Introduction
Lung cancer is the leading cause of
cancer-associated mortality globally, with the highest mortality
rate due to abnormal cell proliferation and a high metastasis rate
in 2014 (1). Chemotherapy is the
standard treatment for advanced lung cancer with high metastatic
ability (2). However, resistance to
drugs is the most notable limitation of chemotherapy (3,4).
Therefore, understanding the mechanisms underlying the cellular
response and drug resistance of cancer is required in order to
improve chemotherapeutic efficacy. Docetaxel (DTX) is a
well-established anti-mitotic chemotherapy agent that functions by
interfering with cell division (5).
It is used to treat a number of cancer types including metastatic
or advanced breast, prostate, thyroid, gastric, ovarian and lung
cancer based on its pro-apoptosis potential and its inhibitory
effect on angiogenesis and cell viability (6–14). DTX has
also been demonstrated to upregulate growth/differentiation factor
15 and results in chemoresistance in prostate cancer (15). However, DTX-induced cellular
resistance responses in lung cancer remain unknown.
Inflammation is involved in the progression and
development of malignancies (16).
Balance between pro- and anti-inflammatory mechanisms is
responsible for tissue homeostasis (17). However, an abnormal imbalance is often
associated with the development of cancer accompanied by an
inflammatory response (18–21). Inflammatory responses are mediated by
specific cytokines and chemokines commonly expressed by immune
cells; however, cancer cells have also been reported to produce
cytokines and chemokines in order to promote tumorigenesis in an
inflammatory microenvironment (22).
C-C motif chemokine ligand 2 (CCL2) is a small chemokine, also
known as monocyte chemoattractant protein 1, belonging to the CC
chemokine family (23). CCL2 has been
reported to be secreted and to recruit immune cells to the sites of
inflammation produced by either tissue injury or infection
(24,25) and may also stimulate angiogenesis
through a direct effect on endothelial cells (26). CCL2 has additionally been revealed to
be associated with the development of cancer and to be highly
expressed in cancer cells (27). The
majority of studies of CCL2 focus on its effects on the migration
and metastasis of tumor cells, whereas its effects on cell
proliferation and viability have not been sufficiently explored. It
has been revealed that a CCL2 blockade may enhance the effect of
therapeutic methods on CCL2-induced inflammatory diseases and
cancer (28). Targeting CCL2 with
blocking antibodies has been demonstrated to suppress lung and bone
metastases in vivo, exhibiting a novel therapeutic approach
for cancer (29,30). Furthermore, it has been demonstrated
that CCL2 inhibition decreases the volume of subcutaneous tumor
xenografts in mice (31).
Additionally, CCL2 is involved in DTX-induced chemoresistance in
prostate cancer (32), and CCL2
inhibition augments DTX-induced inhibitory effects on the growth of
prostate cancer (33). CCL2 blockades
have also been demonstrated to inhibit tumor growth in lung cancer
(34,35). However, the role of CCL2 in the
sensitivity of lung cancer to DTX remains unclear.
In the present study, lung cancer A549 cells were
treated with DTX, and then CCL2 expression prior to and following
exposure to DTX was investigated in order to identify whether CCL2
is involved in the molecular mechanism underlying DTX-induced
resistance.
Materials and methods
Cell culture and reagents
The human non-small cell lung cancer cell line A549
was purchased from the American Type Culture Collection (Manassas,
VA, USA). Cells were cultured in Kaighn's modification of Ham's F12
(F12K) medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (FBS; Lonza Group,
Ltd., Basel, Switzerland), 5 mM non-essential amino acids, 5 mM
L-glutamine, 100 U/ml penicillin and streptomycin (Invitrogen;
Thermo Fisher Scientific, Inc.), in a humidified 5% CO2
incubator at 37°C. For serum-starved culture, cells were first
maintained in F12K medium with FBS for 24 h, and then PBS was used
to wash the cells three times and was replaced with F12K without
FBS. DTX was purchased from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany), dissolved in dimethylsulfoxide (DMSO). The
phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) inhibitor
LY294002 was purchased from Enzo Life Sciences, Inc. (Farmingdale,
NY, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from A549 cells using an RNA
isolation kit (A&A Biotechnology, Gdynia, Poland) according to
the manufacturer's protocol. cDNA was obtained by RT using a
RevertAid™ First Strand cDNA synthesis kit (Fermentas; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol
and was amplified using a TaqMan® Gene Expression assay
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with
fluorigenic carboxyfluorescein-labeled probes using the specific
primers for the target protein CCL2. PCR involved 40 amplification
cycles of 94°C for 10 sec, 53°C for 30 sec and 72°C for 40 sec,
followed by final extension at 72°C for 10 min. Primer sequences
for CCL2 were: 5′-GAACACACTCAGCGCAGTTA-3′ (forward primer) and
5′-CACCCACCCTCTCTTTGATTAC-3′ (reverse primer). Primer sequences for
GAPDH were 5′-CATGGCCTTCCGTGTTCCTA-3′ (forward primer) and R,
5′-CCTGCTTCACCACCTTCTTGAT-3′ (reverse primer). Fluorescence
detection was performed using the ABI PRISM 7700 Sequence Detector
(PerkinElmer; Applied Biosystems; Thermo Fisher Scientific, Inc.).
The mRNA expression of CCL2 was calculated using the formula
2−ΔΔCq (36) and was
normalized to the level of GAPDH. The relative level of CCL2 mRNA
was presented as a percentage of the control.
Western blot analysis
A total of 5×105 A549 cells were plated
in a culture dish and grown to 80% confluence, and then treated
with 0, 2 and 4 nM of DTX. The cells were then washed twice with
PBS and homogenized with M-PER Mammalian Protein Extraction reagent
(Pierce; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Following centrifugation at 12,000 × g for
10 min at 4°C, the supernatant was collected and quantified using a
bicinchoninic acid quantification kit (Beyotime Institute of
Biotechnology, Haimen, China). The proteins (50 µg) were separated
by SDS-PAGE (12% gel; Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) and transferred onto polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 5% non-fat dried milk in Tris-buffered
saline with 0.1% Tween-20 for 1 h at room temperature, and
incubated with the following specific primary antibodies overnight
at 4°C: Anti-CCL2 antibody (cat. no. MAB679; 1:500; R&D Systems, Inc.,
Minneapolis, MN, USA), anti-B-cell lymphoma 2 (Bcl-2, cat. no.
sc7382; 1:500) and anti-GAPDH antibodies (cat. no. sc-365062;
1:3,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
anti-phospho-Bcl-2 (pSer70, cat. no. 2871; 1:1,000),
anti-Bcl-2-associated death promoter (Bad; cat. no. 9292; 1:1,000),
anti-phospho-Bad (pSer112, cat. no. 9291; 1:1,000),
anti-protein kinase B (AKT; cat. no. 9272; 1:1,000) and
anti-phospho-AKT (pSer473; cat. no. 9271; 1:1,000)
antibodies (Cell Signaling Technology, Inc., Danvers, MA, USA).
