Introduction
Breast cancer is one of the most common and
malignant tumor types among females globally, which accounts for
30% all new cancer diagnoses in females (1). Additionally, with a global annual
increase of ~200 million patients, the mortality rate is increasing
each year (2). On average there is a
female diagnosed with breast cancer every three minutes globally
(3). In China, the annual incidence
of female breast cancer has experienced a sharp increase from 3 to
4% of the female population, which is notably higher than the
average global growth rate for the diagnosis of breast cancer
(4). Chemotherapy remains an
important breast cancer treatment; however, clinical practice has
confirmed that 30–50% of patients with breast cancer are either not
sensitive to the treatment or the treatment does not produce
effective results (5). Rather, they
demonstrate heart and kidney side effects, which frequently cause
extensive physical and mental harm to patients (6). Thus, it is a common goal of doctors and
patients to discover novel drugs that improve the efficacy and
reduce the toxicity of cancer treatments.
An increasing number of studies have focused on
histone deacetylation, which is an important epigenetic
modification involved in the development of numerous malignant
tumor types, including melanoma, leukemia, prostate cancer, lung
cancer and colon cancer (7–10). In the case of breast cancer, histone
deacetylation is closely associated with the apoptosis,
differentiation and down-regulation of tumor suppressor gene
expression and cell sensitivity to drugs (11,12). In
the previous study, it was determined that the histone deacetylase
(HDAC) regulator breast cancer metastasis-suppressor 1 like can
regulate the activity of HDAC1/2 and inhibit the transcription of
frizzled class receptor 10 and its downstream pathway, thus
inhibiting the occurrence of epithelial-mesenchymal transition
(EMT) in breast cancer (13).
Inhibition of histone acetylase activity can induce breast cancer
cell apoptosis, promote cancer cell differentiation, reduce drug
resistance and inhibit tumor cell proliferation and the occurrence
of EMT in breast cancer cells (14);
therefore, targeting the specific inhibition of protein acetylation
of enzymes may present an alternative treatment strategy for breast
cancer.
Apoptosis serves an important role in cancer
treatment and is a popular target of numerous treatment strategies
due to its disorder being closely associated with tumor development
(15,16). In terms of cell growth arrest and
apoptosis regulation, p53 serves an important role as a tumor
suppressor (17,18). By inactivating p53, cancer cells can
avoid arrest despite carrying genetic damage (18). Previous studies demonstrated that the
apoptosis-stimulating proteins
phorbol-12-myristate-13-acetate-induced protein 1, p21 and PUMA may
affect the progression of breast cancer through mediating the p53
pathway (19–21); therefore, studying the p53 pathway may
identify novel therapeutic methods for breast cancer.
Recent advances in HDAC inhibitors have been
encouraging. This is a class of compounds that target HDAC and
focus on the malignant proliferation of cells through selective
inhibition of growth and induction of apoptosis (14). Additionally, a recent study also
determined that inhibitors may reverse multidrug resistance of
tumors, and significantly reverse cisplatin resistance in ovarian
cancer and colorectal cancer cells (22,23). This
demonstrates the potential research and developmental value of
multidrug resistance drug reversal agents.
AR-42 is a novelly discovered class of
phenylbutyrate protein deacetylase inhibitors that display
localized enrichment in tumor tissues (24). AR-42 was initially determined to be
effective in various blood tumor types, including leukemia,
lymphoma and other blood tumor types, and it serves a role in the
inhibition of tumor growth (25,26).
Previous studies demonstrated AR-42 to have antitumor effects in
solid tumor types, including hepatocellular carcinoma, ovarian
cancer and pancreatic cancer (27–29). In
addition, AR-42 was determined to have a synergistic effect with
cisplatin (30), indicating a
potential antitumor effect of AR-42. The role of AR-42 in breast
cancer remains unclear; therefore, the aim of the present study was
to investigate the antitumor effects of AR-42 and its associated
mechanisms in breast cancer.
Materials and methods
Cell culture
The human breast cancer MCF-7 cell line was provided
by the American Type Culture Collection (Manassas, VA, USA). MCF-7
cells were cultured with Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal bovine serum (Thermo Fisher Scientific, Inc.) and incubated
in a 5% CO2 atmosphere at 37°C.
Antibodies and chemicals
AR-42 (Arno Therapeutics, Flemington, NJ, USA) and
cycloheximide (CHX) were prepared in dimethyl sulfoxide
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The CHX chase
assay identified the p53 degradation half-life. Anti-β-actin (cat.
no. sc-47778; 1:1,000), rabbit anti-p53 (cat. no. sc-6243; 1:1,000)
and anti-Ac-lysine (cat. no. sc-81623; 1:1,000) antibodies were
purchased from Santa Cruz Biotechnology (New York, USA).
Horseradish peroxidase (HRP)-conjugated anti-mouse (cat. no. 7076;
1:2,000) and anti-rabbit (cat. no. 7074; 1:2,000) IgG were
purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Cell viability assay
Cells were plated in 96-well plates at density of
5,000 cells/well and were treated with AR-42 (0.025, 0.05, 0.1,
0.2, 0.4 and 0.8 µmol/l; cat. no. S2244; Selleck Chemicals,
Houston, TX, USA) and/or 5-FU (0, 0.25, 0.5, 0.1 and 0.2 µmol/l;
cat. no. F6627; Sigma Aldrich; Merck KGaA). After 72 h of drug
exposure at 37°C, cells were treated with MTT solution (5 mg/ml),
to dissolve the purple formazan, for an additional 4 h at 37°C, and
the optical density (OD)490 value was detected using enzyme
labeling apparatus (ELx800 Strip Reader; BioTek Instruments, Inc.,
Winooski, VT, USA), reflecting the number of viable cells.
