Introduction
Acute heat stroke causes thermal injury of cells and
changes to the circulatory system, which can in turn trigger
extensive tissue injury to the blood coagulation system, the liver,
heart and kidneys, and, most importantly, to the central nervous
system (CNS) (1). According to the
report of a cohort study covering 16 emergency departments, only
57% patients survived following heat stroke during the August 2003
heat wave in Paris (2); even
following discharge from the hospital, survivors continued to
suffer neurological damage during the 1995 heat wave in Chicago
(3). The mortality rate may increase
as a result of global warming and the expected universal increase
in the intensity and frequency of heat waves (4,5).
Combining prior studies, a previous review reported
that high temperatures can cause certain CNS malformations, and
that heat induces programmed cell death, which causes teratogenic
damage to the developing brain (6).
However, the mediation between metabolic changes and cell apoptosis
remains poorly characterized.
Mitogen-activated protein kinase (MAPK) signaling
pathways are involved in heat-stress-induced organ injury and human
umbilical vein endothelial cell (HUVEC) apoptosis (7,8).
Activation of MAPK signaling pathways in responding to stressors,
such as oxidative stress, also induces neuronal apoptosis through
the mitochondria-dependent pathway (9). The representative mammalian MAPK
pathways, which include p38 MAPK, c-Jun N-terminal kinase (JNK),
and extracellular signal-regulated kinase 1/2 (ERK1/2), are
concatenated via three consecutive phosphorylation events, which
are mediated by a MAPK, a MAPK kinase, and a MAPK kinase kinase
(10–12). Substrates with distinct functions are
phosphorylated by MAPKs, including other protein kinases that are
recognized as MAPK-activated protein kinases (MAPKAPKs; also known
as MKs) (13). MK5 and MK2 exhibit
considerable structural, functional and evolutional similarities,
and are substrates for p38 (14,15). The
function of the p38 stress-activated protein kinase/MK2 axis in
apoptosis and inflammation has been recorded in detail (16), but whether MK2 and MK5 participate in
heat-stress-induced neuron cell apoptosis remains unclear.
Reactive oxygen species (ROS) are involved in cell
apoptosis in response to thermal injury (17). The overproduction of ROS may cause
necrosis or apoptosis in various pathological conditions, including
ischemia (18). To assess the roles
of MK5 and MK2 in cell apoptosis mediated by heat-stress-induced
ROS accumulation, the present study utilized the application of
adenoviral dominant-negative and -positive constructs of MK2 and
MK5 into F98 cells. Dominant-negative MK2 [Ad-MK2(A)], or a
specific inhibitor of MK2, impeded heat stress-induced ROS
production and cell apoptosis, while Ad-MK5(A) did not. These
findings indicate that p38 MAPK-MK2 plays a part in suppressing
apoptosis following heat stress. Hence, p38 MAPK-MK2 signaling
could be a treatment target for use in improving the outcomes of
heatstroke patients.
Materials and methods
Cell culture and treatments
Rat malignant glioma F98 cells were procured from
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). F98 cells were incubated in Dulbecco's modified
Eagle's medium containing 100 U/ml of penicillin, 100 µg/ml of
streptomycin (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and 10% (v/v) fetal bovine serum (Invitrogen; Thermo
Fisher Scientific, Inc.). The temperature was controlled at 37°C
and the atmosphere was humidified, comprising 95% air and 5%
CO2. Culture dishes were incubated in a circulating
water bath for 60 min at 43±0.5°C for heat stress, or at 37±0.5°C
for control. Cells were further cultured at 37°C for the designated
time following the replacement of culture media with fresh
media.
Infection with adenoviral
constructs
Generation of recombinant adenoviral constructs was
performed as previously described (19,20).
Briefly, the present study used Ad-MKK6(E), Ad-MK2(E), Ad-MK5(E),
the constitutively active forms of MKK6, MK2 and MK5 encoded by
recombinant adenovirus. The present study also used Ad-MKK6(A),
Ad-MK2(A) and Ad-MK5(A), the constitutively negative forms of MKK6,
MK2 and MK5. These adenoviruses were constructed by Vigene
Biosciences (Jinan, China). F98 cells were seeded in 35-mm Petri
dishes (1×105/dish), 72 h prior to infection
[103 inclusion-forming units (IFU)/cell], and were
exposed to heat stress after a 24-h incubation period.
Flow cytometric analysis of cell
apoptosis using Annexin V-fluorescein isothiocyanate (FITC)/PI
staining
The proportions of apoptotic cells in each group
were analyzed by flow cytometry using an Annexin V-FITC apoptosis
kit (Shanghai Lianke Biology Co., Ltd., Shanghai, China), according
to the manufacturer's protocol. Approximately 1×106
cells were collected, and were washed with ice-cold PBS prior to
resuspension in binding buffer containing 5 µl FITC-Annexin V and
10 µl propidium iodide. All samples were analyzed using a flow
cytometer (FACSCanto™ II; BD Biosciences, San Jose, CA, USA) and
analyzed using FlowJo software version 9.0 (Tree Star, Inc.,
Ashland, OR, USA).
Assay for caspase-3 activity
Caspase-3 activity was determined using a caspase-3
activity assay kit, according to the manufacturer's protocol
(Biovision, Inc., Milpitas, CA, USA). Briefly, the cells were lysed
in caspase-3 sample lysis buffer (Biovision, Inc.). The homogenates
were then centrifuged at 10,000 × g at 4°C for 10 min and the
supernatant was collected for protein estimation using
bicinchoninic acid for the caspase-3 assay. The cell lysates were
then exposed to the DEVD substrate conjugate provided in the kit
for 1 h at 37°C. The sample was measured using an automatic
microplate reader (SpectraMax M5; Molecular Devices, LLC,
Sunnyvale, CA, USA) at an excitation of 400 nm and emission of 505
nm.
Measurements of ROS
ROS levels were assessed using
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Molecular
Probes; Thermo Fisher Scientific, Inc.), which is converted to
highly fluorescent 2′,7′-dichlorofluorescein (DCF) in the presence
of ROS. F98 cells (3×105) were incubated at 43°C for 1 h
before incubation at 37°C for 0, 3, 6 or 12 h; cells were then
incubated with 5 mM DCF-DA at 37°C for 30 min in darkness.
