Introduction
Glioblastoma multiforme (GBM; WHO glioma grade IV)
is the most common primary malignant brain tumour in adults
characterised by a very poor prognosis with a median survival of
15–18 months (1) and less than 5% of
patients alive at 5 years (2,3). Standard treatment of newly diagnosed GBM
is maximal safe resection followed by local radiotherapy with
concomitant and adjuvant temozolomide (TMZ) (4). Despite standard therapy, GBM tends to
infiltrate brain parenchyma with a high local recurrence rate
(~90%) and a very low response to successive therapy. According to
the 2007 WHO guidelines for brain tumours (5), the new knowledges on molecular biology
and cytogenetics have allowed to define emerging GBM variants
(2,6).
GBM with primitive neuroectodermal tumour-like components
(GBM/PNET) is an emerging variant of GBM, which represent nearly
0.5% of GBM cases (6–8). PNET of the brain is an aggressive
neoplasm which commonly occur in children and rarely in adults.
Compared to GBM, PNET has a similar poor prognosis but has better
response to therapy and long term survival with a 4-year survival
rate of 38% (9,10). Furthermore this tumour showed a lower
rate of local recurrence following total resection and a high risk
to disseminate into the cerebrospinal fluid (CSF). Extracranial
metastasis in PNET are rare but more frequent than in GBM. PNET is
generally treated with surgery followed by craniospinal irradiation
(CSI) combined with platinum-based chemotherapy in order to prevent
CSF dissemination (11).
In May 2016 the latest WHO Classification of Tumors
of the Central Nervous System was published. According to this new
classification the embryonal tumors other than medulloblastoma
underwent reclassification, with removal of the term ‘PNET’ from
the diagnostic lexicon. The term GBM/PNET was then replaced with
the term ‘Glioblastoma with primitive neuronal component’
(GBM-PNC).
GBM/PNET tumour presents more in adults and have two
different histological architectures including the traditional
astrocytic GBM areas with high expression of GFAP (7) and the hypercellular undifferentiated
PNET areas, with lower expression of GFAP and neuronal
immunophenotype (S-100, Synaptophysin, NeuN, NSE and NFP) (7,12,13). In magnetic resonance imaging (MRI) the
PNET areas of hypercellularity are seen as reduced apparent
diffusion coefficient (ADC) (14).
Regarding genetic alterations it's possible to observe
‘glioma-like’ characteristics, such as 10q deletion, EGFR
amplification or 1p/19q deletions and ‘PNET-like’ characteristics,
such as high Ki-67 index, N-myc or C-myc amplifications (7,11). The
role of mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2
remains controversial. They commonly occur in low grade gliomas and
secondary GBMs, rarely in primary GBMs. IDH1 mutation have been
observed also in a small percentage of adult PNETs (15–17). The
PNET components are explained by two main hypotheses (8): PNET-like foci arise from pre-existing
gliomas, most often a secondary GBM (neuroblastic or neuronal
metaplasia) and the clonal expansion of tumour stem cells or
progenitor cells resulting in PNET-like nodules (7,15,18).
Since most of the data concerning the management of
GBM/PNETs are based on single case reports or case series (2,7–9,11,12,15,18–20)
and despite therapeutic strategies have been identified for GMBs
and PNETs, a common therapeutic protocol for GBM/PNETs has not yet
been defined. In addition no evidence on treatment survival
outcomes are published (11).
Therefore GBM/PNETs constitute a challenging diagnostic and
therapeutic dilemma, since GBM is typically treated with alkylating
agents while PNETs respond to platinum-based chemotherapy.
Recent studies showed that GBM/PNETs have a
PNET-like clinical behaviour with increased risk of CSF
dissemination (7,8) and a possible benefit with ‘PNET-like’
platinum-based chemotherapy upfront or after standard therapy for
GBM has failed (7,14).
We reported our experience with a recurrent GBM/PNET
patient, which developed a rare extracranial metastasis, and then a
review of the main papers reporting this rare GBM variant.
Case report
In February 2016, a 60-year-old Caucasian male
patient referred to our emergency department for ideomotor slowdown
and occasional speech impairment. Patient was not affected by major
comorbidities, except for mild emphysema due to cigarette
smoking.