Cytotoxicity (%)=(1-OD490 of experimental well)/OD490 of control
well.
Colony formation assays
The cells were digested with 0.25% trypsin solution
at 37°C for 2 mins and prepared for single cell suspension. They
were seeded into 6-well plates (Wuxi Nest Biotechnology, Co. Ltd.,
Jiangsu, China) with 1,000 cells/per well cultured with Dulbecco's
modified Eagle's medium supplemented with 10% fetal bovine serum
(Thermo Fisher Scientific, Inc.), then gently shook to evenly
disperse in the cells normal culture conditions. Following
incubation at 37°C for two weeks, the cells were carefully washed
twice with PBS, fixed for 15 min with 100% methanol and stained for
15 min with 0.1% crystal violet dye at room temperature. Water was
then used to slowly wash away the dyeing liquid, and the cells were
left to dry naturally in the air. The 0 µM AR-42 was the negative
control group. Each experiment was repeated at least three
times.
Apoptosis assay
MCF-7 cells were treated with 0, 0.025, 0.05, 0.1 or
0.2 µM AR-42 at 37°C for 48 h in culture conditions, and then
harvested and washed with PBS twice. The cells were collected and
stained with an Annexin V/PI double flow cytometry kit (Nanjing
KeyGen Biotech, Co., Ltd., Nanjing, China), according to the
manufacturer's protocol, to detect the cell apoptosis rate. The 0
µM AR-42 were the negative control. Each experiment was repeated at
least three times.
Analysis of in vitro drug
interaction
Analysis of the drug's synergistic inhibitory effect
was determined using the coefficient of drug interaction (CDI)
metric. CDI was calculated as follows: CDI=AB/(A*B). A or B is the
ratio of the single drug group to the control group in OD490 and AB
represents the ratio of the two-drug combination group to the
control group in OD490. CDI>1 signifies antagonism, CDI=1
indicates additivity, and CDI<1 indicates synergism. A
CDI<0.7 was considered to indicate a statistically significant
synergistic effect.
Western blotting and
immunoprecipitation
Procedures were conducted as previously described
(31). Following extraction of total
cellular protein, the protein concentration was determined by the
BCA method. The sample was mixed with loading buffer (cat. no.
P0015; Beyotime Institute of Biotechnology, Shanghai, China) and
placed in a boiling water bath for 10 min, and following 10%
SDS-PAGE electrophoresis (30 µg/lane) was transferred to a
polyvinylidene fluoride membrane. The membranes were incubated in
5% skim milk at room temperature for 2 h. The membranes were
incubated with antibodies against human p53 or β-actin overnight at
4°C and incubated at room temperature for 2 h with anti-mouse and
anti-rabbit IgG HRP-conjugated antibodies. The chemiluminescence
system was exposed to enhanced chemiluminescent (Advansta, Inc.,
Menlo Park, CA, USA; R-03025-D25), and the experiment was repeated
three times. ImageJ 1.50f software (National Institutes of Health,
Bethesda, MD, USA) was used for densitometric analysis of the
experimental data.
To investigate the interaction between AR-42 and
Ack-p53 at the endogenous level, the clarified supernatants were
first incubated with anti-p53 or anti-Ac-lysine for 2 h at 4°C.
Protein A/G-agarose was then added and incubated for 2 h to
overnight. Precipitates were washed four times with RIPA buffer
(Beyotime Institute of Biotechnology) and analyzed by western
blotting as aforementioned. ImageJ 1.50f software was used for
densitometric analysis of the experimental data.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocols. cDNA was synthesized with the MLv
transcriptase kit (Invitrogen; Thermo Fisher Scientific, Inc.). The
quantitative analysis of p21 and puma expression was assayed using
a SYBR® green kit (Takara Bio, Inc., Otsu, Japan) with
gene-specific primers. Primer sequences for p21, puma, and β-actin
were as follows: p21, forward,. 5′-GCTCGGCTCTTCACCAAG-3′, and
reverse, 5′-GTCACTGTCTTGTACCCTTGTG-3′; puma, forward,
5′-CGACCTCAACGCACAGTACGA-3′, and reverse,
5′-AGGCACCTAATTGGGCTCCAT-3′; β-Actin, forward,
5′-GGTGGCTTTTAGGATGGCAAG-3′, and reverse,
5′-ACTGGAACGGTGAAGGTGACAG-3′. β-Actin was used as a normalization
control. The standard PCR conditions were: 95°C for 15 mins,
followed by 40 cycles of 95°C for 5 sec, 60°C for 30 sec, and 72°C
for 40 sec. The fold changes were calculated through relative
quantification with 2−ΔΔCq (32). All of the reactions were performed in
a 20 µl reaction volume in triplicate. These experiments were
repeated at least three times independently.