H2O2 was used as a positive control;
H2O2 (200 µM) was added to F98 cells and
incubated at 37°C for 1 h. In addition, cells were pretreated with
or without MnTBAP for 1 h at 37°C and were then incubated at 43°C
(HS) or 37°C (control) for 60 min. Analysis of the fluorescence
intensity of DCF was subsequently performed using a flow cytometer,
and images were obtained via laser scanning confocal microscopy
(OLS4100; magnification, ×40; Olympus Corporation, Tokyo, Japan) at
an excitation of 488 nm and emission of 530 nm.
Western blot analysis
F98 cells (~1×106) were maintained at
37°C (control) or 43°C (HS) for 60 min and further incubated for 0,
3, 6 or 12 h at 37°C. Cells were pretreated with CMPD-1 (10 µM;
cat. no. 263847-55-8; Tocris Bioscience, Bristol, UK), MnTBAP (10
µM; cat. no. ab141496; Abcam, Cambridge, MA, USA), SP600125 (10 µM;
cat. no. ab120065; Abcam), SB203580 (5 µM; cat. no. ab120162;
Abcam), PD98059 (10 µM; cat. no. ab120234; Abcam), SB202474 (10 µM;
cat. no. IMA1013; Jingke Science and Technology Co., Ltd.,
Shanghai, China), the specific inhibitors of MK2, ROS, JNK, p38,
ERK and the negative control, respectively, at 37°C for 30 min.
Cells were then exposed to heat stress or control heat treatment,
followed by recovery at 37°C for 12 h. Control cells were treated
with PBS instead of LPS and were constantly incubated at 37°C.
Cells were homogenized in radioimmunoprecipitation assay lysis
buffer with phenylmethylsulfonyl fluoride (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). Following centrifugation at 14,000 × g
at 4°C for 10 min, the supernatants were used for western blot
analysis. Protein concentration was determined using a
Bicinchoninic Acid Protein Assay kit (Thermo Fisher Scientific,
Inc.). Proteins (20 µg/well) were separated by SDS-PAGE using 10%
SDS polyacrylamide gels and transferred onto polyvinylidene
difluoride membranes. Membranes were blocked with blocking solution
(5% skimmed milk diluted with PBS) at room temperature for 2 h,
followed by incubation with primary antibodies. The following
rabbit primary antibodies were used at a 1:2,000 dilution: GAPDH
(cat. no. ab70699; Abcam), phosphorylated (p)-JNK (cat. no. 4668;
Cell Signaling Technology, Inc., Danvers, MA, USA), p-p38 (cat. no.
4511; Cell Signaling Technology, Inc.), p-ERK1/2 (cat. no. 4370;
Cell Signaling Technology, Inc.), JNK (cat. no. 9252; Cell
Signaling Technology, Inc.), p38 (cat. no. 8690; Cell Signaling
Technology, Inc.), ERK1/2 (cat. no. 4695; Cell Signaling
Technology, Inc.), MK2 (cat. no. ab131531; Abcam), p-MK2 (cat. no.
ab63378; Abcam), MK5 (cat. no. 7419; Cell Signaling Technology,
Inc.), p-MK5 (cat. no. 9771; Cell Signaling Technology, Inc.),
caspase-3 (cat. no. 14220S; Cell Signaling Technology, Inc.) and
cleaved caspase-3 (cat. no. 9654S; Cell Signaling Technology, Inc.)
overnight at 4°C. A horseradish peroxidase-conjugated goat
anti-rabbit IgG antibody functioned as the secondary antibody
(1:5,000; cat. no. 7074; Cell Signaling Technology, Inc.) for
incubation at room temperature for 2 h. Proteins were visualized
using an Enhanced Chemiluminescence Reagent (Pierce; Thermo Fisher
Scientific, Inc.). Membranes were exposed to light-sensitive film
and quantified using Image J software version 1.3.4.67 (National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
was used to analyze all data for statistical significance. Data are
expressed as the mean ± standard error from no less than three
independent duplicate experiments. One-way analysis of variance was
performed followed by Fisher's least significant difference post
hoc test for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
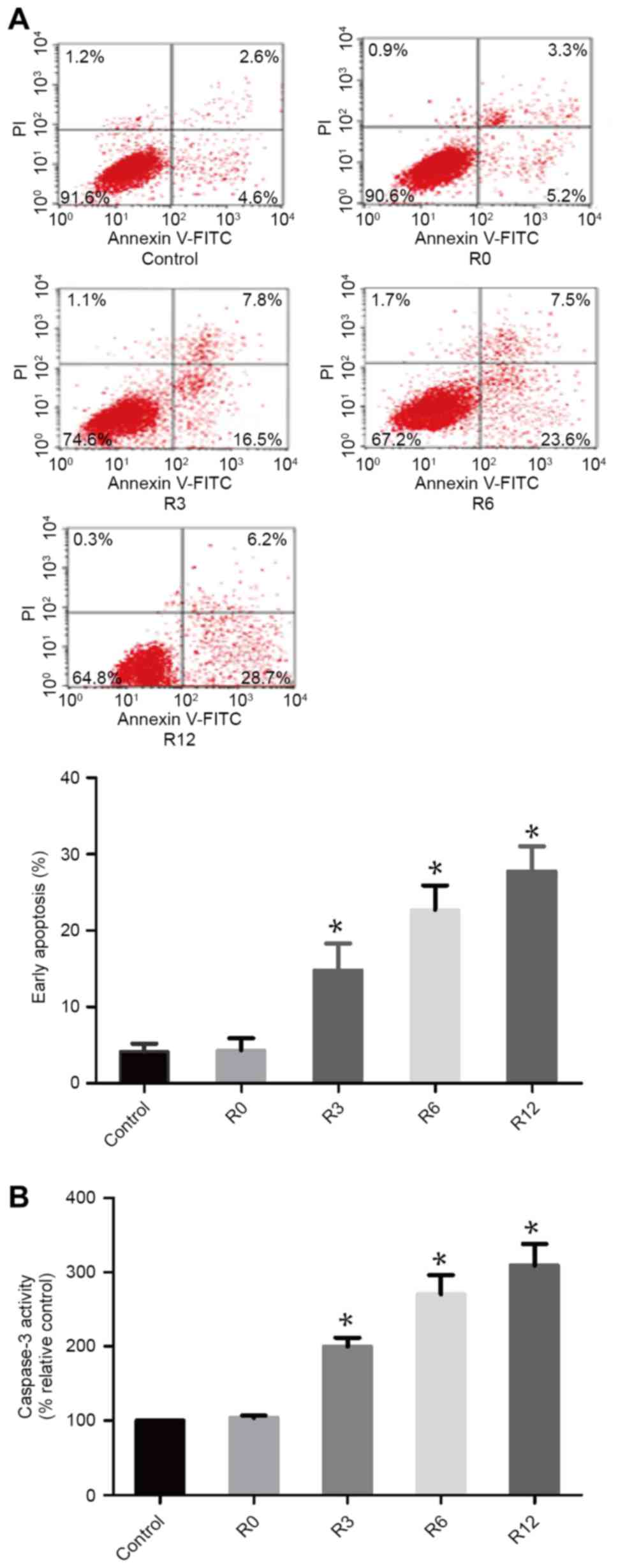
Heat stress increases cell apoptosis
in F98 cells
To assess whether heat stress could induce F98 cells
to apoptose in a heat-stress cell model, the proportion of
apoptotic cells were quantitatively evaluated using flow cytometry.