On admission, neurologic examination evidenced a
mild right downward drop to the antigravitary Mingazzini test, a
flattening of the nasolabial fold and a slight ideomotor slowdown
with Karnofsky Performance Status (KPS) 100%. No speech disorders
were detected. Computed tomography (CT) scan of the head and
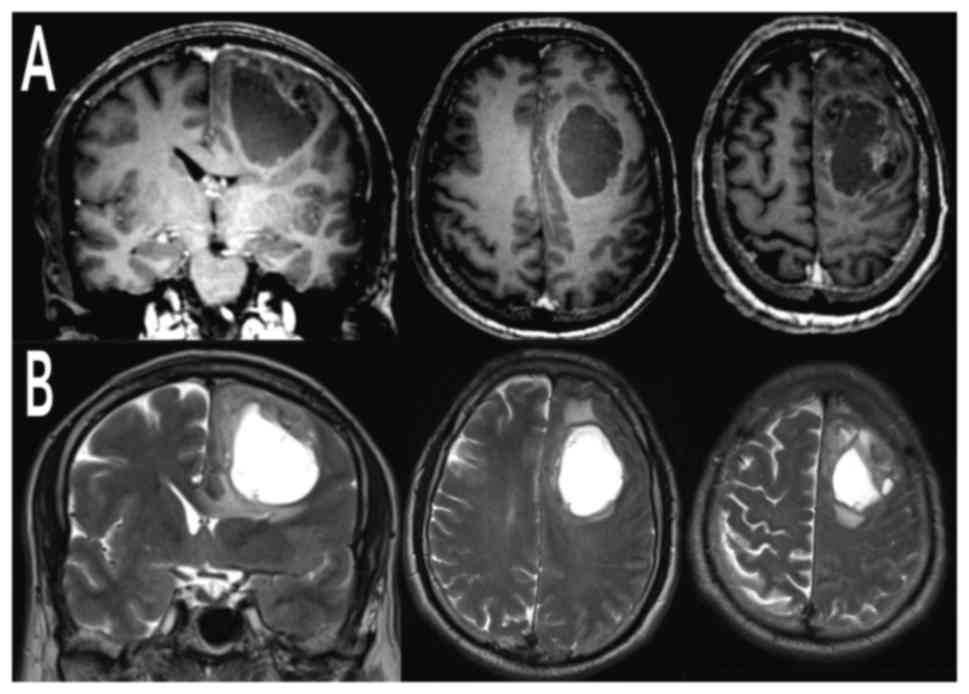
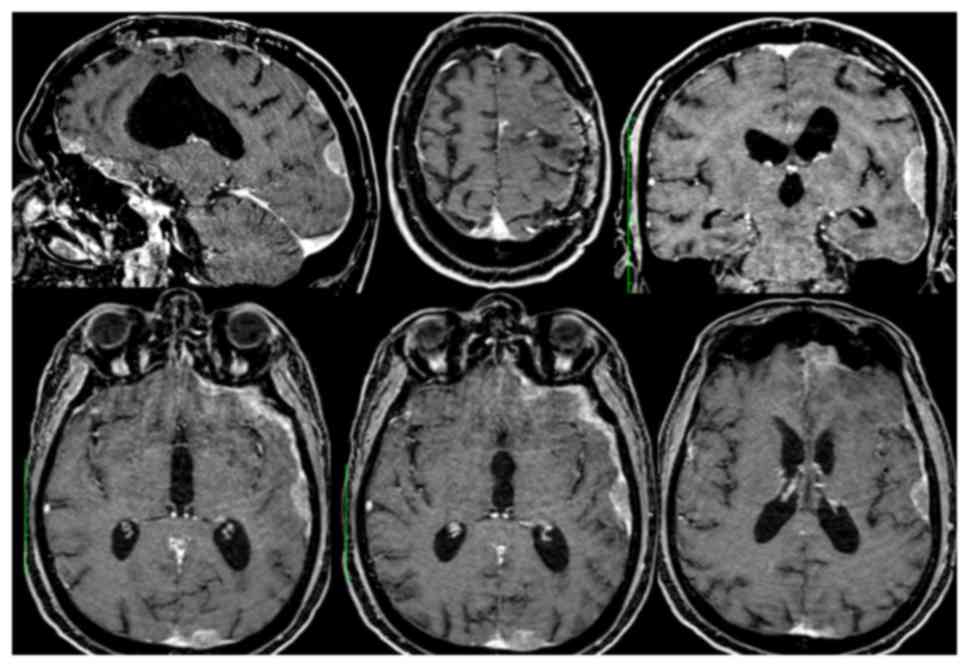
contrast-enhanced MRI of the brain uncovered a voluminous left
frontal cystic lesion (AP 54 mm × LL 37 mm) with compressive effect
and perilesional edema. The lesion presented heterogeneous
post-contrastografic enhancement of the cystic walls and
multinodular component anchored to the anterosuperior portion of
the cyst lesion (Fig. 1). Total body
(TB) CT scan did not show any extracranial localization.
A paramedian frontal-parietal craniotomy with gross
total resection of the neoplasia was performed under local
anestesia (awake surgery). The lesion, removed en-bloc, presented a
central liquid component and extremely vascularized margins (glioma
aspects). Postoperative course was uneventful. The
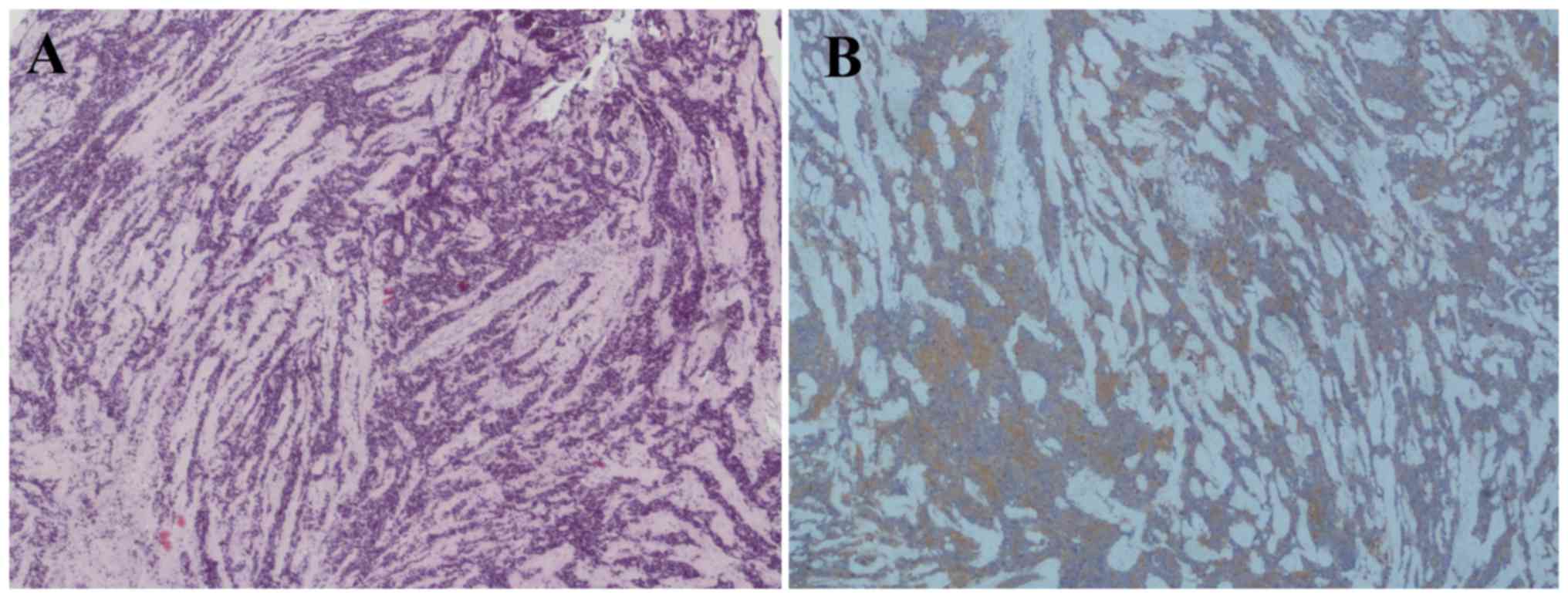
histopathological examination revealed that the tumor tissue was
composed of two distinct components. An area with severe cellular
pleomorphism in a fibrillar background, typical of malignant
gliomas and an area composed of markedly hypercellular mass and