Statistical analysis
SPSS 16 software (SPSS, Inc., Chicago, IL, USA) was
used for statistical analysis of the experimental data. Values are
expressed in triplicate and presented as the mean ± standard
deviation. Comparisons were conducted using One-way analysis of
variance followed by Tukey's post hoc test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
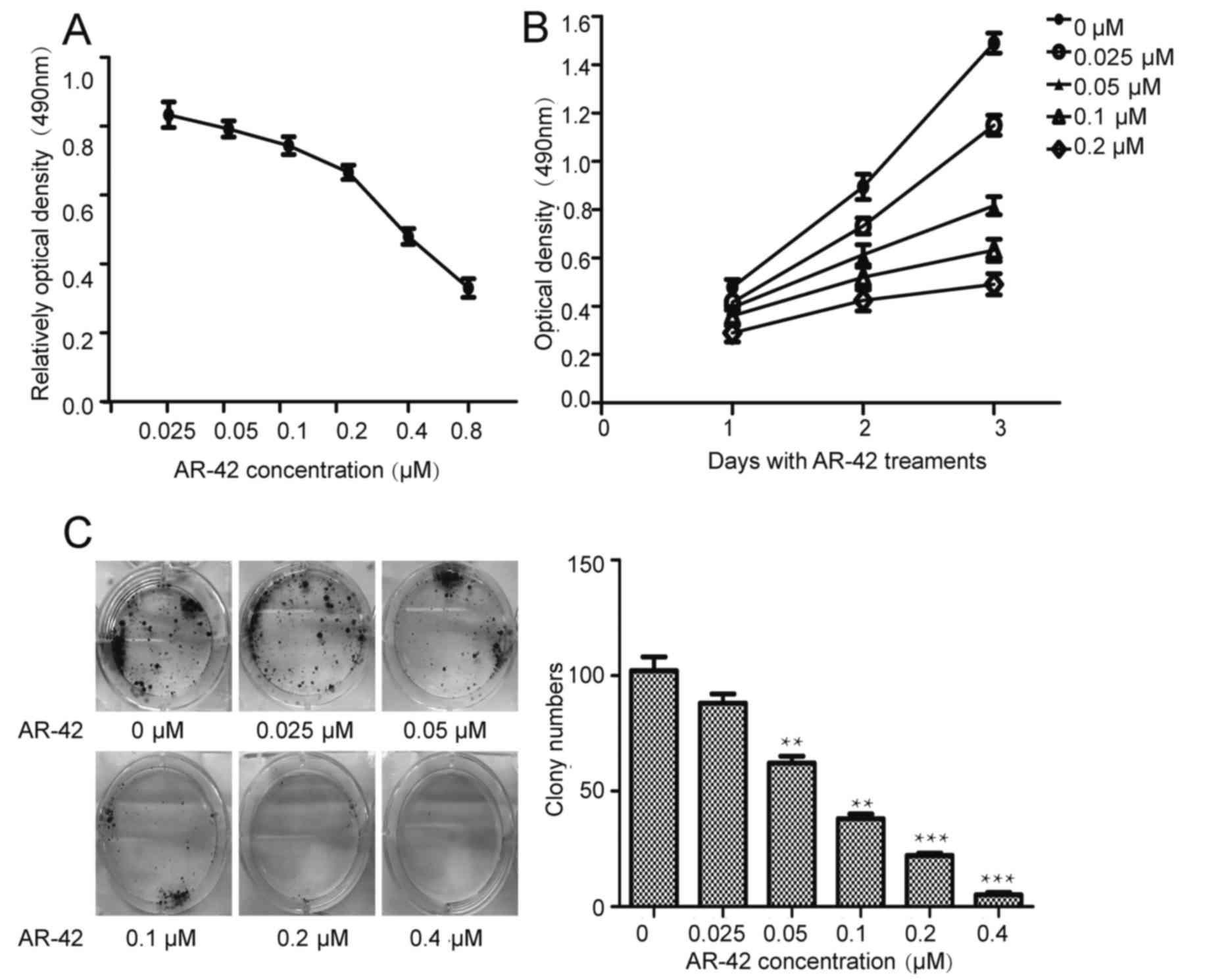
AR-42 inhibits the growth of the MCF-7
cell line in a dose- and time-dependent manner
Previous studies have demonstrated the antitumor
effects of AR-42 in blood tumor and solid tumor types (25–29). To
investigate the antitumor effects of AR-42 in breast cancer, the
MCF-7 cell line was treated with AR-42 at concentrations of 0–0.8
µM for different periods of time. The MTT assay was used to
evaluate cell viability. As depicted in Fig. 1A and B, AR-42 inhibits MCF-7 cell
growth in a dose- and time-dependent manner. Furthermore, the
colony formation assays (Fig. 1C)
indicated that AR-42 inhibited colony formation in a dose-dependent
manner.