This was followed by incubation with horseradish
peroxidase-conjugated secondary antibodies goat anti-mouse (cat.
no. sc-2005; 1:2,000) and anti-rabbit immunoglobulin G (cat. no.
sc-2004; 1:2,000; both Santa Cruz Biotechnology, Inc.) for 2 h at
room temperature. Then the membranes were washed with Tris-buffered
saline with 0.1% Tween-20 three times for 5 min each time.
Development was performed using an enhanced
chemiluminescence-detecting reagent (GE Healthcare, Chicago, IL,
USA). The protein blots were quantified by densitometry using
QuantityOne software (version 4.6.2; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), and the amounts were expressed relative to the
internal reference GAPDH.
Coomassie blue stain
The staining solution containing 0.1%
Coomassie® R-250 (Thermo Fisher Scientific Inc.,
Rochester, NY, USA) was prepared in 40% ethanol and 10% acetic
acid. Following SDS-PAGE as described above, gel is incubated in a
staining container containing 100 ml Coomassie® Blue
R-250 staining solution at 95°C for 10 min and then gently agitated
for 15 min at room temperature. The gel was rinsed once with
deionized water. A destaining solution containing 10% ethanol and
7.5% acetic acid was prepared. The stained gel was placed in a
staining container containing 100 ml destaining solution, gently
agitated at room temperature on an orbital shaker until the desired
background was achieved.
CCL2 siRNA and overexpression
CCL2 small interfering RNA (siRNA) and control siRNA
were obtained from GE Healthcare Dharmacon, Inc. (Lafayette, CO,
USA). The sequence of CCL2 siRNA: 5′-CTCGCGAGCTATAGAAGAA-3′. The
small interference RNA of negative control (siCtrl): Sense,
5′-UUCUCCGAACGUGUCACGUTT-3′; Antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of 2×104 A549 cells
were seeded in 12-well plate and cultured for 24 h, and then
transfected using 100 nmol siRNA and 5 µl Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h according to
the manufacturer's protocol. Cells were grown for a further 48 h
following transfection, and then were lysed in lysis buffer [50 mM
Tris-HCl (pH 7.4), 1 mM EDTA, 1% NP40, 150 mM NaCl, 10 mM NaF, 1 mM
Na3VO4] containing a protease inhibitor cocktail (Roche, NJ, USA)
and used for protein expression analysis. A recombinant lentiviral
vector containing CCL2 and control vector were obtained from the
Department of Medicine, Oregon Health & Science University
(Portland, OR, USA). The lentiviral infection and establishment of
A549 cells with stable CCL2 expression was performed as previously
described (37).
Cell viability assay
A549 cell viability was evaluated using an MTT assay
(Sigma Aldrich; Merck KGaA). MTT assay was performed using 96-well
plate according to the manufacturer's protocol. In total,
5×104 cells/well were cultured at 37°C with 5%
CO2 overnight and 0, 2, 4 or 15 nM DTX was added to the
culture for 6 days or 4 nM for 48 h. Then cells were incubated with
20 µl MTT (5 mg/ml in PBS) for 4 h at 37°C, and then cells were
lysed for 10 min by the addition of 200 µl DMSO (OriGene
Technologies, Inc., Rockville, MD, USA) used to dissolve the
formazan crystals. Absorbance was measured at 570 nm using a
Rainbow microplate reader (Tecan Group, Ltd., Mannedorf,
Switzerland). Cell viability was expressed as a percentage of the
untreated control.
Caspase-3 activity assay
Caspase-3 activity was evaluated in A549 cells using
the human active caspase-3 ELISA kit (cat. no. KM300; R&D
Systems, Inc.) according to the manufacturer's protocol. In total,
5×105 cells/well were cultured in 6-well plates for 24 h
at 37°C. Cells were treated with or without 4 nM DTX/5 µM LY294002
for 48 h at 37°C. Subsequently, the cells were lysed with
Extraction Buffer (included in the human active caspase-3 ELISA
kit) and analyzed according to the manufacturer's protocol.
Absorbance was measured at 570 nm using a microplate reader.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical analyses were performed using SPSS software
(version 11.0; SPSS, Inc., Chicago, IL, USA). All experiments were
performed at least three times. One-way analysis of variance
(ANOVA) was used to assess differences between groups. Duncan's new
multiple range test as a post hoc test was used following ANOVA for
pairwise comparison followed by Bonferroni's correction. P<0.05
was considered to indicate a statistically significant
difference.