high nuclear/cytoplasmic ratios with small, round, monomorphic and
hyperchromatic nuclei, typical of neuroendocrine neoplasia
(Fig. 2A and B). Immunohistochemistry
assay showed positivity for glial fibrillary acidic protein (GFAP)
in the 60% of the cells and for Synaptophysin (Syn) in the 40% of
the cells (Fig. 2C and D).
overexpression of EGFR and positivity for P53 protein was aslo
observed. Ki67 was 70%. Histologic diagnosis, according to the
current WHO classification (2007) was GBM PNET-like IDH1
wild-type.
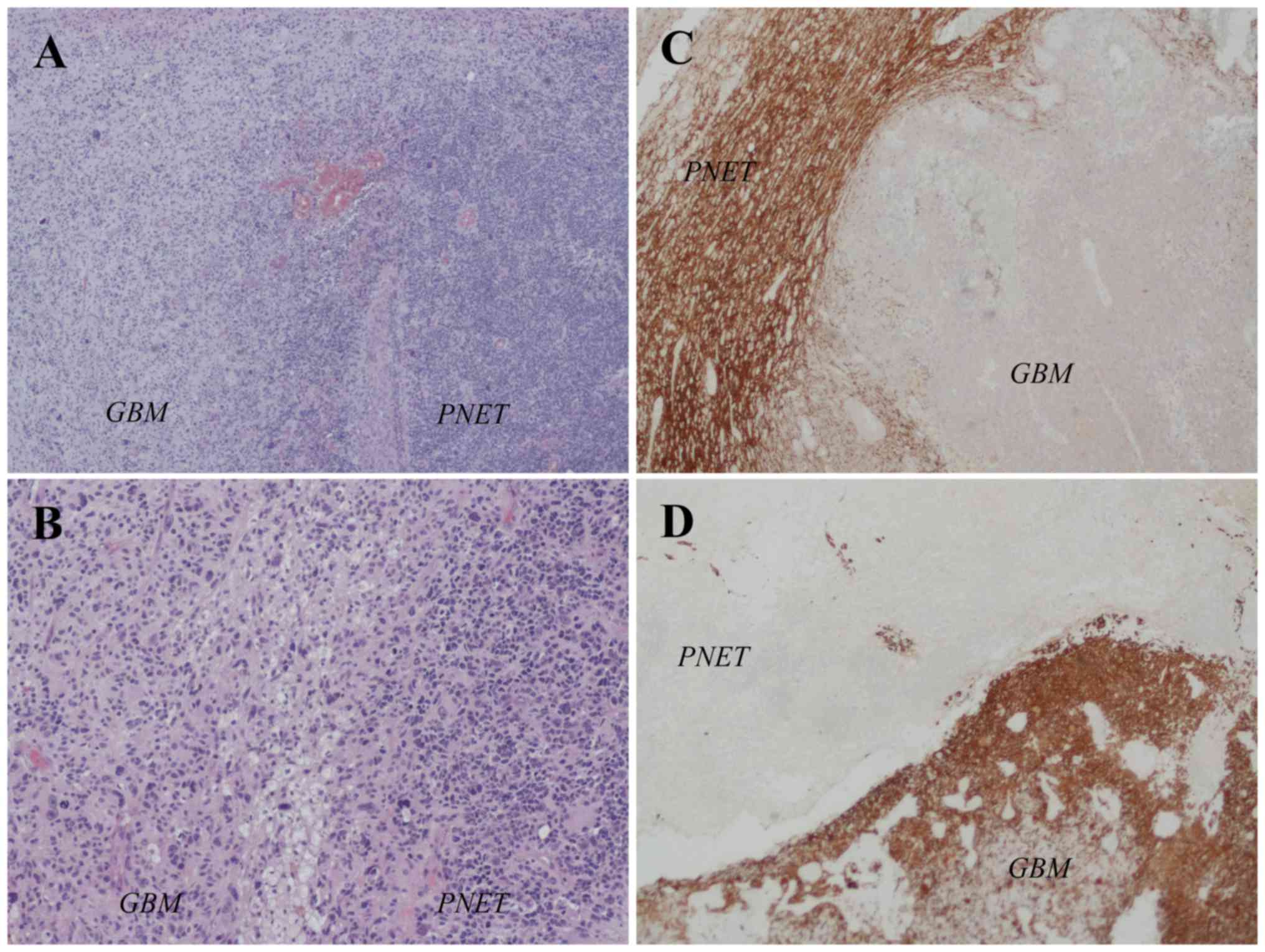 | Figure 2.Interface between the GBM and PNET
components in the primitive tumor. In the (A) H&E
(magnification, ×40) and (B) H&E (magnification, ×100) staining
images, on the left there is a typical area of GBM with severe
cellular pleomorphism in a fibrillar background and on the right
there is an area of PNET composed of a high cellular mass with
small, round, monomorphic and hyperchromatic nuclei. (C) Syn
staining (magnification, ×40) revealed a PNET component, while (D)
the glial fibrillary acidic protein immunohistochemical staining
(magnification, ×40) defined the GBM counterpart. GBM, glioblastoma
multiforme; PNET, primitive neuroectodermal tumour; H&E,
hematoxylin and eosin. |
All control brain MRI scans were obteined with 3
Tesla MRI using gadolinium-enhanced T1-weighted, T2/FLAIR-weighted,
perfusion-weighted, diffusion-weighted and MR spectroscopy
sequences.