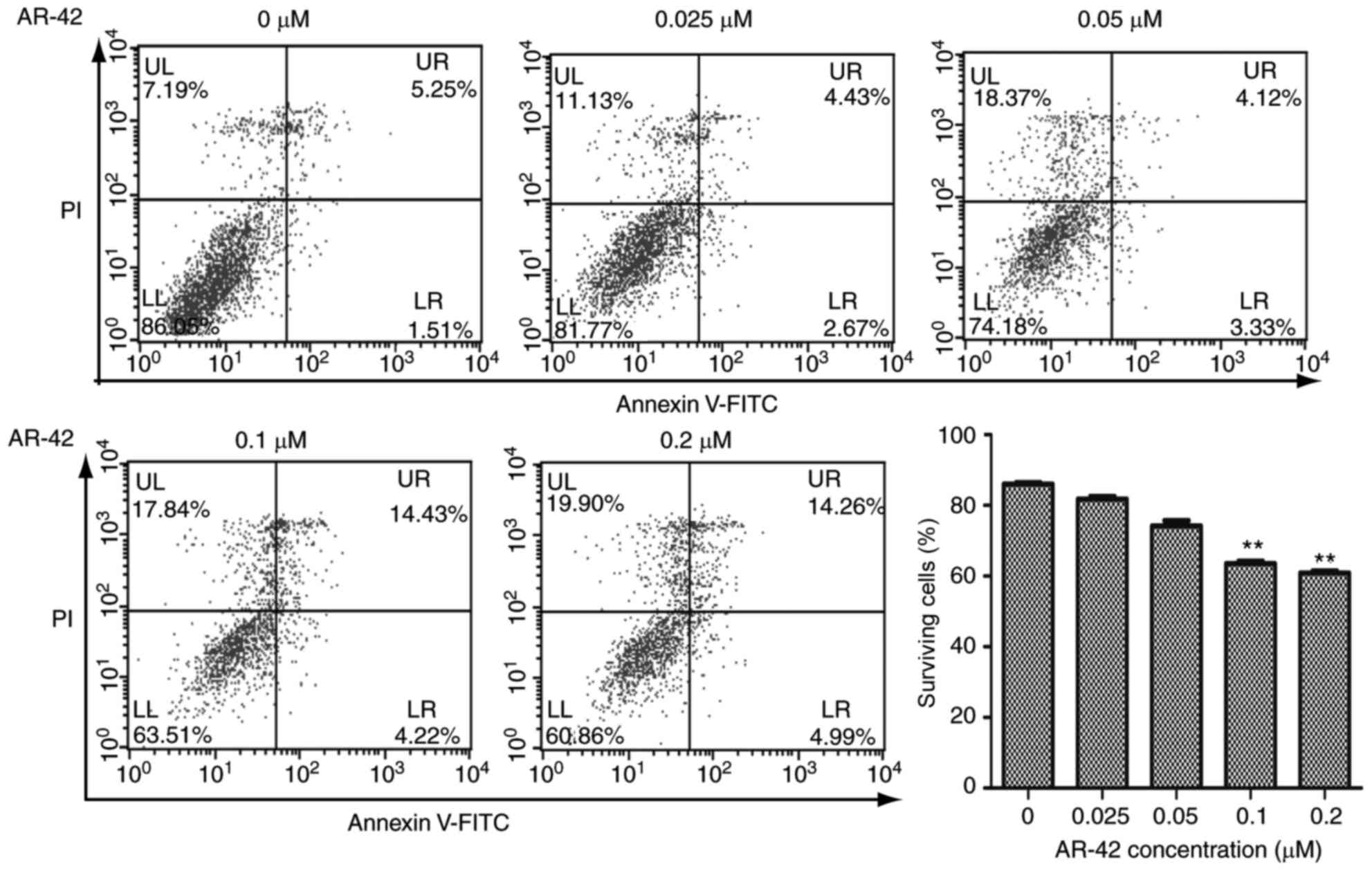
AR-42 inhibits the growth of MCF-7 cells by
promoting apoptosis. Inhibition of cell growth may be the result of
apoptosis, induction of necrosis or cell cycle arrest (15). Apoptosis is a programmed cell death
process regulated by multiple genes, and its disorder is closely
associated with the development of tumors (16). To detect whether AR-42 affects the
apoptotic rate of MCF-7 cells, flow cytometry was performed on the
cells. As depicted in Fig. 2,
following treatment with 0–0.2 µM AR-42 for 48 h, the survival
ratios of MCF-7 cells were decreased from 86.05% to 81.77, 74.18,
63.51 and 60.86% as the AR-42 concentration increased from 0.0 to
0.2 µm (Fig. 2. P<0.01 vs. control
group). These results indicated that AR-42 inhibits the growth of
MCF-7 cells by promoting cell apoptosis.
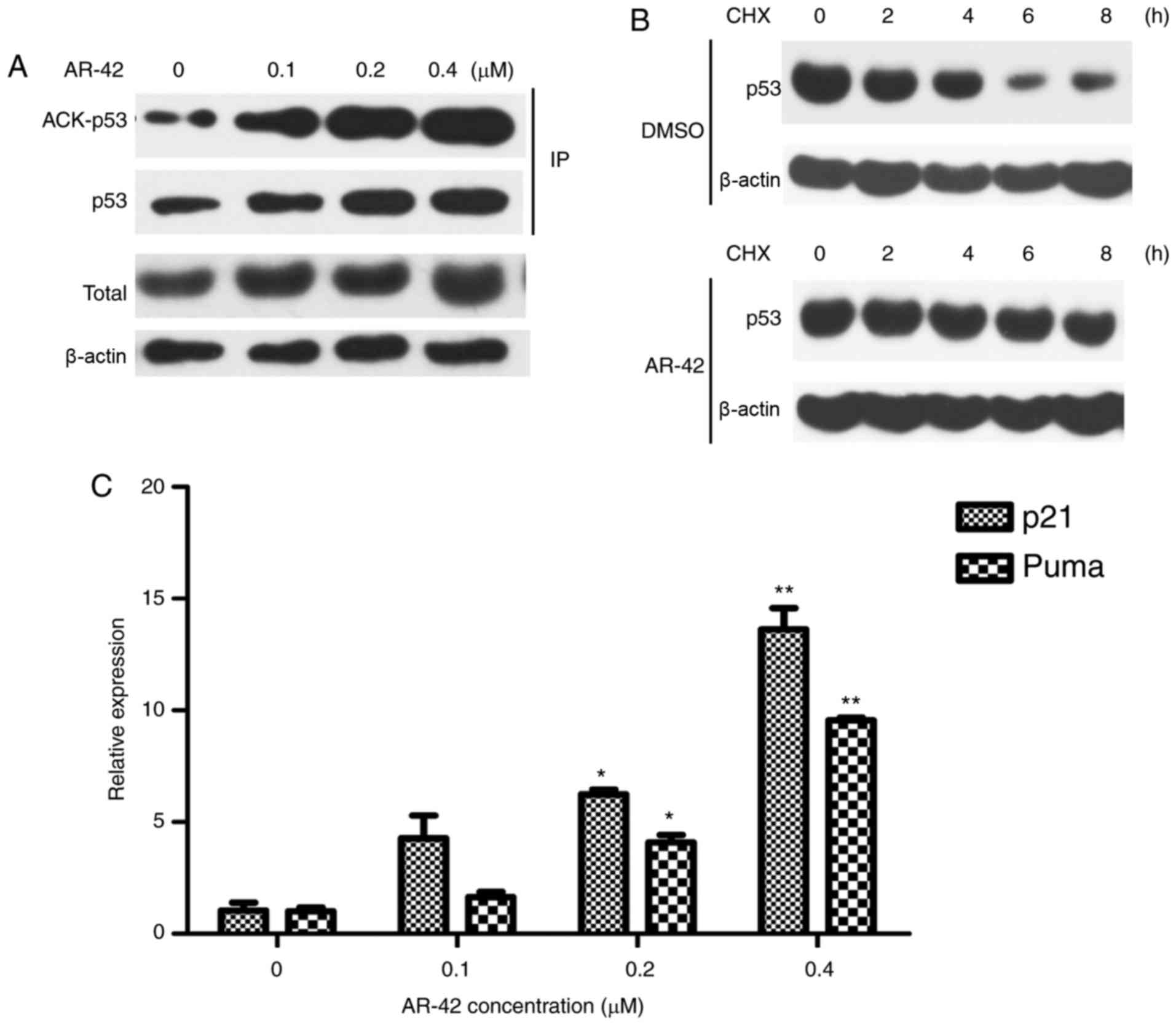
AR-42 induces MCF-7 cell apoptosis by
increasing the acetylation level of p53
Acetylation has been reported to increase the
activity and stability of p53 (33,34). To
understand the mechanisms underlying AR-42 mediated apoptosis in
MCF-7 cells, whether AR-42 affects p53 acetylation level was
investigated. As depicted in Fig. 3A and
B, the expression of acetylated p53 was elevated following