After F98 cells were subjected to heat stress at 43°C for 60 min,
the quantity of early apoptotic cells increased markedly over time,
with early apoptosis induced in 28.97±1.855 of cells by 12 h, which
was significantly higher compared with that in the control
treatment group 4.178±0.5901 (P<0.05; Fig. 1A). Caspase-3 activity was observed
using the fluorogenic substrate Ac-DEVD-AMC at 0, 3, 6 and 12 h
following 60 min of 43°C heat stress (Fig. 1B). The activity of caspase-3 mirrored
the occurrence of cell apoptosis.
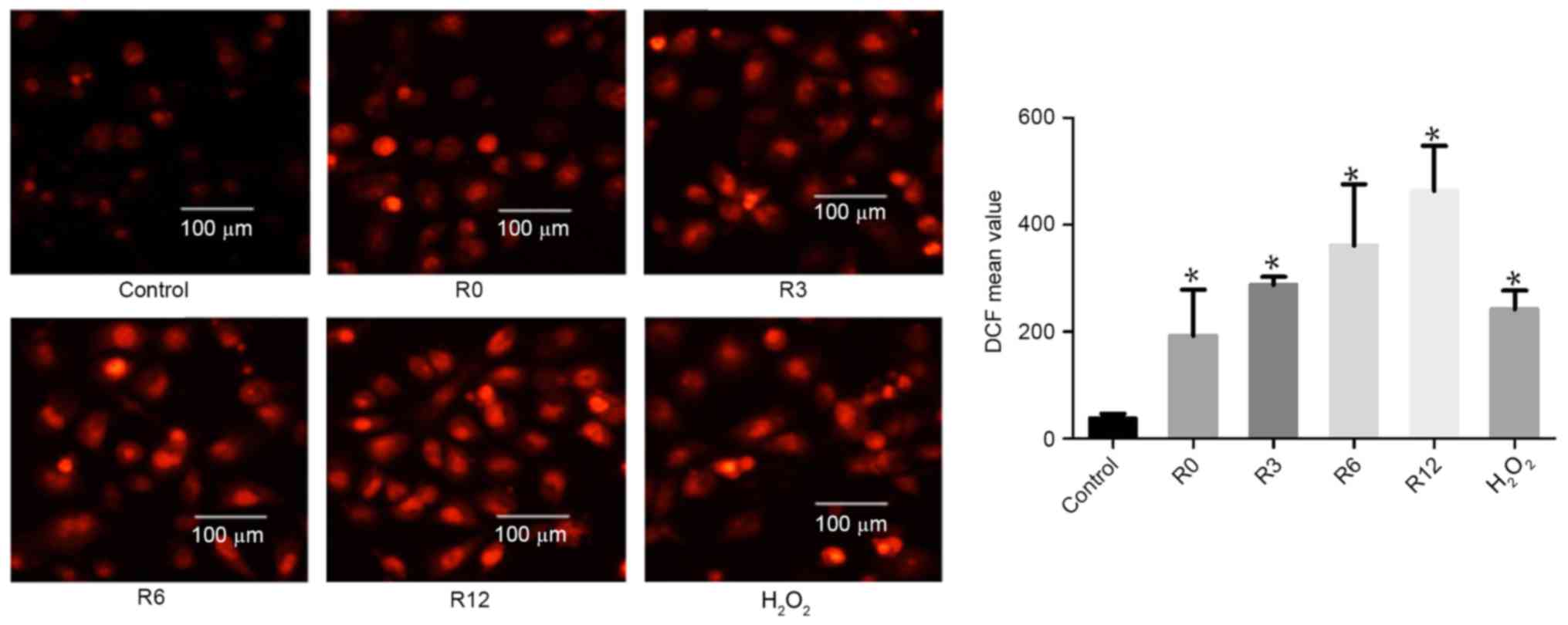
Heat stress increases ROS generation
in F98 cells
On the basis of evidence that ROS generation plays a
crucial part in the heat-stress response, whether accumulation of
ROS in F98 cells could be influenced by heat stress was
investigated. Cells were subjected to heat stress at 43°C for 60
min. Culture medium was substituted for fresh medium and the cells
received further incubation at 37°C for 0, 3, 6 or 12 h. The levels
of intracellular ROS increased in a time-dependent manner:
192.9±49.83 at 0 h, 287.7±8.718 at 3 h, 361.0±66.16 at 6 h and
463.6±48.52 at 12 h, compared with the control treatment group
(38.44±4.83) respectively at 0, 3, 6 and 12 h, as demonstrated by
flow cytometric analysis of DCF fluorescence.
H2O2 was used as a positive control (Fig. 2).