On the 1st postoperative month, at the end of March,
the patient referred to our emergency department for tonic-clonic
seizure with loss of consciousness. A brain MRI was performed
showing the presence of pathological tissue of 5×4 cm size
involving the corpus callosum. From April to May the patient
underwent hypofractionated radiotherapy (40 Gy in 15 fractions)
with concomitant oral TMZ 75 mg/mq/day (Stupp protocol).
In May, on the 3rd postoperative month, after the
end of radio-chemotherapy, a control brain MRI showed a
contrast-enhancing centronecrotic nodule in left anterior frontal
site, adjacent to the previous surgical site, and one in the body
of the corpus callosum, as for satellite expression of the primary
tumor. Neurologic examination and KPS were stable. The patient
started metronomic TMZ 150 mg/day orally for 5 days every 28 days
until the end of the 4th postoperative month.
On the 5th postoperative month, after one cycle of
TMZ 150 mg/day, a control brain MRI showed a context of stable
disease. The patient underwent two additional cycles of oral TMZ
300 mg/day for 5 days every 28 days.
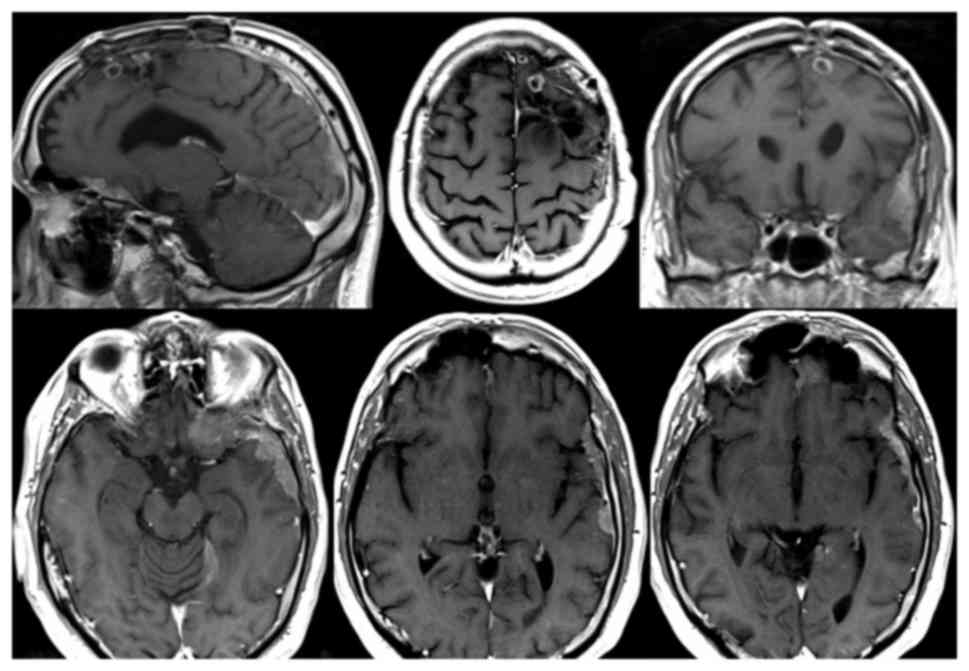
In August, on the 6th postoperative month, a control
brain MRI documented slight volumetric reduction of the two left
frontal centronecrotic nodules (10 mm vs. 13 and 5 mm vs. 10 mm)
and of the lesion of the corpus callosum (7 mm vs. 20 mm)
(intracerebral partial response). However it also showed
contrast-enhancing leptomeningeal thickening in the left temporal
lobe (maximum 14 mm), left ethmoido-sphenoidal emiplanum (maximum
12 mm) and left portion of the tentorium (maximum 10 mm) (Fig. 3). Moreover, a subcutaneous nodule was
clinically evident in the left frontal region, at the level of the
previous craniotomy. Despite the new findings the patient was still
clinically stable.
In September (7th postoperative month) a surgical
excision of the subcutaneous nodule, under local anesthesia, was
performed. Histologic examination confirmed a GBM PNET-like
extracranial metastasis. Histological and immunohistochemical
staining showed that the metastasis was almost completely composed
of the neuroendocrine tumor (95%) while the GBM component was
limited to 5% of the tumor mass (Fig.
4). A TB CT scan of stadiation did not show any other
extracranial localization, but a further increased of the left
cerebral leptomeningeal thickening (23 mm vs. 14 mm in the temporal
lobe).