treatment with AR-42. Furthermore, the CHX chase assay results
indicated that AR-42 prolonged the half-life of the p53 protein,
indicating that AR-42 increased the stability of the p53 protein.
p21 and PUMA are important downstream transcriptional targets of
p53 (20); their mRNA levels
following AR-42 treatment in MCF-7 cells was examined. As depicted
in Fig. 3C, AR-42 treatment induced
p21 and PUMA expression in MCF-7 cells in a dose-dependent
manner.
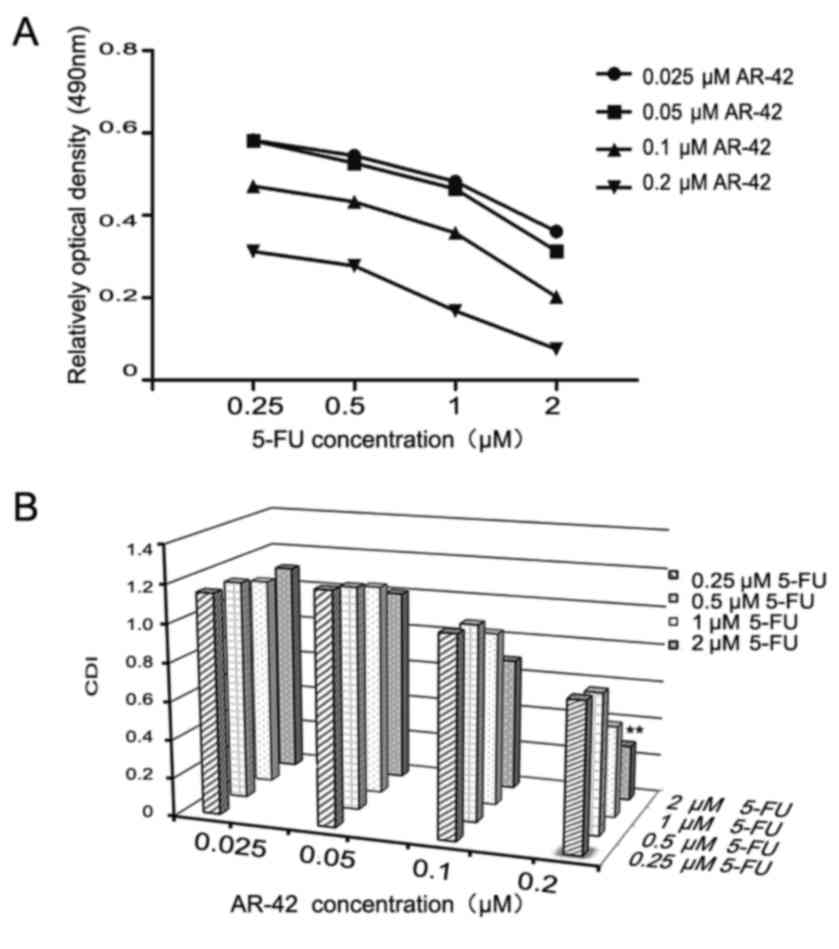
AR-42 combined with 5-FU has a
synergistic inhibitory effect on MCF-7 cells
Chemotherapy remains an important breast cancer
treatment. As a conventional chemotherapy drug, 5-FU can
effectively improve the survival rate of clinical patients
(35). Previous study demonstrated
that 5-FU has serious side effects, such as cytotoxicity, leading
to a narrow therapeutic effect (35);
therefore, it's necessary to investigate possible synergistic
effects of combining 5-FU and AR-42 in MCF-7 cells. As depicted in
Fig. 4A, the results revealed that
AR-42 increased the cytotoxicity of 5-FU towards MCF-7 cells. The
synergistic effect is indicated as a CDI value. As presented in
Fig. 4B, 0.25 µM 5-FU combined with
0.2 µM AR-42 had the most significant synergistic effect
(CDI<0.7).