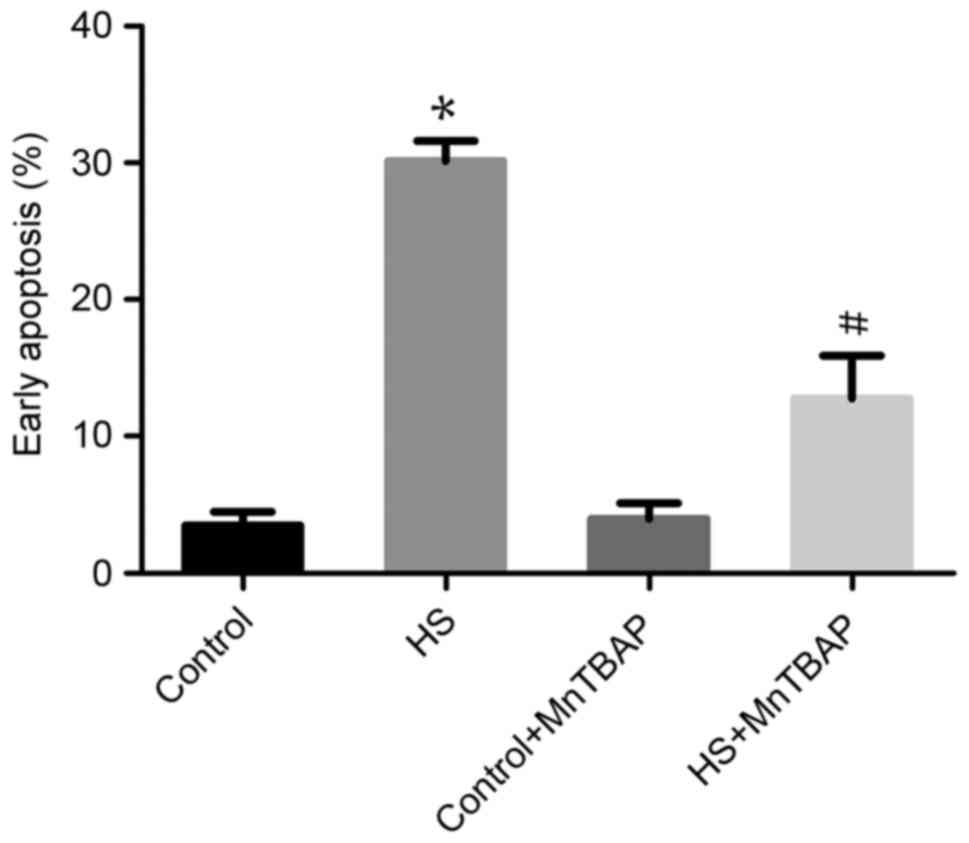
Function of ROS in heat-stress-treated
F98 cells
The role of ROS overproduction in
heat-stress-induced apoptosis was investigated using the
antioxidant MnTBAP to pretreat cells. As the cell-permeable ROS
scavenger MnTBAP depleted ROS, the proportion of F98 cells heat
stress-induced early apoptosis was significantly reduced compared
with heat-stressed cells not treated with MnTBAP (P<0.05;
Fig. 3). These data indicate that the
apoptosis of neurons induced by heat stress is mediated by ROS
release.
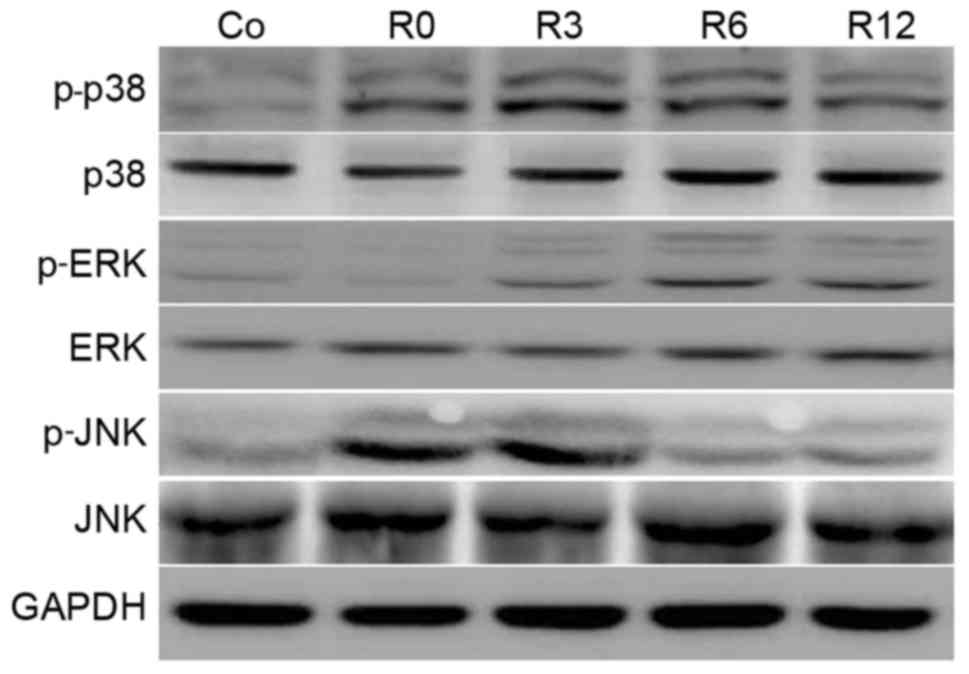
Heat stress induces MAPK activation in
neurons
Activation of MAPK is implicated in enhanced
cellular sensitivity to oxidative-stress-induced neuronal apoptosis
(9). Therefore, the time course of
MAPK phosphorylation in F98 cells stimulated by heat stress was
investigated. The phosphorylation of p38 MAPK was rapid, peaking at
3 h and sustaining to 12 h. Phosphorylation of JNK sustained to 3 h
and declined following shock recovery. In contrast to p38 and JNK,
ERK activation increased more gradually and peaked (Fig. 4).
p38 activation is involved in
heat-stress-induced apoptosis
To examine the function of MAPK activation in
heat-stress-induced apoptosis, F98 cells underwent heat stress with
or without MAPK inhibitors. As shown in Fig. 5, incubation with SB203580 (a
p38-specific inhibitor) alone substantially decreased ROS
production and the number of cells in early apoptosis. Incubation
with PD98059 (an ERK-specific inhibitor) and SP600125 (a
JNK-specific inhibitor) exerted no effect on ROS production or cell
apoptosis compared with the heat-shock group that did not receive
additional treatment.