At the end of September, adjuvant chemotherapy was
adopted according to the scheme: Carboplatin AUC5 IV day 1 every 21
days and Etoposide 100 mg/mq IV day 1–3 every 21 days. After 2
cycles of chemotherapy, which the patient well-tolerated, a control
brain MRI was performed and documented a significant regression of
the diffuse leptomeningeal thickening with maximum residual of 4–5
mm (Fig. 5). It also showed a
stability of left frontomesial nodules and of the lesion of the
corpus callosum. Patient clinical condition and KPS were
stable.
On the 9th postoperative month (november) the
patient underwent 3rd chemotherapy cycle. After several days he
experienced occasional upper and inferior limb tremors and an
episode of generalized seizure. Successively he reported the onset
of weakness and drowsiness followed by aphasia and dysphagia.
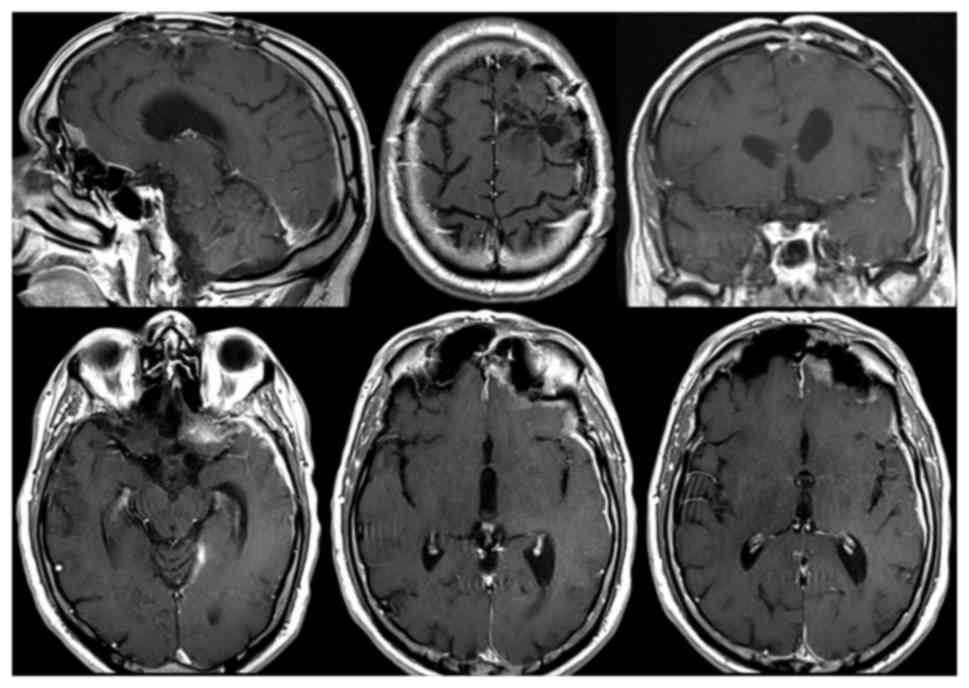
In December, on the 10th post-operative month, a
brain MRI performed shows a massive disease progression with
increased of leptomeningeal thickening of the left cerebral
hemisphere, which presents a multinodular contrast-enhancing aspect
(Fig. 6). The nodulations have a size
between 0.7 and 2.9 cm and the major nodules are those of the
temporal-insular (2.9 cm) and parietal lobe (2.6 and 1.8 cm) with
loco-regional parenchymal spread. In the left-frontal area, at the
level of the previous craniotomy, a contrast-enhancing
leptomeningeal thickening was detected, attributable to local
recurrence of the disease. At the end of the 10th postoperative
month the patient died for the evolution of the disease. Informed
consent was obtained from the parents of the subject for
participation in the present study.
Discussion
GBM and PNETs of the brain showed different pattern
of recurrence and this is reflected in the different standard of
treatment. GBM has a local infield pattern of recurrence so the
standard therapy consists of surgery followed by chemoradiotherapy
in order to improve local control. For recurrent GBM different
therapeutic approaches are provided such as resurgery,
reirradiation and systemic chemotherapy based on alkylating agents,
bevacizumab, carboplatin, etoposide and their combination (21). On the other hand PNETs of the brain
show a high tendency of leptomeningeal dissemination and
extracranial metastasization (22),
so the standard therapy consists of surgery followed by CSI
combined with concurrent platinum-based chemotherapy, particularly
carboplatin, or other concomitant chemotherapeutic agents, such as
etoposide and TMZ (11).
Some genetic alterations are expressed more
frequently in secondary GBMs including O6-methylguanine-DNA
methyltransferase (MGMT) promoter methylation (36% of primary vs.
75% of secondary GBMs) and IDH1 and IDH2 mutations (70–80% of low
grade gliomas and secondary GBMs), which correlate to an improved
prognosis (15).
MGMT promotor methylation is associated with better
response rate, progression-free survival (PFS) and overall survival
(OS) in GBMs treated with radiotherapy and/or chemotherapy with
alkylation agents (TMZ) in newly diagnosed and recurrent GBMs
(23).
Also IDH1/IDH2 mutations are correlated with better
OS and PFS. In addition, IDH1 mutated GBM patients undergone
complete resection have an improved survival. IDH1 mutations have
been observed in a small percentage of adult PNETs and of GBM/PNETs
and seem to have a positive prognostic value (15). The possibility that the emergence of
PNET-like elements may preferentially occur in secondary GBMs with
wild-type IDH genes should be evaluated (20). EGFR mutation/amplification correlates
with better prognosis when associated with methylated MGMT promotor
(23).
In contrast to GBMs and PNETs, there is no standard
treatment for GBM/PNETs because the majority of the available data
on their therapeutic approach derived from case reports and case
series (Table I).
 | Table I.Summary of the main published case
reports and case series on GBM/primitive neuroendorcrine tumor. |
Table I.