Results
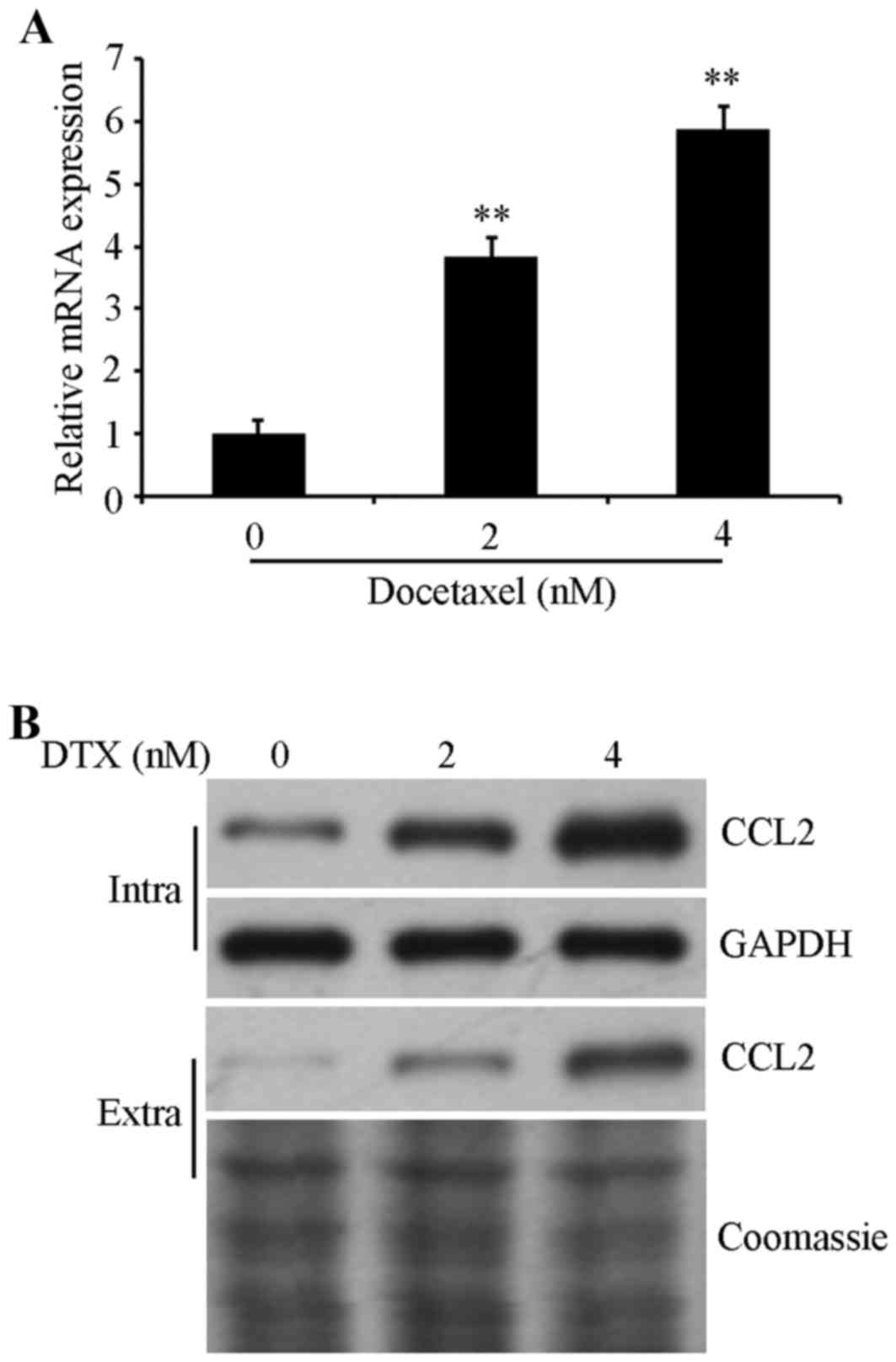
CCL2 is increased in DTX-treated A549
cells
To investigate the role of CCL2 in regulating the
DTX resistance of lung cancer, CCL2 expression in was detected in
A549 cells prior to and following DTX treatment. Cells were exposed
to 0, 2 and 4 nM DTX for 72 h, and then the mRNA expression level
was determined using RT-qPCR and protein expression was examined
using western blot analysis. The results confirmed that the mRNA
expression levels of CCL2 were significantly increased (P<0.01;
Fig. 1A) and the
intracellular/extracellular protein expression of CCL2 were
increased in DTX-treated cells in a dose-dependent manner compared
with non-treated control cells (Fig.
1B), suggesting that CCL2 is associated with DTX-induced
resistance in lung cancer A549 cells.
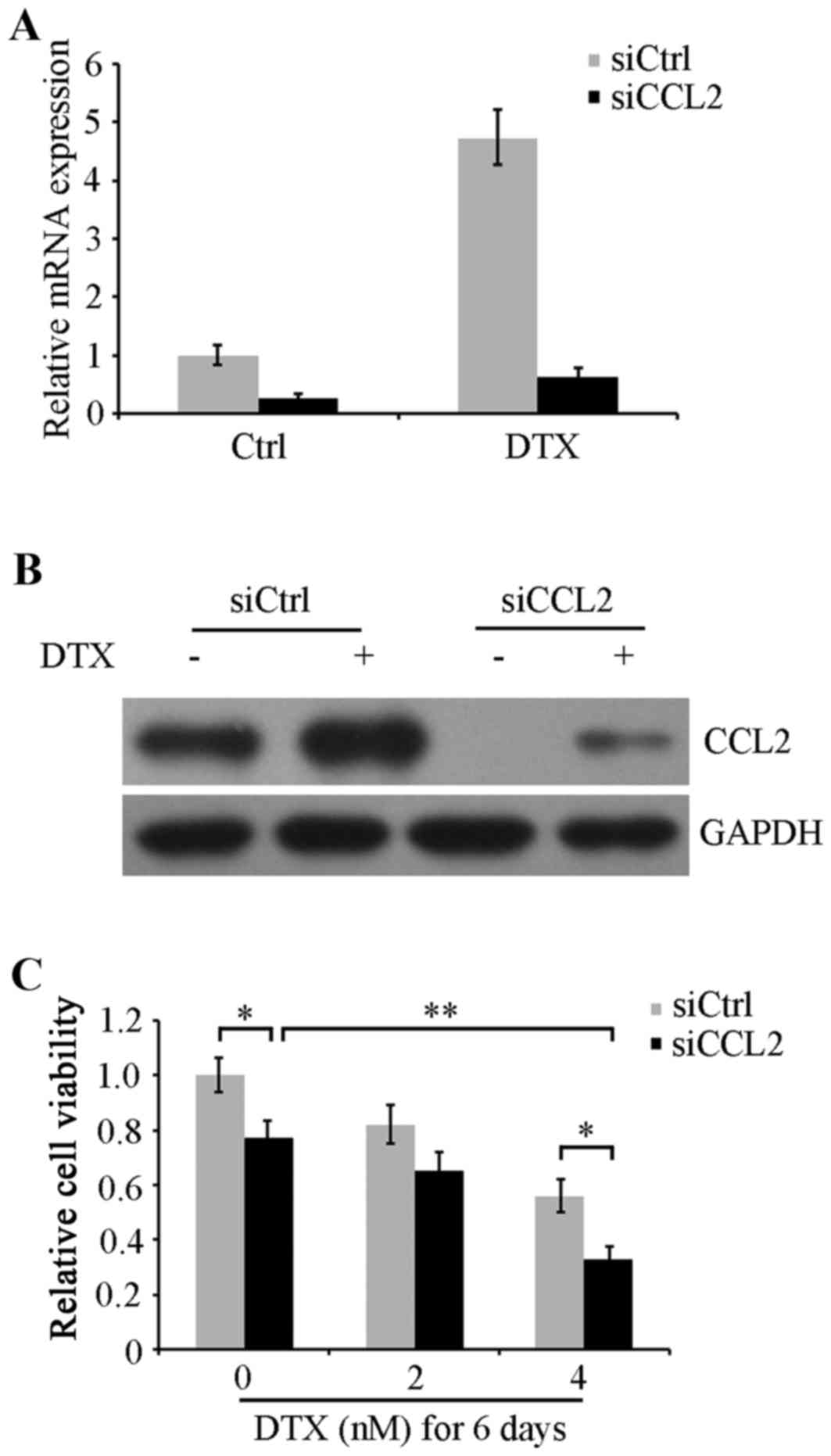
CCL2 downregulation augments the
inhibition of cell viability by DTX
To confirm the role of DTX-induced CCL2 expression
in the chemoresistance of lung cancer, CCL2 expression was knocked
down using siRNA in A549 cells. Cells were transfected with CCL2
siRNA or control siRNA for 48 h followed by 24 h recovery, and then
were treated with or without 4 nM DTX for another 48 h. RT-qPCR and
western blot analysis were performed to determine CCL2 mRNA
expression levels and protein expression. The data revealed that
DTX-induced CCL2 expression was substantially decreased in CCL2
siRNA treated cells compared with siRNA control-transfected cells
(Fig. 2A and B). Next, following
transfection, CCL2-silenced and non-silenced A549 cells were
maintained in serum-free medium with 0, 2 and 4 nM DTX for 6 days.