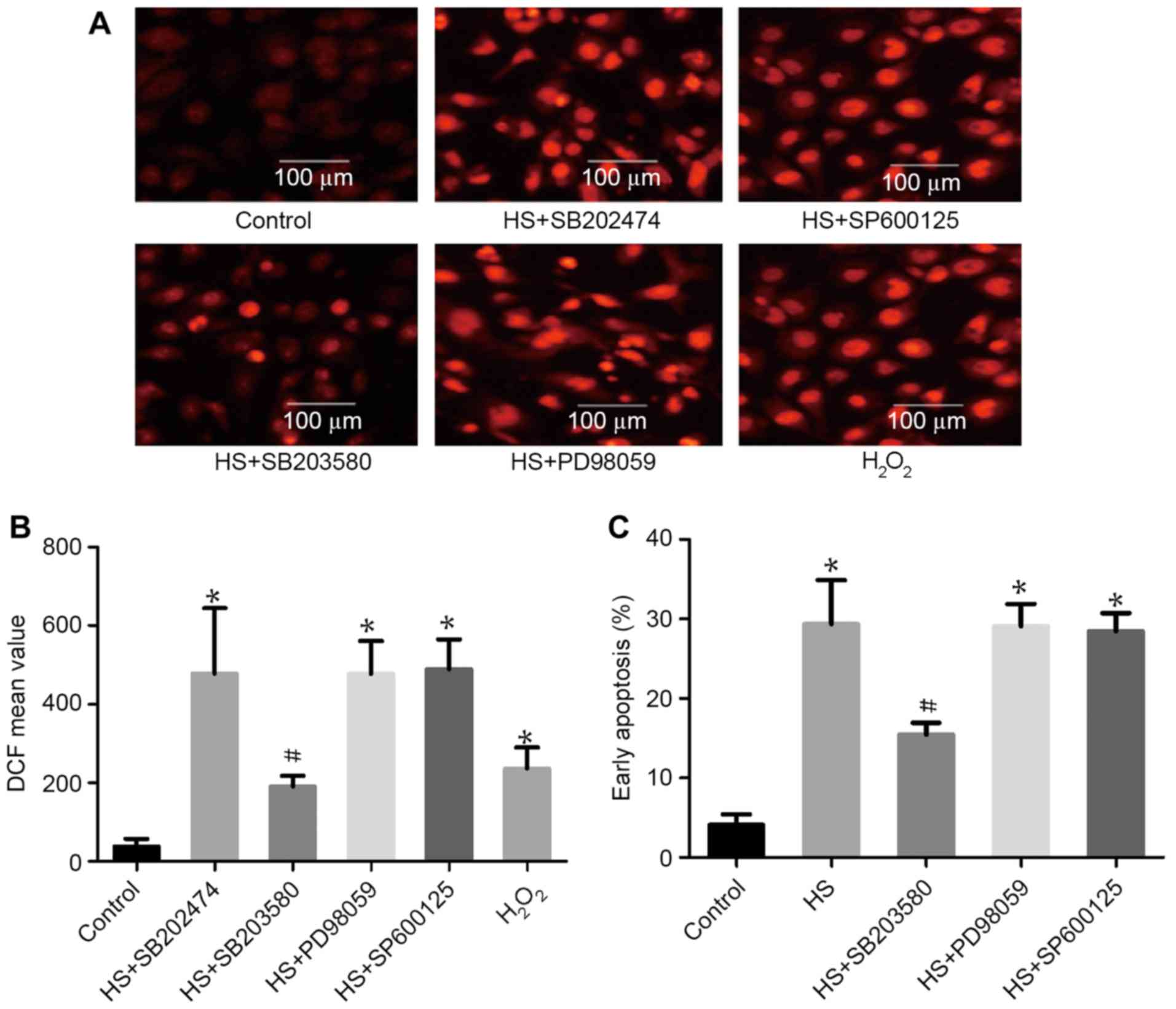 | Figure 5.The role of mitogen-activated protein
kinases in heat-stress-triggered apoptosis in neurons. F98 cells
were pretreated with or without the indicated inhibitors prior
exposure to heat stress or control heat treatment, and cells were
further incubated for 12 h at 37°C. H2O2 was
used as a positive control for ROS. (A and B) ROS quantity was
assessed by 2′,7′-dichlorodihydrofluorescein diacetate staining;
(A) images were obtained via laser scanning confocal microscopy and
(B) analysis of the fluorescence intensity of ROS probes was
performed using flow cytometry. (C) Analysis of apoptosis was
performed through flow cytometry using Annexin V-fluorescein
isothiocyanate/propidium iodide staining. #P<0.05 vs.
HS group, *P<0.05 vs. control group. ROS, reactive oxygen
species; HS, heat stress; SB203580, p38 inhibitor; SP600125, JNK
inhibitor; PD98059, ERK inhibitor; DCF,
2′,7′-dichlorofluorescein. |
SB203580 inhibits heat stress-induced
MK2 and MK5 activation
To investigate whether MAPKs have an effect on
downstream MK2 and MK5 in heat-stressed F98 cells, MAPK inhibitors
were used pretreat cells, with SB202474 used as a control. SB203580
(the specific inhibitor of p38) suppressed the heat-stress-induced
activation of MK5 and MK2, whereas PD98059 (the inhibitor of ERK)
and SP600125 (the inhibitor of JNK) exerted no such effect
(Fig. 6). These results indicate that
MK2 and MK5 activation occurs downstream of p38 activation.