Summary of the main published case
reports and case series on GBM/primitive neuroendorcrine tumor.
| Author, year | No. of
patients | MGMT
methylation | IDH mutation | Surgery | Adjuvant
radiotherapy | Adjuvant GBM-like
chemotherapy |
Radiochemotherapy | TMZ | CSI | ‘PNET-like’
Platinum-based chemotherapy | OS (months) | (Refs.) |
|---|
| Perry et al,
2009 | 53 | NA | NA | 12/19=63% | 18/23=78% | 16/23=70% | 14/23=61% | 10/16=63% | 1/23=4% | 31% | 1–40 mOS=9.1 | (7) |
| Song et al,
2011 | 10 | NA | 2/8=25% | 100% | – | – | 9/10=90% | 1/10=10% | No | No | 2–31 mOS=10 | (15) |
| Karina et al,
2012 | 1 | NA | No | Yes | – | – | Yes, Stupp
protocol | – | No | No | NA | (20) |
| Lee et al,
2012 | 3 | NA | NA | 100% | – | – | 100%
TMZ+carboplatin | 100% | No | 100% ifosfamide,
carboplatin and etoposide | 32–56 | (24) |
| Kimbason et al,
2015 | 5 | 3/5 pt | 2/3 pt | 100% | – | 100% | 100% | 100% Stupp
protocol | – | 40% | 12–24 mOS=16 | (18) |
| Chu et al,
2015 | 1 | NA | NA | Yes, multiple | Yes, multiple | Yes, multiple
including TMZ, PCV, BEV | – | Yes | No | Yes, carboplatin
and etoposide | 28 | (8) |
| Forbes and
Vredenburgh, 2016 | 1 | NA | NA | Yes | No | No | No | No | Yes | Alternating:
cisplatin, vincristine, cyclophosphamide, TMZ, etoposide | No recurrence in 3
years | (9) |
| O'Leary et al,
2016 | 6 | NA | NA | 100% | No | No | 100% CSI+TMZ | 100%+CSI | 100%+TMZ | No | 8–43 50% of
response | (11) |
| Konar et al,
2017 | 1 | No | No | Yes | No | – | Yes, Stupp
protocol | No | No | No | About 6 months | (22) |
The largest series published on GBM/PNETs belong to
Varlet et al (n=40) (19) and
Perry et al (n=53) (7), which
showed the clinical, radiological and histopathological differences
of this variant of GBM compared to classic GBM and used for the
first time the term ‘malignant glioma with PNET-like component
(MG-PNET)’.
In 2004 Varlet et al (19) reported that local radiotherapy was an
inadequate method for preventing the tumour spreading and mentioned
chemotherapy as a more efficient therapeutic approach.
The largest multi-institutional case series was
performed by Perry et al (7)
in 2009 which studied 53 GBM/PNET patients treated with radiation
(78%), TMZ (63%) and platinum-based chemotherapy (31%). Nineteen
patients underwent surgical resection with 18 patients (78%)
received adjuvant radiotherapy (17 local and one cranio-spinal), of
which 14 were treated with concomitant chemotherapy (OS from 1
month to 3.3 years, median OS-mOS=12 months). Sixteen patients
(70%) received adjuvant GBM-like chemotherapy, including TMZ and
BCNU (OS from 1 month to 3.3 years, mOS=8 months). Three patients
received ‘PNET-like’ platinum-based chemotherapy upfront with
survival of 10 and 20 months in two patients. In 3 patients,
therapy was switched from TMZ to platinum-based regimens after
signs of progression on imaging, with radiological responses
(1). The mOS of all patients was 9.1
months, a survival similar to that reported for MGs. Perry et
al (7) reported also a higher
frequency of IDH mutations in GBM/PNETs compared to primary GBMs
(7%), supporting the hypothesis of a secondary GBM origin for most
of GBM/PNET cases. They concluded that the addition of
platinum-based chemotherapy should be considered in order to
prevent CSF dissemination, particularly after failure with TMZ, and
appeared to improve survival.
Also Song et al (15) in 2011 observed a high frequency (25%)
of IDH1 mutations in the cases analysed for this mutations.
Moreover, the IDH1 mutated patients were still alive 14 and 31
months after diagnosis, underlying the correlation between IDH1
mutations and improved prognosis.
In 2012 Karina et al (20) described a case of a GBM/PNET patient
which underwent surgery and chemoradiotherapy as Stupp protocol.
Platinum-based chemotherapy was reserved for recurrence or
failure.
In the same year Lee et al (24) analysed 3 GBM/PNET cases treated with
surgery followed by adjuvant radio-chemotherapy including TMZ and
carboplatin. After 4 week break, 3 cycles of consolidation
chemotherapy with ifosfamide, carboplatin and etoposide were
performed followed by adjuvant TMZ. Overall this protocol appears
to improve survival compared to the literature.
In 2015 Kimbason et al (18) reported a case series of 5 GBM/PNET
patients treated with surgical resection followed by
chemoradiotherapy with TMZ and then different chemotherapy
regimens. Two patients received platinum-based therapy with
carboplatin and showed longer survival since diagnosis. The authors
supported that GBM/PNET patients should be treated aggressively
using a multimodal approach including maximal surgical resection,
radiation therapy and platinum-based chemotherapy to better address
the PNET component.
Chu et al (8)
in 2015 described a case of a recurrent GBM/PNET patient treated
with multiple courses of surgery, radiation and chemotherapy
regimens including TMZ, procarbazine with lomustine and vincristine
(PCV), bevacizumab, carboplatin and etoposide. This approach
resulted in modest local responses and control. This case suggested
that aggressive therapies, with the early introduction of CSI and
platinum-based chemotherapy may be utilized in attempts to have a
better disease control and a longer survival.