Cell viability was evaluated using an MTT assay. The results
indicated that CCL2-silenced cells exhibited significantly
decreased viability compared with non-silenced cells in groups
treated with 0 and 4 nM DTX (P<0.05). Cell viability was also
decreased by DTX in a concentration-dependent manner. Notably, CCL2
silencing in combination with 4 nM DTX revealed a significantly
increased viability inhibition compared with DTX alone or CCL2
siRNA alone (P<0.01; Fig. 2C).
These results imply that the DTX resistance response in lung cancer
A549 cells may be caused by DTX-induced CCL2 expression, and CCL2
inhibition may attenuate DTX resistance and augment DTX-induced
cytotoxicity.
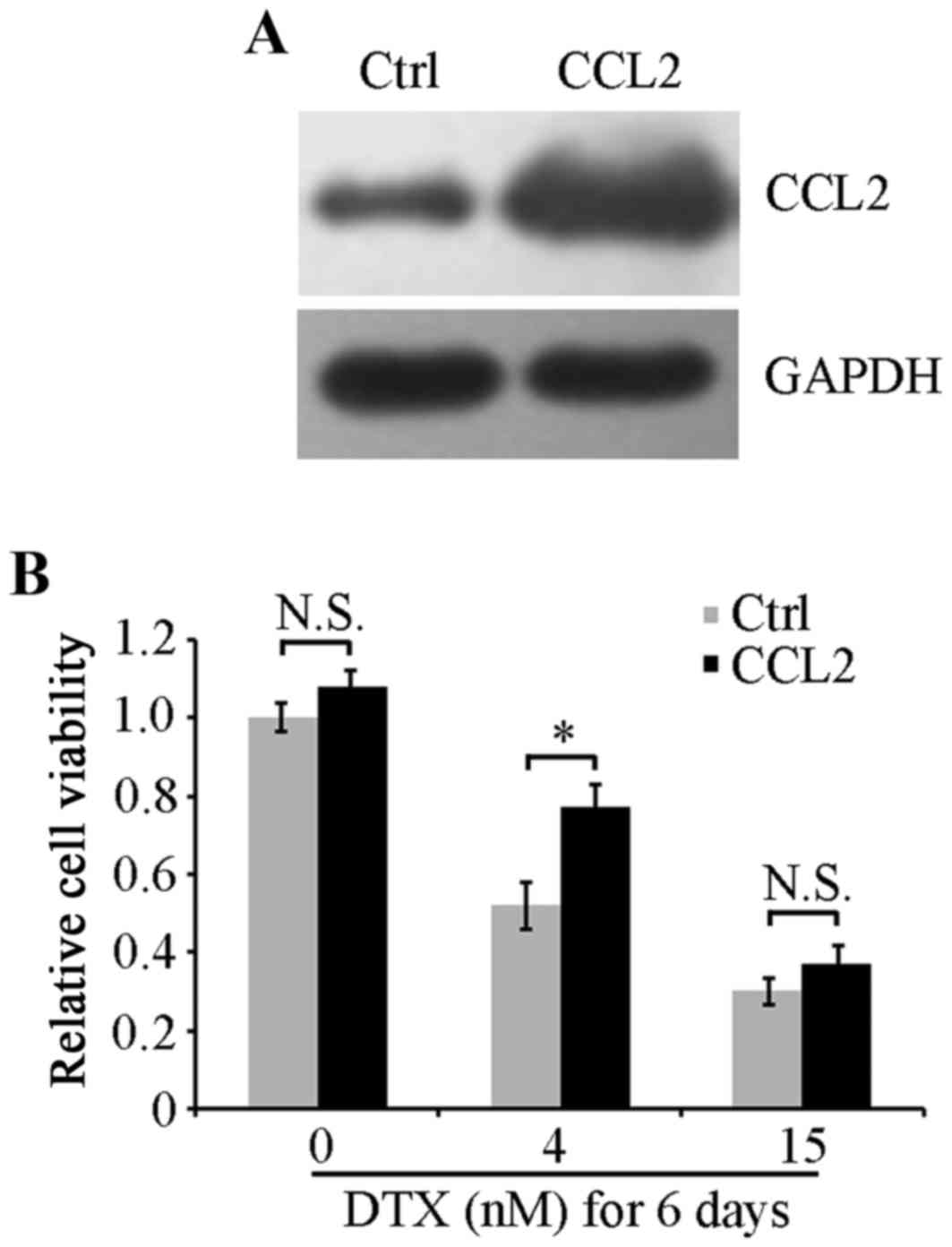
CCL2 upregulation attenuates DTX
cytotoxicity
To verify the role of CCL2 in DTX resistance of lung
cancer A549 cells, an A549 cell line that stably overexpressed CCL2
was established using recombinant CCL2 lentiviruses. Western blot
analysis revealed that CCL2 was effectively overexpressed in CCL2
lentivirus-infected cells and was increased compared with control
lentivirus-infected cells (Fig. 3A).
Viability of cells with CCL2 overexpression or empty control
vectors was examined using an MTT assay. No significant differences
were identified in cell viability between the two cell lines under
serum-free culture conditions (Fig.
3B). When treated with 4 nM DTX for 6 days, CCL2-overexpressed
cells exhibited a significantly increased cell viability compared
with control cells (P<0.05; Fig.
3B). However, when cells were treated with a high concentration
of DTX at 15 nM, CCL2 overexpression did not attenuate the
inhibitory effect on cell viability induced by DTX, suggesting that
the resistance response in A549 cells may be induced by DTX at a
low concentration but not at a high concentration that caused the
suppression of cell viability to sufficiently overcome the positive
effect of CCL2 on cell viability.