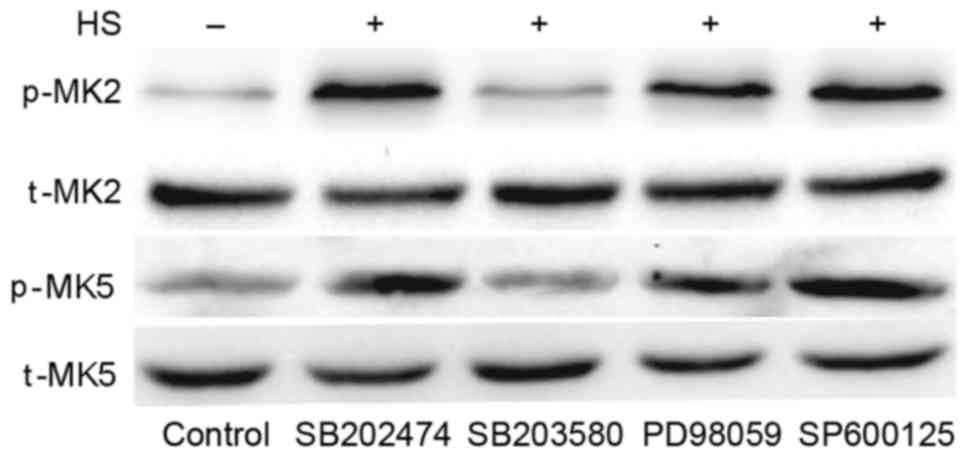 | Figure 6.Inhibition of p38 weakens heat
stress-induced phosphorylation of MK5 and MK2. F98 cells underwent
pretreatment with or without the indicated inhibitors for 30 min
prior to exposure to heat stress or control heat treatment. After
the cells received further incubation for 12 h at 37°C, antibodies
specific for p-MK2, p-MK5, MK2, MK5 were used to assess the protein
levels in the cell lysates. HS, heat stress; p-, phosphorylated;
t-, total; MK, mitogen-activated protein kinase-activated protein
kinase; SB203580, p38 inhibitor; PD98059, ERK inhibitor; SP600125,
JNK inhibitor. |
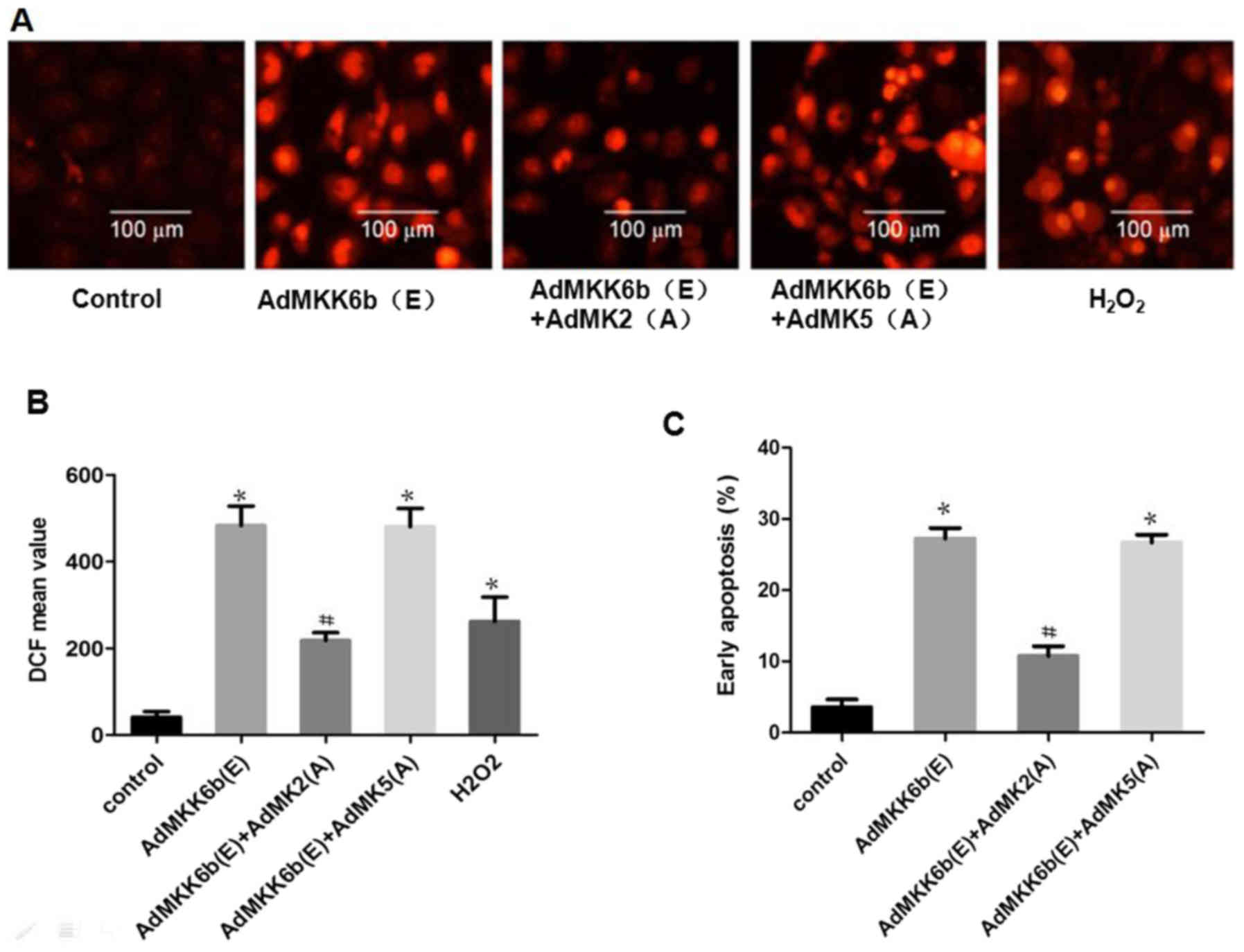
Activation of MK2 induces neuronal ROS
production-mediated apoptosis
To investigate whether MK2 and MK5 have an effect on
p38 MAPK-mediated ROS production and cell apoptosis, F98 cells were
transfected with an adenoviral dominant-active form of p38 or a
dominant-negative MK2 and MK5. ROS production and cell apoptosis
were induced by transfection with Ad-MKK6b(E), the dominant active
kinase MKK6b (upstream of p38), whose effect was strikingly
weakened upon co-transfection with Ad-MK2(A), an adenoviral
construct of the dominant negative MK2; however, co-transfection
with dominant-negative MK5 [Ad-MK5(A)] to inhibit MK5 did not
elicit a similar response (Fig.
7).
Inhibition of MK2 activation
alleviates heat-stress-induced ROS production and glial cell
apoptosis
Whether heat stress-induced ROS production and
apoptosis could be suppressed by inhibition of MK2 or MK5 was
assessed. As shown in Fig. 8A and B,
the results revealed that incubation with CMPD-1 (a specific
inhibitor of MK2) or transfection with Ad-MK2(A) inhibited heat
stress-induced ROS accumulation, whereas transfection with
Ad-MK5(A) exerted no such inhibitory function.
H2O2 was used as a positive control. The
effect of MK2 inhibition on cell apoptosis was further
investigated. The results revealed that transfection with Ad-MK2(A)
or incubation with CMPD-1 decreased heat-stress-induced cell
apoptosis, while transfection with Ad-MK5(A) did not (Fig. 8C).