Recently Forbes and Vredenburgh (9) reported a case of GBM/PNET composed of
predominately PNET with small areas of GBM positive for GFAP (10%)
treated with surgical resection, radiotherapy and chemotherapy
targeting both the PNET and GBM components. The patient underwent
CSI followed by 12 cycles of alternating chemotherapy based on
cisplatin, vincristine, cyclophosphamide to target the PNET
component and TMZ with etoposide to target the GBM component. With
this multimodal approach and combination of chemotherapeutic agents
the patient did not show any sign of recurrence.
In 2016 O'Leary et al (11) have raised the doubt that using a CSI
irradiation with concomitant PNET-like chemotherapy could
under-treat the GMB component of GBM/PNET, especially in those with
MGMT methylation. In their study, they showed that CSI associated
with TMZ could represent a good choice for patients with
TMZ-sensitive tumours with high risk of CSF spread.
In 2017 Konar et al (22) reported a case of GBM/PNET which
developed extraparenchymal metastasis (dural metastasis) of pure
PNET component after surgery and chemoradiotherapy as Stupp
protocol. According to the authors early CSI in GBM/PNET patients
should be performed, even after completion of adjuvant
radio-chemotherapy.
GBM/PNET represents a diagnostic and therapeutic
challenge since the two histological components have distinct
clinico-pathological characteristics, treatment features and
prognosis (20). In GBM/PNETs the
role of surgery and radiotherapy as a first phase approach is
established. The fulcrum of the therapeutic dilemma is represented
by the choice of the best chemotherapy which should target both GBM
and PNET components and should be driven by the their histological
predominance.
In a clinical context, the therapeutic rationale for
GBM/PNET is trying to combine the gold-standard treatment for the
GBM component with an adequate therapeutic coverage for the risk of
CSF dissemination, typical of PNETs. A possible therapeutic
strategy could be represented by surgical resection followed by
radiotherapy, suitable for either the GBM and PNET components,
combining alkylating agents. According to the predominance of GBM
or PNET component, radiotherapy could consist of focal irradiation,
as Stupp protocol, or CSI with concomitant and adjuvant TMZ,
respectively (4,11). An early introduction of ‘PNET-like’
platinum-based chemotherapy should be planned in order to prevent
CSF dissemination or in case of disease recurrence (7,8,14,18,20).
Therefore it's fundamental to assess the two
components of GBM/PNET in order to guarantee the best treatment
choice (20). The preoperative
identification of areas of reduced ADC, typically of PNET-foci, on
MRI should address to consider the diagnosis of GBM/PNET and to
make targeted biopsies (14,15). Regarding the immunohistochemical
findings, the two components of GBM/PNET should be identified with
the use of GFAP and neuronal markers in order to quantify the two
histologies and plan successive therapeutic approaches.
Further studies on different treatment approaches
with long-term follow up are needed to optimize treatment algorithm
of this rare tumour type. Moreover new molecular targets related to
novel targeted therapies and newer biomarkers related to clinical
outcome are currently under investigation for gliomas and PNETs.
Many advances in the understanding of cancer biology are made in
the field of gene sequencing and interaction with tumor
microenvironment. They aim to identify alterations of the immune
system defing immune phenotypes (25,26) and
cancer pathways associated with intracellular and intercellular
signaling important to mediate cancer pathogenesis and progression
(27–33).
In conclusion our case report and the review of the
literature support the view that GBM/PNET should be treated
aggressively using a multimodal approach including surgery,
chemoradiotherapy and/or the early introduction of CSI and
platinum-based chemotherapy upfront or at recurrence, in order to
target both the PNET and GBM components.
References
|
1
|
Ostrom QT, Gittleman H, Farah P, Ondracek
A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the united states in 2006–2010. Neuro Oncol. 15
Suppl 2:ii1–i56. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Han BR, Choi HJ, Yang JS, Kang SH, Cho YJ
and Choi KC: Emerging variant glioma: Glioblastoma with a primitive
neuro-ectodermal tumor (PNET) component. The Nerve. 1:40–43. 2015.
View Article : Google Scholar
|
|
3
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year Analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karsy M, Gelbman M, Shah P, Balumbu O, Moy
F and Arslan E: Established and emerging variants of glioblastoma
multiforme: Review of morphological and molecular features. Folia
Neuropathol. 50:301–321. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perry A, Miller CR, Gujrati M, Scheithauer
BW, Zambrano SC, Jost SC, Raghavan R, Qian J, Cochran EJ, Huse JT,
et al: Malignant gliomas with primitive neuroectodermal tumor-like
components: A clinicopathologic and genetic study of 53 cases.
Brain Pathol. 19:81–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chu A, Bourgeois DJ and Prasad D: A
responsive yet persistently recurrent GBM with PNET features. Appl
Rad Oncol. 4:28–30. 2015.
|
|
9
|
Forbes V and Vredenburgh J: Primitive
neuroectodermal tumor with glioblastoma multiforme components in an
adult: A collision tumor. Cureus. 8:e4562016.PubMed/NCBI
|
|
10
|
Kim DG, Lee DY, Paek SH, Chi JG, Chloe G
and Jung HW: Supratentorial primitive neuroectodermal tumors in
adults. J Neurooncol. 60:43–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O'Leary B, Mandeville HC, Fersht N, Solda
F, Mycroft J, Zacharoulis S, Vaidya S and Saran F: Craniospinal
irradiation with concomitant and adjuvant temozolomide-a
feasibility assessment of toxicity in patients with glioblastoma
with a PNET component. J Neurooncol. 127:295–302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kandemir NO, Bahadir B, Gül S, Karadayi N
and Ozdamar SO: Glioblastoma with primitive neuroectodermal
tumor-like features: Case report. Turk Neurosurg. 19:260–264.
