Falcone R, Landowski TH, Oshiro
MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L,
Fernández-Luna JL, Nuñez G, et al: Constitutive activation of Stat3
signaling confers resistance to apoptosis in human U266 myeloma
cells. Immunity. 10:105–115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martelli AM, Nyåkern M, Tabellini G,
Bortul R, Tazzari PL, Evangelisti C and Cocco L: Phosphoinositide
3-kinase/Akt signaling pathway and its therapeutical implications
for human acute myeloid leukemia. Leukemia. 20:911–928. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yao H, Mi S, Gong W, Lin J, Xu N, Perrett
S, Xia B, Wang J and Feng Y: Anti-apoptosis proteins Mcl-1 and
Bcl-xL have different p53-binding profiles. Biochemistry.
52:6324–6334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Molinete M, Vermeulen W, Bürkle A,
Ménissier-de Murcia J, Küpper JH, Hoeijmakers JH and de Murcia G:
Overproduction of the poly(ADP-ribose) polymerase DNA-binding
domain blocks alkylation-induced DNA repair synthesis in mammalian
cells. EMBO J. 12:2109–2117. 1993.PubMed/NCBI
|
|
30
|
Jamil S, Stoica C, Hackett TL and Duronio
V: MCL-1 localizes to sites of DNA damage and regulates DNA damage
response. Cell Cycle. 9:2843–2855. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Swain U and Subba Rao K: Study of DNA
damage via the comet assay and base excision repair activities in
rat brain neurons and astrocytes during aging. Mech Ageing Dev.
132:374–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou BB and Elledge SJ: The DNA damage
response: Putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stokbroekx RA, Vandenberk J, Van Heertum
AH, Van Laar GM, van der Aa MJ, Van Bever WF and Janssen PA:
Synthetic antidiarrheal agents.
2,2-Diphenyl-4-(4′-aryl-4′-hydroxypiperidino)butyramides. J Med
Chem. 16:782–786. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Awouters F, Niemegeers CJ and Janssen PA:
Pharmacology of antidiarrheal drugs. Annu Rev Pharmacol Toxicol.
23:279–301. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Clay GA, Mackerer CR and Lin TK:
Interaction of loperamide with [3H]naloxone binding sites in guinea
pig brain and myenteric plexus. Mol Pharmacol. 13:533–540.
1977.PubMed/NCBI
|
|
36
|
Harper JL, Shin Y and Daly JW: Loperamide:
A positive modulator for store-operated calcium channels? Proc Natl
Acad Sci USA. 94:pp. 14912–14917. 1997; View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Merritt JE, Brown BL and Tomlinson S:
Loperamide and calmodulin. Lancet. 1:2831982. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gillet JP, Calcagno AM, Varma S, Marino M,
Green LJ, Vora MI, Patel C, Orina JN, Eliseeva TA, Singal V, et al:
Redefining the relevance of established cancer cell lines to the
study of mechanisms of clinical anti-cancer drug resistance. Proc
Natl Acad Sci USA. 108:pp. 18708–18713. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gazdar AF, Gao B and Minna JD: Lung cancer
cell lines: Useless artifacts or invaluable tools for medical
science? Lung cancer. 68:309–318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kirk R: Genetics: Personalized medicine
and tumour heterogeneity. Nat Rev Clin Oncol. 9:2502012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jacobson MD, Weil M and Raff MC:
Programmed cell death in animal development. Cell. 88:347–354.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Westhoff MA, Marschall N and Debatin KM:
Novel approaches to apoptosis-inducing therapies. Adv Exp Med Biol.
930:173–204. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Salvesen GS and Dixit VM: Caspases:
Intracellular signaling by proteolysis. Cell. 91:443–446. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Simbulan-Rosenthal CM, Rosenthal DS, Iyer
S, Boulares AH and Smulson ME: Transient poly(ADP-ribosyl)ation of
nuclear proteins and role of poly(ADP-ribose) polymerase in the
early stages of apoptosis. J Biol Chem. 273:13703–13712. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mills JR, Malina A and Pelletier J:
Inhibiting mitochondrial-dependent proteolysis of Mcl-1 promotes
resistance to DNA damage. Cell Cycle. 11:88–98. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee JH and Paull TT: Activation and
regulation of ATM kinase activity in response to DNA double-strand
breaks. Oncogene. 26:7741–7748. 2007. View Article : Google Scholar : PubMed/NCBI
|