Discussion
Breast cancer is known to be one of the most
widespread and prevalent tumor types globally (1). Breast cancer cases in China account for
12% of the global total novel diagnosed cases of breast cancer each
year (4); therefore, it is necessary
to investigate novel strategies for breast cancer treatment.
HDAC inhibitors represent a novel anticancer
therapeutic strategy (36). A
significant number of HDAC inhibitors have been developed in the
past decade (3). Previous studies
have demonstrated that AR-42, a member of a novelly discovered
class of phenylbutyrate-derived HDAC inhibitors, has been
demonstrated to have antitumor effects in blood tumor and solid
tumor types (24–26). Apoptosis disorders are closely
associated with the development of tumors (15); therefore, the induction of tumor cell
apoptosis may be an efficient strategy to prevent tumor
progression. In the present study, it was demonstrated that AR-42
inhibited the proliferation of MCF-7 breast cancer cells by
indirectly regulating acetylation of the p53 protein, which also
indicated a synergistic effect when combined with 5-FU. The results
indicated that the proliferation of breast cancer cells may be
suppressed by AR-42 treatment, which may prove to be an alternative
therapeutic approach for the future treatment of breast cancer.
Apoptosis is a programmed cell death process
regulated by multiple genes, and its disorder is closely associated
with the development of tumors (15).
In terms of cell growth arrest and apoptosis regulation, p53 serves
an important role as a tumor suppressor by inducing extrinsic and
intrinsic apoptotic pathways to ensure efficient death responses
(17,37). Acetylation is essential for p53
activity, and its function is important in transcriptional
activation and senescence (33,34).
Acetylation of p53 affects cell activity and function, such as
Lys-373 and Lys-382 acetylation, which inhibits p53 degradation,
thereby mediating apoptosis and enhancing cell drug sensitivity
(34). It has been demonstrated that
treatment of colon cancer cells with SIRTI siRNA or the HDACI MS275
can increase p53 acetylation levels and enhance paclitaxel-induced
apoptosis (38). The SIRT1 small
molecule inhibitor Tenovin-1 can inhibit increased p53 expression
and activity levels and inhibit melanoma cell proliferation
(38,39). As depicted in Fig. 3, the results indicated that AR-42
treatment increased the level of p53 protein and its acetylation
formation. In addition, AR-42 treatment significantly prolonged the
half-life of the p53 protein. It is notable that p53 acetylation
itself is effective in stabilizing and increasing the level of
total p53 (40).
As a result, the accumulation of the p53 protein and
its acetylation may be the consequence of AR-42-mediated apoptosis
in MCF-7 cells. The tumor suppressor protein p53 affects a vast
number of downstream targets and performs a braking function by
blocking injured cells from entering the cell cycle and promoting
apoptosis (18). Among these
downstream targets are p21 and PUMA, which are major mediators of
the function of p53 (41). Previous
studies performed on other cancer cell lines have demonstrated that
HDAC inhibition results in the upregulation of p21 and PUMA
expression through increasing the acetylation of p53 (20,21,42). In
the present study, AR-42 increased p21 and PUMA expression,
confirming p53 activation; therefore, AR-42 treatment may
successfully induce molecular mediators of cell cycle arrest and
apoptosis.
Currently, chemotherapy remains an important breast
cancer treatment. As a conventional chemotherapy drug, 5-FU can
effectively improve the survival rate of clinical patients.
However, it has serious limitations, such as cytotoxicity, which
leads to its narrow therapeutic effect (35,43). Thus,
combining 5-FU with other pharmaceutical agents will provide more
effective ways to sensitize cancer cells to chemotherapy, while
reducing toxicity to normal cells (35). Combination therapy may achieve fewer
side effects and greater therapeutic efficacy. The results
indicated that AR-42 increased the cytotoxicity of 5-FU and
indicated a significant synergistic effect when combined with 5-FU,
which indicates that the combined treatment of AR-42 and 5-FU may
be an effective strategy for treating breast cancer.
5-FU serves an important role in early breast cancer
treatment following adjuvant therapy, breast cancer recurrence and
metastasis following palliative care (43,44). It is
an anti-metabolic chemotherapeutic agent, acting mainly through the
irreversible inhibition of thymidylate synthase (TS), which results
in a lack of thymine and the synthesis of non-functional DNA
(35). A previous study demonstrated
that p53 is important for drug sensitivity to TS inhibitors, such
as 5-FU (45). It has been previously
reported that acetylation of p53 is essential for preventing
degradation and opening a conformation that allows binding to DNA
(46). 5-FU mediated DNA damage
activates numerous signaling pathways, such as p53-mediated
apoptosis. The results indicated that AR-42 treatment caused MCF-7
cell apoptosis. It was demonstrated that AR-42 and 5-FU have the
ability to activate p53-mediated apoptosis, which may clarify how
AR-42 synergistically combines with 5-FU to restrain the growth of
MCF-7 cells; however, the specific mechanisms underlying the
synergistic effect of AR-42 and 5-FU require further study.