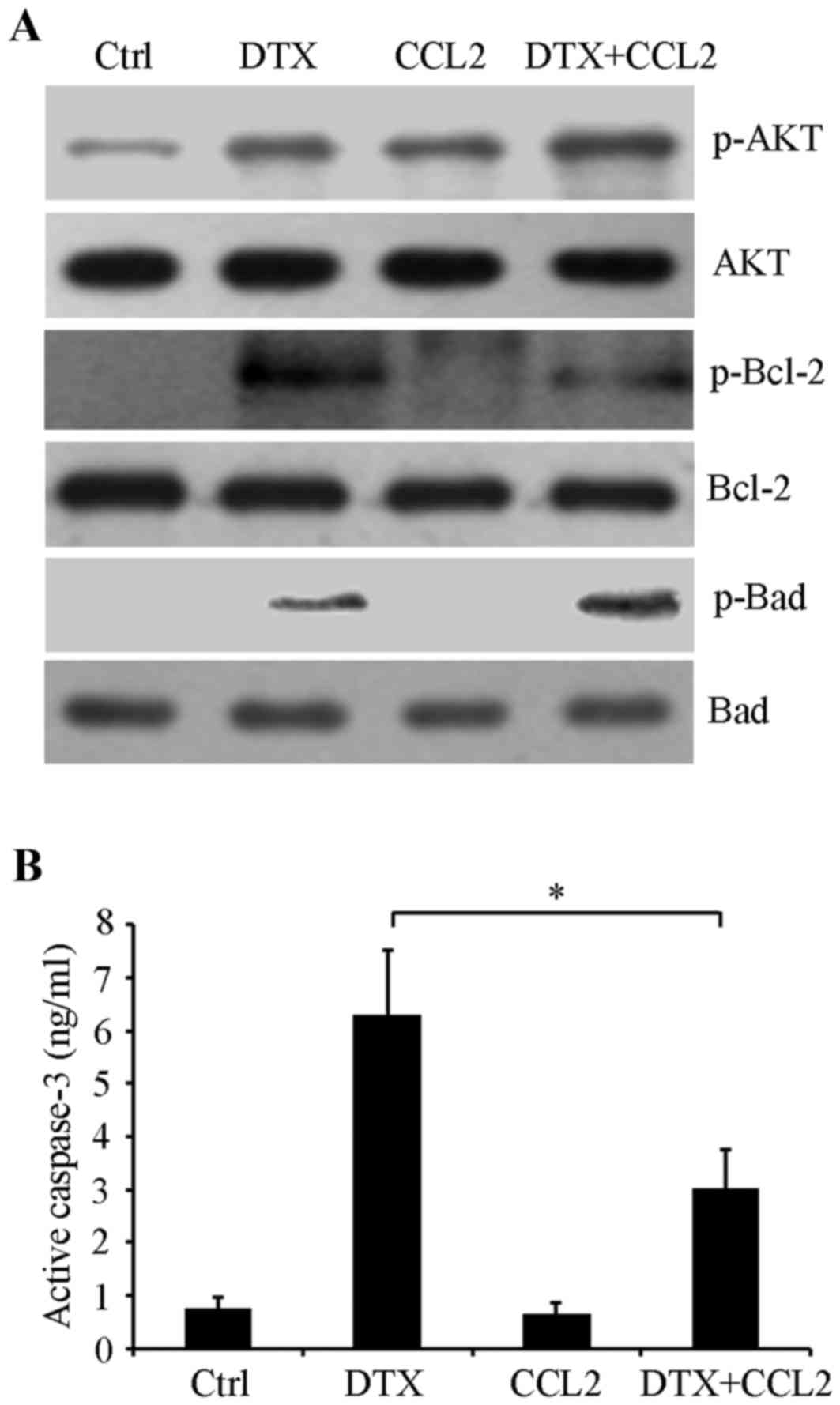
CCL2 influences the activation of
viability and apoptosis-associated signaling
To determine the mechanism underlying the
CCL2-mediated DTX resistance response in lung cancer cells, cells
with CCL2 overexpression or empty control vectors were treated with
or without 4 nM DTX for 48 h in serum-free culture medium. Western
blot analysis results revealed that DTX treatment induced AKT
phosphorylation at Ser473 compared with the control and
CCL2 overexpression also substantially stimulated the activation of
AKT compared with the control by promoting its phosphorylation at
Ser473. DTX in combination with CCL2 notably enhanced
AKT phosphorylation (Fig. 4A),
suggesting that CCL2 mediated PI3K/AKT signaling activation serves
an important function in the DTX induced resistance response. Next,
activities of apoptosis-associated proteins including the
anti-apoptotic protein Bcl-2, pro-apoptotic protein Bad and
caspase-3 were detected. Western blot analysis revealed that the
phosphorylation of Bcl-2 at Ser70 was induced by DTX,
whereas its phosphorylation was attenuated by CCL2. Meanwhile, CCL2
enhanced the phosphorylation of the pro-apoptotic protein Bad at
Ser112 induced by DTX (Fig.
4A). Additionally, a caspase-3 activity assay indicated that
CCL2 significantly inhibited the activation of caspase-3 induced by
DTX (P<0.05; Fig. 4B). These data
suggest that CCL2 attenuates DTX-induced cytotoxicity potentially
by regulating the activities of the PI3K/AKT signaling pathway and
these apoptosis-associated proteins.
Inhibition of PI3K/AKT signaling
augments DTX cytotoxicity
In order to further confirm whether PI3K/AKT
activation participated in regulating the activities of the
aforementioned apoptosis-associated proteins, the PI3K/AKT-specific
inhibitor LY294002 was used to inhibit this signaling pathway.
Control, normal and CCL2-overexpressing cells were treated with or
without 4 nM DTX, or 4 nM DTX combined with 5 µM LY294002 for 48 h.
Western blot analysis revealed that LY294002 restored the
phosphorylation of Bcl-2 compared with CCL2-overexpressing cells
treated with DTX alone, whereas Bad phosphorylation at
Ser112 was unaffected, suggesting that it may be a
target of other signaling molecules (Fig.
5A). Additionally, the activity of caspase-3 in
CCL2-overexpressing cells treated with DTX was significantly
restored by LY294002 (P<0.05; Fig.
5B). Therefore, it may be that DTX-induced CCL2 stimulates the
PI3K/AKT signaling pathway to resist DTX-induced cytotoxicity.
Next, cell viability was determined using an MTT assay. The results
revealed that LY294002 enhanced DTX inhibition on cell viability in
CCL2-overexpressing cells (P<0.05; Fig. 5C), indicating that PI3K/AKT is a
promising target for improving the efficacy of chemotherapy.