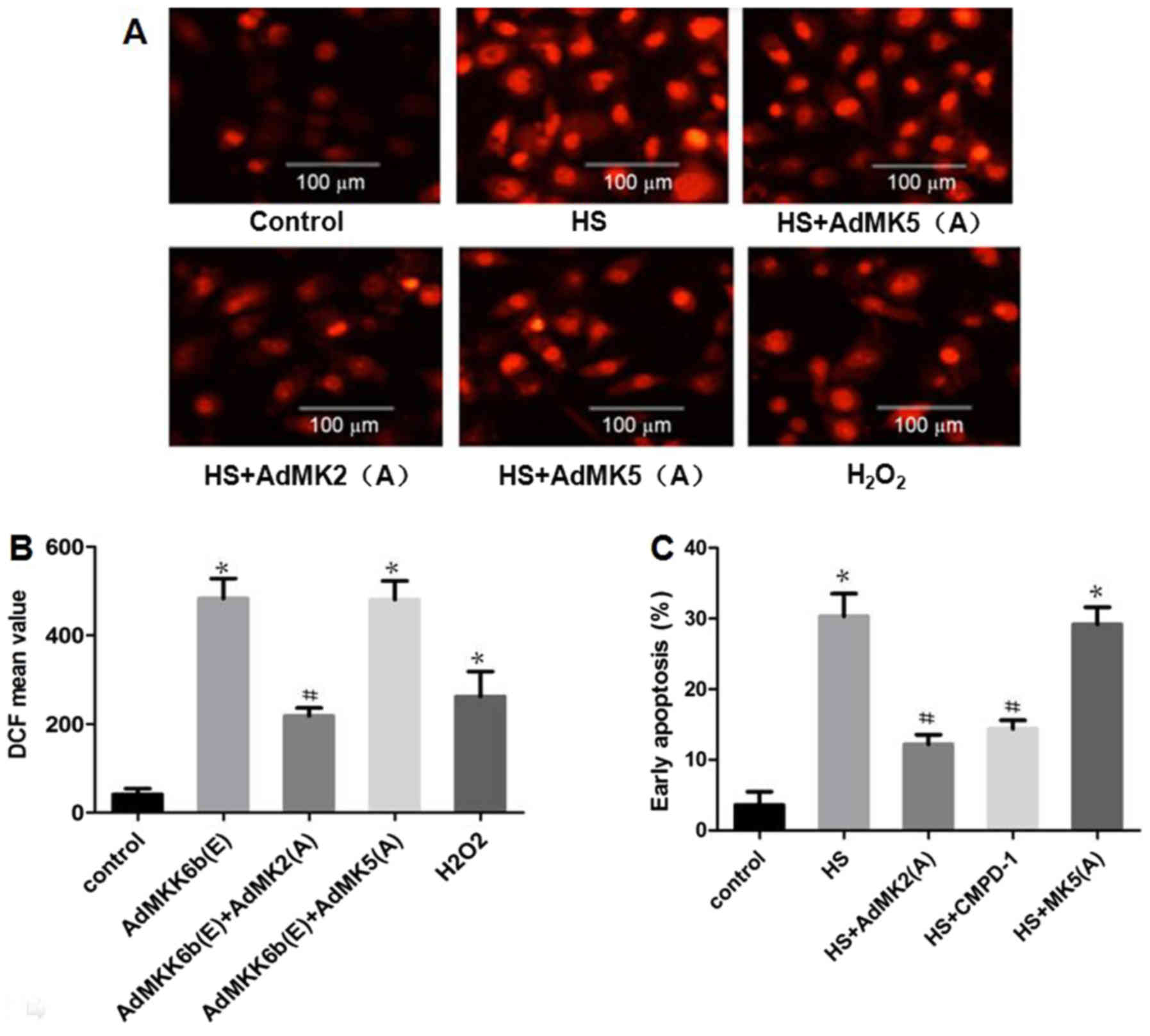 | Figure 8.Inhibiting MK2, but not MK5,
alleviates heat-stress-induced apoptosis. F98 cells were
transfected with adenoviral dominant negative form of MK5 and MK2
prior to exposure to heat stress (43°C, 1 h) or control treatment,
and the cells were incubated for 12 h at 37°C. (A and B) Assay of
ROS quantity was performed using 2′,7′-dichlorodihydrofluorescein
diacetate staining; (A) images were obtained via laser scanning
confocal microscopy and (B) analysis of the fluorescence intensity
of ROS probes was performed by flow cytometry. (C) Analysis of
apoptosis was performed through flow cytometry using Annexin
V-fluorescein isothiocyanate/propidium iodide staining.
#P<0.05 vs. the HS group, *P<0.05 vs. the control
group. ROS, reactive oxygen species; MK, mitogen-activated protein
kinase-activated protein kinase; HS, heat stress; CMPD-1, MK2
inhibitor; DCF, 2′,7′-dichlorofluorescein. |
Discussion
The number of studies investigating the influence of
heat stress continues to rise owing to fluctuations in temperature
being among the most common stressors in biological systems
(5,21). In a previous study, following the
selective heating of hemispheres of the canine brain above 42–43°C
for 30 min, several histopathological changes, including focal
hemorrhages, spotty infarction and edema, were observed (22). Subsequent to heating at 43°C for 50
min, impairments of myelin tracts in the white matter and neurons
in the gray matter of the brains were immediately observed
(22). A cerebral lesion composed of
a central coagulation of necrosis encircled by a hypervascular
section with clear contours was observed by Sneed et al
(19) in 1986, when the canine brain
was subjected to a 30-min single heat treatment at 43–44°C
(19). Heat stress may be the cause
of a reduction in the cellular processes initiated by various
regional neurons in the CNS, and apoptosis may be integrally
involved in the CNS injury of heat-involved diseases ascribed to
heat stress (6).
The present study used the rat glioma F98 cell line
to study the mechanism of CNS injury caused by heat stress. The
results indicated that apoptosis was induced by heat stress in F98
cell lines. The expression of MAPKs in heat stressed F98 cells was
assessed, and it was found that high-intensity heat stress
triggered MAPK activation. p38 activation was implicated in heat
stress-induced ROS accumulation-mediated apoptosis, but neither JNK
nor ERK had any effect on this, which could implicate ROS
associated with intense heat stress. Our and previous reports have
associated oxidative stress with heat stress and suggested
synergistic augmentation of cell death and increased ROS generation
in heat-exposed cells (17,23–25). The
present study indicated that mediation of oxidative stress is
mainly achieved by intense heat stress, inducing an increase in ROS
production. Using the cell-permeable ROS scavenger MnTBAP, it was
further found that heat stress generates ROS. Taken together, the
results suggest that the proportion of F98 cells exposed to heat
stress in early apoptosis is increased by ROS accumulation, whereas
p38, a product of acute heat stress, may function as an upstream
signal that stimulates this accumulation.