In conclusion, the present study investigated the
anti-tumorigenic role of AR-42 in breast cancer cells. The results
demonstrated that AR-42 inhibits the proliferation of MCF-7 breast
cancer cells via the induction of cell apoptosis. Notably,
combination assays demonstrated that joint AR-42 and 5-FU treatment
have a significant synergistic effect on MCF-7 breast cancer
cells.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81201550, 81560452 and
81672866), the Zhujiang Star 2015 of Science and Technology Program
of Guangzhou, China (grant no. 201506010037), the Natural Science
Foundation of Jiangxi Province (grant nos. 20161BAB205192 and
20171ACB21073), the Excellent Youth Foundation of Jiangxi
Scientific Committee (grant no. 20162BCB23001), the Science and
Research Fund of Jiangxi Health and Family Planning Commission
(grant no. 20164002) and The Foundation of Nanchang Science and
Technology Bureau (grant no. 2016 ZSCX 009).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RZ and JW designed the research, analyzed data, and
wrote the manuscript. XT, XW, CJ, FZ performed experiments and
prepared the figures. JS, DS and ZZ were involved in drafting the
manuscript, revising it critically for important intellectual
content, designed the study and acquired the data. XBL and QL
participated in the design of the study and helped finalize the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Turtoi A, Peixoto P, Castronovo V and
Bellahcene A: Histone deacetylases and cancer-associated
angiogenesis: Current understanding of the biology and clinical
perspectives. Crit Rev Oncog. 20:119–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fan L, Strasser-Weippl K, Li JJ, St Louis
J, Finkelstein DM, Yu KD, Chen WQ, Shao ZM and Goss PE: Breast
cancer in China. Lancet Oncol. 15:e279–e289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meattini I, Curigliano G, Terziani F,
Becherini C, Airoldi M, Allegrini G, Amoroso D, Barni S, Bengala C,
Guarneri V, et al: SAFE trial: An ongoing randomized clinical study
to assess the role of cardiotoxicity prevention in breast cancer
patients treated with anthracyclines with or without trastuzumab.
Med Oncol. 34:752017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu L, Wu K, Ma S and Zhang S: HDAC
inhibitors: A new radiosensitizer for non-small-cell lung cancer.
Tumori. 101:257–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marti RM, Sorolla A and Yeramian A: New
therapeutic targets in melanoma. Actas Dermosifiliogr. 103:579–590.
2012.(In English; In Spanish). PubMed/NCBI
|
|
8
|
Eigl BJ, North S, Winquist E, Finch D,
Wood L, Sridhar SS, Powers J, Good J, Sharma M, Squire JA, et al: A
phase II study of the HDAC inhibitor SB939 in patients with
castration resistant prostate cancer: NCIC clinical trials group
study IND195. Invest New Drugs. 33:969–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schech A, Kazi A, Yu S, Shah P and Sabnis
G: Histone deacetylase inhibitor entinostat inhibits
tumor-initiating cells in triple-negative breast cancer cells. Mol
Cancer Ther. 14:1848–1857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chiu HW, Yeh YL, Wang YC, Huang WJ, Chen
YA, Chiou YS, Ho SY, Lin P and Wang YJ: Suberoylanilide hydroxamic
acid, an inhibitor of histone deacetylase, enhances
radiosensitivity and suppresses lung metastasis in breast cancer in
vitro and in vivo. PloS One. 8:e763402013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee
KH, Kim TY, Han SW, Oh DY, Kim TY, et al: Histone deacetylase
inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances
anti-tumor effects of the poly (ADP-ribose) polymerase (PARP)
inhibitor olaparib in triple-negative breast cancer cells. Breast
Cancer Res. 17:332015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi YK, Li ZH, Han XQ, Yi JH, Wang ZH, Hou
JL, Feng CR, Fang QH, Wang HH, Zhang PF, et al: The histone
deacetylase inhibitor suberoylanilide hydroxamic acid induces
growth inhibition and enhances taxol-induced cell death in breast
cancer. Cancer Chemother Pharmacol. 66:1131–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong C, Qu S, Lv XB, Liu B, Tan W, Nie Y,
Su F, Liu Q, Yao H and Song E: BRMS1L suppresses breast cancer
metastasis by inducing epigenetic silence of FZD10. Nat Commun.
5:54062014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Di Martile M, Del Bufalo D and
Trisciuoglio D: The multifaceted role of lysine acetylation in
cancer: Prognostic biomarker and therapeutic target. Oncotarget.
7:55789–55810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bennett M, Macdonald K, Chan SW, Luzio JP,
Simari R and Weissberg P: Cell surface trafficking of Fas: A rapid
mechanism of p53-mediated apoptosis. Science. 282:290–293. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zuckerman V, Wolyniec K, Sionov RV, Haupt
S and Haupt Y: Tumour suppression by p53: The importance of
apoptosis and cellular senescence. J Pathol. 219:3–15.
2009.PubMed/NCBI
|
|
19
|
Vousden KH: Apoptosis. p53 and PUMA: A
deadly duo. Science. 309:1685–1686. 2005.
|
|
20
|
Hikisz P and Kilianska ZM: PUMA, a
critical mediator of cell death-one decade on from its discovery.
Cell Mol Biol Lett. 17:646–669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alaee M, Khaghani S, Behroozfar K, Hesari
Z, Ghorbanhosseini SS and Nourbakhsh M: Inhibition of nicotinamide
phosphoribosyltransferase induces apoptosis in estrogen
receptor-positive MCF-7 breast cancer cells. J Breast Cancer.