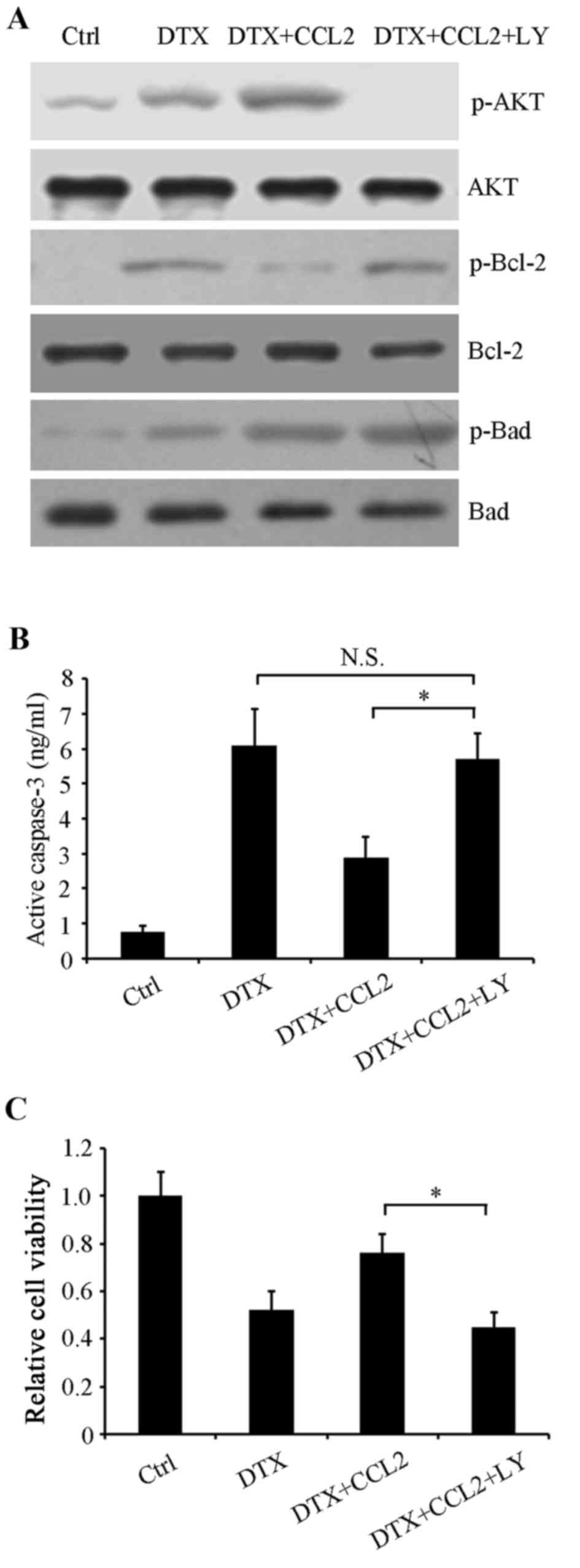 | Figure 5.Phosphoinositide 3-kinase/AKT
signaling pathway participates in the CCL2-mediated DTX-resistance
response. A549 cells were treated with or without 4 nM DTX or 5 µM
LY, or a combination, for 48 h. (A) Western blot analysis was
performed to examine the phosphorylation of AKT, Bcl-2 and Bad
using their respective specific antibodies. Total AKT, Bcl-2 and
Bad were used as loading controls. (B) Caspase-3 activity was
detected using a human active caspase-3 ELISA. (C) Cell viability
was evaluated using an MTT assay. *P<0.05 with comparisons shown
by lines. N.S., not significant; AKT, protein kinase B; CCL2, C-C
motif chemokine ligand 2; DTX, docetaxel; LY, LY294002; Ctrl,
control; p-, phosphorylated; AKT, protein kinase B; Bcl-2, B-cell
lymphoma 2; Bad, Bcl-2-associated death promoter. |
Discussion
Lung cancer is a common malignancy with the highest
mortality rate in the United States in 2014 (1). DTX is widely used as a cancer treatment
(6–14). However, the emergence of
chemotherapeutic resistance iTHs universal and DTX-resistance
responses have been observed in a number of patients (15). In the present study, it was
demonstrated that DTX induced a resistance response in lung cancer
A549 cells and the underlying molecular mechanism was investigated.
A comparison of mRNA and protein levels of the chemokine CCL2 prior
to and following DTX treatment identified an increase in CCL2 in
DTX-treated A549 cells. Additionally, it was revealed that CCL2
silencing augmented the inhibition of DTX on viability, whereas
CCL2 upregulation protected A549 cells from DTX-induced
cytotoxicity.
PI3K/AKT signaling has been reported to be
positively associated with cell viability and apoptosis resistance,
and PI3K may promote tumor cell survival by triggering AKT
activation (38–40). In the present study, it was indicated
that DTX treatment or CCL2 overexpression, or the two combined,
activated AKT by inducing AKT phosphorylation at Ser473,
suggesting the involvement of PI3K/AKT signaling in a DTX-induced
resistance response. AKT is able to directly control apoptosis by
regulating phosphorylation to affect the activities of
apoptosis-associated proteins including the pro-apoptotic Bad and
caspase cascades (41–43). AKT may phosphorylate Bad at several
sites to inactivate the pro-apoptotic function (44). It was revealed that DTX induced CCL2
and activated AKT, and CCL2 enhanced DTX-induced Bad
phosphorylation at Ser112 leading to Bad inactivation,
which blocked apoptosis. However, the PI3K/AKT inhibitor LY294002
did not alter Bad phosphorylation at Ser112, suggesting
that Bad Ser112 may not be the target of PI3K/AKT in the
resistance response. Additionally, DTX has been demonstrated to
promote the phosphorylation of anti-apoptotic protein Bcl-2 at
Ser70 (45,46). It has also been revealed that Bcl-2
phosphorylation abolishes its anti-apoptosis effect, resulting in
caspase-3 activation and cellular apoptosis (47,48). It
was revealed that DTX induced Bcl-2 phosphorylation at
Ser70 resulting in its inactivation and the activation
of caspase-3 that triggered cell apoptosis, whereas CCL2
overexpression attenuated Bcl-2 phosphorylation and caspase-3
activation induced by DTX, suggesting that CCL2 decreased
DTX-induced cytotoxicity by blocking cell apoptosis. Additionally,
it was indicated that the inhibition of PI3K/AKT by the inhibitor
LY294002 augmented DTX treatment toxicity, implying that
chemotherapy combined with PI3K/AKT inhibition may be more
efficient for the treatment of lung cancer.