2009.PubMed/NCBI
|
|
13
|
Karina A, Jonker BP, Morey A, Selinger C,
Gupta R and Buckland ME: Glioblastoma with primitive
neuroectodermal tumour-like components. Pathology. 44:270–273.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ali S, Joseph NM, Perry A, Barajas RF Jr
and Cha S: Apparent diffusion coefficient in glioblastoma with
PNET-like components, a GBM variant. J Neurooncol. 119:353–360.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song X, Andrew Allen R, Terence Dunn S,
Fung KM, Farmer P, Gandhi S, Ranjan T, Demopoulos A, Symons M,
Schulder M and Li JY: Glioblastoma with PNET-like components has a
higher frequency of isocitrate dehydrogenase 1 (IDH1) mutation and
likely a better prognosis than primary glioblastoma. Int J Clin Exp
Pathol. 4:651–660. 2011.PubMed/NCBI
|
|
16
|
Gessi M, Setty P, Bisceglia M, zur Muehlen
A, Lauriola L, Waha A, Giangaspero F and Pietsch T: Supratentorial
primitive neuroectodermal tumors of the central nervous system in
adults: Molecular and histopathologic analysis of 12 cases. Am J
Surg Pathol. 35:573–582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayden JT, Frühwald MC, Hasselblatt M,
Ellison DW, Bailey S and Clifford SC: Frequent IDH1 mutations in
supratentorial primitive neuroectodermal tumors (sPNET) of adults
but not children. Cell Cycle. 8:1806–1807. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kimbason T, Turner SG, Kazmi SA, Fourgas
E, Gergel T, Belles L, Whitmire A, Lacroix M and Toms SA: Malignant
glioma with primitive neuroectodermal components: Clinical and
pathologic features with treatment modalities of five cases. J Clin
Exp Pathol. 5:2552015. View Article : Google Scholar
|
|
19
|
Varlet P, Soni D, Miquel C, Roux FX, Meder
JF, Chneiweiss H and Daumas-Duport C: New variants of malignant
glioneuronal tumors: A clinicopathological study of 40 cases.
Neurosurgery. 55:1377–1391. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karina A, Jonker BP, Morey A, Selinger C,
Gupta R and Buckland ME: Glioblastoma with primitive
neuroectodermal tumour-like components. Pathology. 44:270–273.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davis ME: Glioblastoma: Overview of
disease and treatment. Clin J Oncol Nurs. 20:S2–S8. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Konar SK, Bir SC, Maiti TK, Patra DP,
DiPoto Brahmbhatt AC, Jacobsohn JA and Nanda A: Early dural
metastasis from a case of glioblastoma with primitive
neuroectodermal differentiation: A case report and literature
review. J Clin Neurosci. 35:78–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Szopa W, Burley TA, Kramer-Marek G and
Kaspera W: Diagnostic and therapeutic biomarkers in glioblastoma:
Current status and future perspectives. Biomed Res Int.
2017:80135752017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee APS, Brewer J, Back M and Wheeler H:
Combination therapy for glioblastoma multiforme with primitive
neuroectodermal tumor components: Case series. J Clin Oncol. 30
Suppl 15:e125072012.
|
|
25
|
Mostafa H, Pala A, Högel J, Hlavac M,
Dietrich E, Westhoff MA, Nonnenmacher L, Burster T, Georgieff M,
Wirtz CR and Schneider EM: Immune phenotypes predict survival in
patients with glioblastoma multiforme. J Hematol Oncol. 9:772016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xue S, Hu M, Iyer V and Yu J: Blocking the
PD-1/PD-L1 pathway in glioma: A potential new treatment strategy. J
Hematol Oncol. 10:812017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ho WL, Hsu WM, Huang MC, Kadomatsu K and
Nakagawara A: Protein glycosylation in cancers and its potential
therapeutic applications in neuroblastoma. J Hematol Oncol.
9:1002016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Z, Guo Q, Wang R, Xu G, Li P, Sun Y,
She X, Liu Q, Chen Q, Yu Z, et al: The D domain of LRRC4 anchors
ERK1/2 in the cytoplasm and competitively inhibits MEK/ERK
activation in glioma cells. J Hematol Oncol. 9:1302016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Chong Y, Tu Y, Liu N, Yue C, Qi Z,
Liu H, Yao Y, Liu H, Gao S, et al: CRM1/XPO1 is associated with
clinical outcome in glioma and represents a therapeutic target by
perturbing multiple core pathways. J Hematol Oncol. 9:1082016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li B, Huang MZ, Wang XQ, Tao BB, Zhong J,
Wang XH, Zhang WC and Li ST: TMEM140 is associated with the
prognosis of glioma by promoting cell viability and invasion. J
Hematol Oncol. 8:892015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mao JM, Liu J, Guo G, Mao XG and Li CX:
Glioblastoma vasculogenic mimicry: Signaling pathways progression
and potential anti-angiogenesis targets. Biomark Res. 3:82015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Braoudaki M and Lambrou GI: MicroRNAs in
pediatric central nervous system embryonal neoplasms: The known
unknown. J Hematol Oncol. 8:62015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kelleher FC and Thomas DM: Molecular
pathogenesis and targeted therapeutics in ewing sarcoma/primitive
neuroectodermal tumours. Clin Sarcoma Res. 2:62012. View Article : Google Scholar : PubMed/NCBI
|