20:20–26. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zuco V, Cassinelli G, Cossa G, Gatti L,
Favini E, Tortoreto M, Cominetti D, Scanziani E, Castiglioni V,
Cincinelli R, et al: Targeting the invasive phenotype of
cisplatin-resistant non-small cell lung cancer cells by a novel
histone deacetylase inhibitor. Biochem Pharmacol. 94:79–90. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Shang YP, Chen HY and Li J:
Histone deacetylases function as novel potential therapeutic
targets for cancer. Hepatol Res. 47:149–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin TY, Fenger J, Murahari S, Bear MD,
Kulp SK, Wang D, Chen CS, Kisseberth WC and London CA: AR-42, a
novel HDAC inhibitor, exhibits biologic activity against malignant
mast cell lines via down-regulation of constitutively activated
Kit. Blood. 115:4217–4225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zimmerman B, Sargeant A, Landes K,
Fernandez SA, Chen CS and Lairmore MD: Efficacy of novel histone
deacetylase inhibitor, AR42, in a mouse model of, human
T-lymphotropic virus type 1 adult T cell lymphoma. Leuk Res.
35:1491–1497. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang S, Suvannasankha A, Crean CD, White
VL, Chen CS and Farag SS: The novel histone deacetylase inhibitor,
AR-42, inhibits gp130/Stat3 pathway and induces apoptosis and cell
cycle arrest in multiple myeloma cells. Int J Cancer. 129:204–213.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Balch C, Naegeli K, Nam S, Ballard B,
Hyslop A, Melki C, Reilly E, Hur MW and Nephew KP: A unique histone
deacetylase inhibitor alters microRNA expression and signal
transduction in chemoresistant ovarian cancer cells. Cancer Biol
Ther. 13:681–693. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen YJ, Wang WH, Wu WY, Hsu CC, Wei LR,
Wang SF, Hsu YW, Liaw CC and Tsai WC: Novel histone deacetylase
inhibitor AR-42 exhibits antitumor activity in pancreatic cancer
cells by affecting multiple biochemical pathways. PloS One.
12:e01833682017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu YS, Chou CH, Tzen KY, Gao M, Cheng AL,
Kulp SK and Cheng JC: Radiosensitizing effect of a
phenylbutyrate-derived histone deacetylase inhibitor in
hepatocellular carcinoma. Int J Radiat Oncol Biol Phys.
83:e181–e189. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li DR, Zhang H, Peek E, Wang S, Du L, Li G
and Chin AI: Synergy of histone-deacetylase inhibitor AR-42 with
cisplatin in bladder cancer. J Urol. 194:547–555. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lv XB, Wu W, Tang X, Wu Y, Zhu Y, Liu Y,
Cui X, Chu J, Hu P, Li J, et al: Regulation of SOX10 stability via
ubiquitination-mediated degradation by Fbxw7α modulates melanoma
cell migration. Oncotarget. 6:36370–36382. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wagner T, Brand P, Heinzel T and Krämer
OH: Histone deacetylase 2 controls p53 and is a critical factor in
tumorigenesis. Biochim Biophys Acta. 1846:524–538. 2014.PubMed/NCBI
|
|
34
|
Tang Y, Zhao W, Chen Y, Zhao Y and Gu W:
Acetylation is indispensable for p53 activation. Cell. 133:612–626.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
de Souza C and Chatterji BP: HDAC
inhibitors as novel anti-cancer therapeutics. Recent Pat Anticancer
Drug Discov. 10:145–162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Coates AS, Millar EK, O'Toole SA, Molloy
TJ, Viale G, Goldhirsch A, Regan MM, Gelber RD, Sun Z,
Castiglione-Gertsch M, et al: Prognostic interaction between
expression of p53 and estrogen receptor in patients with
node-negative breast cancer: Results from IBCSG Trials VIII and IX.
Breast Cancer Res. 14:R1432012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wilking MJ, Singh C, Nihal M, Zhong W and
Ahmad N: SIRT1 deacetylase is overexpressed in human melanoma and
its small molecule inhibition imparts anti-proliferative response
via p53 activation. Arch Biochem Biophys. 563:94–100. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim JH, Yoon EK, Chung HJ, Park SY, Hong
KM, Lee CH, Lee YS, Choi K, Yang Y, Kim K and Kim IH: p53
acetylation enhances Taxol-induced apoptosis in human cancer cells.
Apoptosis. 18:110–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ito A, Kawaguchi Y, Lai CH, Kovacs JJ,
Higashimoto Y, Appella E and Yao TP: MDM2–HDAC1-mediated
deacetylation of p53 is required for its degradation. EMBO J.
21:6236–6245. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zheng S, Koh XY, Goh HC, Rahmat SAB, Hwang
LA and Lane DP: Inhibiting p53 acetylation reduces cancer
chemotoxicity. Cancer Res. 77:4342–4354. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Taieb J, Tabernero J, Mini E, Subtil F,
Folprecht G, van Laethem JL, Thaler J, Bridgewater J, Petersen LN,
Blons H, et al: Oxaliplatin, fluorouracil, and leucovorin with or
without cetuximab in patients with resected stage III colon cancer
(PETACC-8): An open-label, randomised phase 3 trial. Lancet Oncol.
15:862–873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Han Q, Chen R, Wang F, Chen S, Sun X, Guan
X, Yang Y, Peng B, Pan X Li J, et al: Pre-exposure to 50
Hz-electromagnetic fields enhanced the antiproliferative efficacy
of 5-fluorouracil in breast cancer MCF-7 cells. PloS One.
13:e01928882018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Di Gennaro E, Bruzzese F, Pepe S, Leone A,
Delrio P, Subbarayan PR, Avallone A and Budillon A: Modulation of
thymidilate synthase and p53 expression by HDAC inhibitor
vorinostat resulted in synergistic antitumor effect in combination
with 5FU or raltitrexed. Cancer Biol Ther. 8:782–791. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bode AM and Dong Z: Post-translational
modification of p53 in tumorigenesis. Nat Rev Cancer. 4:793–805.
2004. View Article : Google Scholar : PubMed/NCBI
|