CCL2 may be regulated by multiple mechanisms. It has
been reported that CCL2 expression is upregulated by DTX via c-Jun
N-terminal kinases (JNKs) and nuclear factor-κB (NF-κB) pathways in
prostate cancer (32). Whether CCL2
was induced by DTX via JNK or NF-κB signaling pathways requires
further studies. In addition, it has also been revealed that tumor
protein p53 binds to CCL2, consequently significantly
downregulating CCL2 promoter activity, and thus suppressing
CCL2-induced subcutaneous tumor xenografts (49). CCL2 may be regulated by myeloid
differentiation primary response 88 in murine mammary carcinomas
and thus affect cell viability and metastasis (50). CCL2 was revealed to be involved in
visfatin-mediated lung cancer NCI-H446 cells transendothelial
migration and visfatin-induced CCL2 was attenuated by a specific
inhibitor of PI3K/AKT signaling (51). CCL2 was also been revealed to be
inhibited by atypical chemokine receptor D6 and then regulated cell
viability in lung cancer A549 cells (52). The identity of the molecules involved
in the modulation of CCL2 induced by DTX is yet to be
determined.
Monoclonal antibodies against CCL2 have been
administered in lung cancer immunotherapy models and have been
identified to exhibit antitumor activity (34,35). The
results of the present study confirm this the effect of targeting
CCL2 by siRNA in DTX treatment. These results also imply that
targeting DTX-induced CCL2 expression combined with DTX may
effectively decrease chemotherapy-induced resistance and improve
drug efficacy in lung cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TW, XP, ZQ and TZ were the major contributors in
conception and design of the research and revision of manuscript
for important intellectual content. Acquisition of data was
performed by QZ. TW and XP were the major contributors in analysis
and interpretation of data and statistical analysis. Drafting the
manuscript was performed by TW and XP.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
PDQ Adult Treatment Editorial Board:
Non-small cell lung cancer treatment (PDQ®Patient
version. NCI. 2002–2015 May 12.
|
|
2
|
Evans TL: Chemotherapy in advanced
non-small cell lung cancer: Optimal treatment approach for elderly
and patients with poor performance status. Lung cancer. Am J Hemat
Oncol. 7:12–16. 2015.
|
|
3
|
Du Y, Su T, Zhao L, Tan X, Chang W, Zhang
H and Cao G: Associations of polymorphisms in DNA repair genes and
MDR1 gene with chemotherapy response and survival of non-small cell
lung cancer. PLoS One. 9:e998432014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen WL, Kuo KT, Chou TY, Chen CL, Wang
CH, Wei YH and Wang LS: The role of cytochrome c oxidase subunit Va
in non-small cell lung carcinoma cells: Association with migration,
invasion and prediction of distant metastasis. BMC Cancer.
12:2732012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McKeage K: Docetaxel: A review of its use
for the first-line treatment of advanced castration-resistant
prostate cancer. Drugs. 72:1559–1577. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lyseng-Williamson KA and Fenton C:
Docetaxel: A review of its use in metastatic breast cancer. Drugs.
65:2513–2531. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bayet-Robert M, Morvan D, Chollet P and
Barthomeuf C: Pharmacometabolomics of docetaxel-treated human MCF7
breast cancer cells provides evidence of varying cellular responses
at high and low doses. Breast Cancer Res Treat. 120:613–26. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Niu L, Deng J, Zhu F, Zhou N, Tian K, Yuan
H and Lou H: Anti-inflammatory effect of Marchantin M contributes
to sensitization of prostate cancer cells to docetaxel. Cancer
Lett. 348:126–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim E, Matsuse M, Saenko V, Suzuki K,
Ohtsuru A, Mitsutake N and Yamashita S: Imatinib enhances
docetaxel-induced apoptosis through inhibition of nuclear factor-κB
activation in anaplastic thyroid carcinoma cells. Thyroid.
22:717–724. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu H, Xin Y, Zhao J, Sun D, Li W, Hu Y and
Wang S: Metronomic docetaxel chemotherapy inhibits angiogenesis and
tumor growth in a gastric cancer model. Cancer Chemother Pharmacol.
68:879–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Hu YL and Cheng YY: Docetaxel
influences autocrine of transforming growth factors and induces
apoptosis in human ovarian cancer cell line AO. Chin Med Sci J.
21:2042006.PubMed/NCBI
|
|
12
|
He X, Li C, Wu X and Yang G: Docetaxel
inhibits the proliferation of non-small-cell lung cancer cells via
upregulation of microRNA-7 expression. Int J Clin Exp Pathol.
8:9072–9080. 2015.PubMed/NCBI
|
|
13
|
Indo K, Hoshikawa H, Kamitori K, Yamaguchi
F, Mori T, Tokuda M and Mori N: Effects of D-allose in combination
with docetaxel in human head and neck cancer cells. Int J Oncol.
45:2044–2050. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
http://www.cancer.gov/cancertopics/druginfo/fda-docetaxelNational
Cancer Institute. Last Updated. 3:282014.
|
|
15
|
Huang CY, Beer TM, Higano CS, True LD,
Vessella R, Lange PH, Garzotto M and Nelson PS: Molecular
alterations in prostate carcinomas that associate with in vivo
exposure to chemotherapy: Identification of a cytoprotective
mechanism involving growth differentiation factor 15. Clin Cancer
Res. 13:5825–5833. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
de Marzo AM, Platz EA, Sutcliffe S, Xu J,
Gronberg H, Drake CG, Nakai Y, Isaacs WB and Nelson WG:
Inflammation in prostate carcinogenesis. Nat Rev Cancer. 7:256–269.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seruga B, Zhang H, Bernstein LJ and
Tannock IF: Cytokines and their relationship to the symptoms and
outcome of cancer. Nat Rev Cancer. 8:887–899. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiba T, Marusawa H and Ushijima T:
Inflammation-associated cancer development in digestive organs:
Mechanisms and roles for genetic and epigenetic modulation.
Gastroenterology. 143:550–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kawanishi S, Ohnishi S, Ma N, Hiraku Y and
Murata M: Crosstalk between DNA damage and inflammation in the
multiple steps of carcinogenesis. Int J Mol Sci. 18:18082017.
View Article : Google Scholar
|
|
20
|
Ding N, Maiuri AR and O'Hagan HM: The
emerging role of epigenetic modifiers in repair of DNA damage
associated with chronic inflammatory diseases. Mutation
Research-Reviews in Mutation Research: Online September 28, 2017.