Inhibition of different MAPKs with specific
inhibitors indicated that the p38 inhibitor (SB203580), but not the
JNK inhibitor (SP600125) or ERK inhibitor (PD98059) could suppress
activation of the p38 downstream kinases MK2 and MK5 in neurons.
Inhibiting MK2 by transfection with Ad-MK2(A) or incubation with
its specific inhibitor markedly decreased normal and heat
stress-induced ROS accumulation and cell apoptosis, whereas
inhibition of another downstream p38 MAPK kinase, MK5, by
transfection with Ad-MK5(A), did not exert the same effects.
The p38 MAPK-MK2 family has been demonstrated to
modulate apoptosis in response to various stimuli (26,27),
including in neurons (16). A
previous study reported that the apoptosis induced by doxorubicin
in human hepatoma cells, alongside the cleavage of caspase-3 and
poly(ADP-ribose) polymerase, can be diminished by the constant
overexpression of MK5, which is also referred to as
p38-regulated/activated protein kinase or PRAK (28). In the present study, MK5 was
recognized as a downstream target of p38 MAPK in heat-stressed F98
cells, but exerted no effect on cell apoptosis. On the basis of
these results, it was concluded that heat stress stimulation
induced p38-MK2 pathways activation, which served a pro-apoptotic
role by regulating ROS accumulation in glial cells.
To conclude, the data obtained in the present study
revealed that heat stress rapidly leads to apoptosis of F98 cells.
Early apoptosis induced by intense heat stress is associated with
p38MAPK-MK2 signaling, which is in turn mediated by ROS generation.
The present study provides novel strategies for treatment of
heat-associated CNS injury in which glial cell apoptosis
occurs.
Acknowledgements
The present study was supported by the project team
of Natural Science Foundation of Guangdong Province (grant no.
2013030013217).
References
|
1
|
Malamud N, Haymaker W and Custer RP: Heat
stroke: A clinicoorgan pathological study of 125 fatal cases. Mil
Surg. 99:394–449. 1946.
|
|
2
|
Hausfater P, Megarbane B, Dautheville S,
Patzak A, Andronikof M, Santin A, André S, Korchia L, Terbaoui N,
Kierzek G, et al: Prognostic factors in non-exertional heatstroke.
Intensive Care Med. 36:272–280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dematte JE, O'Mara K, Buescher J, Whitney
CG, Forsythe S, McNamee T, Adiga RB and Ndukwu IM: Near-fatal heat
stroke during the 1995 heat wave in Chicago. Ann Intern Med.
129:173–181. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mastrangelo G, Fedeli U, Visentin C, Milan
G, Fadda E and Spolaore P: Pattern and determinants of
hospitalization during heat waves: An ecologic study. BMC Public
Health. 7:2002007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kovats RS and Kristie LE: Heatwaves and
public health in Europe. Eur J Public Health. 16:592–599. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Edwards MJ, Walsh DA and Li Z:
Hyperthermia, teratogenesis and the heat shock response in
mammalian embryos in culture. Int J Dev Biol. 41:345–358.
1997.PubMed/NCBI
|
|
7
|
Yu J, Liu F, Yin P, Zhao H, Luan W, Hou X,
Zhong Y, Jia D, Zan J, Ma W, et al: Involvement of oxidative stress
and mitogen-activated protein kinase signaling pathways in heat
stress-induced injury in the rat small intestine. Stress.
16:99–113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y, Zhou G, Wang Z, Guo X, Xu Q, Huang
Q and Su L: NF-κB signaling is essential for resistance to heat
stress-induced early stage apoptosis in human umbilical vein
endothelial cells. Sci Rep. 5:135472015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
11
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gaestel M: MAPKAP kinases-MKs-two's
company, three's a crowd. Nat Rev Mol Cell Biol. 7:120–30. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
New L, Jiang Y, Zhao M, Liu K, Zhu W,
Flood LJ, Kato Y, Parry GC and Han J: PRAK, a novel protein kinase
regulated by the p38 MAP kinase. EMBO J. 17:3372–3384. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu R, Wu CX, Zhou D, Yang F, Tian S,
Zhang L, Zhang TT and Du GH: Pinocembrin protects against
β-amyloid-induced toxicity in neurons through inhibiting receptor
for advanced glycation end products (RAGE)-independent signaling
pathways and regulating mitochondrion-mediated apoptosis. BMC Med.
10:1052012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo
SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces
apoptosis through transcription-independent p53-mediated
mitochondrial pathways in human umbilical vein endothelial cell.
Sci Rep. 4:44692014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fiers W, Beyaert R, Declercq W and
Vandenabeele P: More than one way to die: Apoptosis, necrosis and
reactive oxygen damage. Oncogene. 18:7719–7730. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sneed PK, Matsumato K, Stauffer PR, Fike
JR, Smith V and Gutin PH: Interstitial microwave hyperthermia in a
canine brain model. Int J Radiat Oncol Biol Phys. 12:1887–1897.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu W, Huan Q, Miao J, Xiao M, Liu H, Zhao
K and Zhao M: MK2 plays an important role for the increased
vascular permeability that follows thermal injury. Burns.
39:923–934. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dorozynski A: Chirac announces
investigation into heat wave's death toll. BMJ. 327:4652003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harris AB, Erickson L, Kendig JH, Mingrino
S and Goldring S: Observations on selective brain heating in dogs.
J Neurosurg. 19:514–521. 1962. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Burdon RH, Gill VM and Rice-Evans C:
Oxidative stress and heat shock protein induction in human cells.
Free Radic Res Commun. 3:129–139. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Skibba JL, Powers RH, Stadnicka A,
Cullinane DW, Almagro UA and Kalbfleisch JH: Oxidative stress as a
precursor to the irreversible hepatocellular injury caused by
hyperthermia. Int J yperthermia. 7:749–761. 1991. View Article : Google Scholar
|
|
25
|
McAnulty SR, McAnulty L, Pascoe DD,
Gropper SS, Keith RE, Morrow JD and Gladden LB: Hyperthermia
increases exercise-induced oxidative stress. Int J Sports Med.
26:188–192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signaling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou J, Wan B, Liu XM, Li R, Wang Y and Yu
L: MK5 is degraded in response to doxorubicin and negatively
regulates doxorubicin-induced apoptosis in hepatocellular carcinoma
cells. Biochem Biophys Res Commun. 427:581–586. 2012. View Article : Google Scholar : PubMed/NCBI
|