In Press.
|
|
21
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sica A, Wang JM, Colotta F, Dejana S,
Mantovani A, Oppenheim JJ, Larsen CG, Zachariae CO and Matsushima
K: Monocyte chemotactic and activating factor gene expression
induced in endothelial cells by IL-1 and tumor necrosis factor. J
Immunol. 144:3034–3038. 1990.PubMed/NCBI
|
|
24
|
Carr MW, Roth SJ, Luther E, Rose SS and
Springer TA: Monocyte chemoattractant protein 1 acts as a
T-lymphocyte chemoattractant. Proc Natl Acad Sci USA. 91:3652–3656.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu LL, Warren MK, Rose WL, Gong W and Wang
JM: Human recombinant monocyte chemotactic protein and other C-C
chemokines bind and induce directional migration of dendritic cells
in vitro. J Leukoc Biol. 60:365–371. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barcelos LS, Talvani A, Teixeira AS,
Cassali GD, Andrade SP and Teixeira MM: Production and in vivo
effects of chemokines CXCL1-3/KC and CCL2/JE in a model of
inflammatory angiogenesis in mice. Inflamm Res. 53:576–584. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Lu Y and Pienta KJ: Multiple
roles of chemokine (C-C motif) ligand 2 in promoting prostate
cancer growth. J Natl Cancer Inst. 102:522–528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ajuebor MN, Swain MG and Perretti M:
Chemokines as novel therapeutic targets in inflammatory diseases.
Biochem Pharmacol. 63:1191–1196. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Said N, Sanchez-Carbayo M, Smith SC and
Theodorescu D: RhoGDI2 suppresses lung metastasis in mice by
reducing tumor versican expression and macrophage infiltration. J
Clin Invest. 122:1503–1518. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qian BZ, Li J, Zhang H, Kitamura T, Zhang
J, Campion LR, Kaiser EA, Snyder LA and Pollard JW: CCL2 recruits
inflammatory monocytes to facilitate breast-tumour metastasis.
Nature. 475:222–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Abangan RS Jr, Williams CR, Mehrotra M,
Duncan JD and Larue AC: MCP1 directs trafficking of hematopoietic
stem cell-derived fibroblast precursors in solid tumor. Am J
Pathol. 176:1914–1926. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qian DZ, Rademacher BL, Pittsenbarger J,
Huang CY, Myrthue A, Higano CS, Garzotto M, Nelson PS and Beer TM:
CCL2 is induced by chemotherapy and protects prostate cancer cells
from docetaxel-inducedcytotoxicity. Prostate. 70:433–442.
2010.PubMed/NCBI
|
|
33
|
Kirk PS, Koreckij T, Nguyen HM, Brown LG,
Snyder LA, Vessella RL and Corey E: Inhibition of CCL2 signaling in
combination with docetaxel treatment has profound inhibitory
effects on prostate cancer growth in bone. Int J Mol Sci.
14:10483–10496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fridlender ZG, Buchlis G, Kapoor V, Cheng
G, Sun J, Singhal S, Crisanti MC, Wang LC, Heitjan D, Snyder LA and
Albelda SM: CCL2 blockade augments cancer immunotherapy. Cancer
Res. 70:109–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fridlender ZG, Kapoor V, Buchlis G, Cheng
G, Sun J, Wang LC, Singhal S, Snyder LA and Albelda SM: Monocyte
chemoattractant protein-1 blockade inhibits lung cancer tumor
growth by altering macrophage phenotype and activating CD8+ cells.
Am J Respir Cell Mol Biol. 44:230–237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qian DZ, Wei YF, Wang X, Kato Y, Cheng L
and Pili R: Antitumor activity of the histone deacetylase inhibitor
MS-275 in prostate cancer models. Prostate. 67:1182–1193. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Annovazzi L, Mellai M, Caldera V, Valente
G, Tessitore L and Schiffer D: mTOR, S6 and AKT expression
inrelation to proliferation and apoptosis/autophagy in glioma.
Anticancer Res. 29:3087–3094. 2009.PubMed/NCBI
|
|
39
|
Falasca M: PI3K/Akt signaling pathway
specific inhibitors: A novel strategy to sensitize cancer cells to
anti-cancer drugs. Curr Pharm Des. 16:1410–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mabuchi S, Ohmichi M, Kimura A, Hisamoto
K, Hayakawa J, Nishio Y, Adachi K, Takahashi K, Arimoto-Ishida E,
Nakatsuji Y, et al: Inhibition of phosphorylation of BAD and Raf-1
by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol
Chem. 277:33490–33500. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo
J and Rosen N: The BAD protein integrates survival signaling by
EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor
cells. Cancer Cell. 8:287–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Basu A and Haldar S: Microtubule-damaging
drugs triggered bcl2 phosphorylation-requirement of phosphorylation
on both serine-70 and serine-87 residues of bcl2 protein. Int J
Oncol. 13:659–664. 1998.PubMed/NCBI
|
|
46
|
Haldar S, Basu A and Croce CM: Serine-70
is one of the critical sites for drug-induced Bcl2 phosphorylation
in cancer cells. Cancer Res. 58:1609–1615. 1998.PubMed/NCBI
|
|
47
|
Wang TH, Wang HS, Ichijo H, Giannakakou P,
Foster JS, Fojo T and Wimalasena J: Microtubuleinterfering agents
activate c-Jun N-terminal kinase/stress-activated protein kinase
through both Ras and apoptosis signal-regulating kinase pathways. J
Biol Chem. 273:4928–4936. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yamamoto K, Ichijo H and Korsmeyer SJ:
BCL-2 is phosphorylated and inactivated by an ASK1/Jun nterminal
protein kinase pathway normally activated at G(2)/M. Mol Cell Biol.
19:8469–8478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tang XR and Amar S: p53 suppresses
CCL2-induced subcutaneous tumor xenograft. Tumour Biol.
36:2801–2808. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Egunsola AT, Zawislak CL, Akuffo AA,
Chalmers SA, Ewer JC, Vail CM, Lombardo JC, Perez DN and Kurt RA:
Growth, metastasis, and expression of CCL2 and CCL5 by murine
mammary carcinomas are dependent upon Myd88. Cell Immunol.
272:220–229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu T, Miao Z, Jiang J, Yuan S, Fang W, Li
B and Chen Y: Visfatin mediates SCLC cells migration across brain
endothelial cells through upregulation of CCL2. Int J Mol Sci.
16:11439–11451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wu FY, Fan J, Tang L, Zhao YM and Zhou CC:
Atypical chemokine receptor D6 inhibits human non-small cell lung
cancer growth by sequestration of chemokines. Oncol Lett. 6:91–95.
2013. View Article : Google Scholar : PubMed/NCBI
|