Introduction
The most notable trait of cancer cells is their
marked survival advantage compared with normal cells, which allows
them to continue proliferating despite apoptotic signals. In this
context, wingless-type (Wnt) signaling is understood to serve a
pivotal role in connecting extracellular survival signals and
intracellular apoptotic signals. Hyperactivation of Wnt signaling
is frequently observed in numerous types of carcinomas (1–3). β-catenin
is a major transcription factor in the Wnt signaling pathway that
is also responsible for controlling cell-cell adhesion through
intracellular binding with α-catenin and E-cadherin (4). When Wnt signaling is activated,
β-catenin translocates to the nucleus to promote cell proliferation
and induce tumor cell migration, invasion and metastasis through
loss of cell-cell contacts (5).
5′-Adenosine monophosphate-activated protein kinase
(AMPK) is a serine/threonine kinase understood to function as an
energy sensor to maintain cellular-energy homeostasis by inhibiting
energy-consuming anabolic pathways and activating energy-generating
catabolic energy pathways when a cell is depleted of energy
(6). When cells are exposed to
stress, cellular adenosine 5′-triphosphate (ATP) consumption is
increased and AMPK is activated, thereby inducing apoptosis
(7). AMPK has also attracted
attention as an important tumor suppressor due to its association
with a number of molecules, including liver kinase B1 (LKB1) and
tuberous sclerosis (8,9). Furthermore, AMPK has been examined for
its role as a cancer regulator, as LKB1 is an upstream activator of
AMPK in colon cancer cells (10).
Metformin can block gluconeogenesis and inhibit
metabolic syndromes by activating AMPK in the liver, implying that
it may be useful for treating type 2 diabetes (11). A previous study has demonstrated that
metformin may be used as a cancer therapy, as it can inhibit
mitochondrial complex I for cellular energy production and induce
apoptosis (12). Metformin-mediated
reduction of cellular energy production promotes AMPK activation,
and activated AMPK can suppress cancer cell proliferation and cause
cancer cell death by inhibiting cancer cell growth signaling
pathways, particularly the mechanistic target of rapamycin (mTOR)
and Wnt signaling pathways (13).
However, to the best of our knowledge, the detailed mechanism
regarding the inhibitory effect of metformin on cell proliferation
is currently unclear. Therefore, the objective of the present study
was to determine whether active AMPK could bind to β-catenin and
suppresses Wnt signaling, thus potentiating metformin-induced
suppression of cell proliferation.
Materials and methods
Cells and reagents
Human colon carcinoma RKO cells were purchased from
the American Type Culture Collection (Manassas, VA, USA) and
cultured in Dulbecco's modified Eagle's medium (Serana Europe GmBH,
Pessin, Germany) containing 10% fetal bovine serum (Biotechnics
Research, Inc., Lake Forest, CA, USA; catalog no. 7101). Metformin
was obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany;
catalog no. D150959). Compound C (catalog no. 171260) and MG132
(catalog no. 474790) were purchased from EMD Millipore (Billerica,
MA, USA). Wnt3a was purchased from R&D Systems Inc.
(Minneapolis, MN, USA; catalog no. 5036-WN).
Cell proliferation measurement
Cells were seeded into 96-well microplates at a
density of 1×104 cells/well and incubated with 0, 0.05,
0.1, 0.5, 1, 2.5, 5, 10 or 20 mM metformin for 24 h at 37°C in an
atmosphere containing 5% CO2. Following incubation with
the test compound, the medium was removed and the cells were
incubated with 5 µl MTT solution (5 mg/ml MTT in PBS) for 1 h
followed by solubilization in dimethyl sulfoxide. The purple
formazan dye converted from MTT by viable cells was quantified by
measuring the absorbance at a wavelength of 595 nm.
The 5-bromodeoxyuridine (BrdU) incorporation assay
was performed using a BrdU Cell Proliferation ELISA kit (Roche
Applied Science, Rotkreuz, Switzerland), according to the
manufacturer's protocol. Absorbance at 370 nm was measured using an
ELISA reader (SpectraMax plus 384; Molecular Devices, LLC,
Sunnyvale, CA, USA).
ATP assay
Intracellular ATP levels were measured using a
Luminescence ATP Detection assay system (ATPlite; PerkinElmer,
Inc., Waltham, MA, USA, catalog no. 6016941), according to the
manufacturer's protocol. Cells were cultured into 96-well
microplates at a density of 1×104 cells/well for 16 h at
37°C. The cells were then treated with 100 µl culture medium
containing 0, 5, 10 or 20 mM metformin for 24 h at 37°C. After 24 h
of incubation, 50 µl mammalian cell lysis solution from the
Luminescence ATP Detection assay system was added to each well and
the plate was placed in an orbital shaker at 55 × g for 5 min.
Subsequently, 50 µl substrate solution from the Luminescence ATP
Detection assay system was added and the plate was placed in an
orbital shaker at 55 × g for 5 min. ATP amount was normalized by
the cell number, which was assessed by BrdU assay.
Immunoblotting
RKO cells were washed with ice-cold PBS. Total
protein was then extracted using protein lysis buffer (20 mM
HEPES-KOH (pH 7.4), 1 mM EGTA, 50 mM KCl and 2 mM MgCl2)
with protease inhibitor cocktail and 1 mM dithiothreitol. The
protein concentration was determined with a DC protein assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA; catalog no.
500-0113). Proteins (10 µg) were loaded on 9% SDS-PAGE gels and
then electrophoretically transferred to polyvinylidene fluoride
membranes. The membranes were blocked with 10% skim milk at room
temperature for 1 h. The proteins were then subjected to immunoblot
analysis using specific antibodies. Primary antibodies were added
at 4°C for 16 h, followed by horseradish peroxidase-conjugated
secondary antibodies, including goat anti-rabbit IgG (Bio-Rad
Laboratories, Inc.; 1:3,000; catalog no. 170-6515) or
goat-anti-mouse IgG (Bio-Rad Laboratories, Inc.; 1:3,000; catalog
no. 172-1011) at room temperature for 1 h. Proteins were visualized
using enhanced chemiluminescent reagent (PerkinElmer, Inc; catalog
no. NEL120001EA). The following primary antibodies were used:
phosphorylated (p)-AMPKα1 (Thr172) (1:1,000; catalog no. 2535s;
Cell Signaling Technology, Inc., Danvers, MA, USA), p-β-catenin
(Ser33/37) (1:500; catalog no. 9561s; Cell Signaling Technology,
Inc.), p-β-catenin (Ser552) (1:1,000; catalog no. 9566s; Cell
Signaling Technology, Inc.), β-catenin (1:1,000; catalog no.
610154; BD Biosciences, San Jose, CA, USA), lamin A/C (1:1,000;
catalog no. 612163; BD Biosciences), AMPKα1/2 (1:1,000; catalog no.
sc-74461; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); β-actin
(1:1,000; catalog no. sc-sc-47778; Santa Cruz Biotechnology, Inc.)
and GAPDH (1:1,000; catalog no. sc-32233; Santa Cruz Biotechnology,
Inc.).
Measurement of oxygen consumption rate
(OCR)
RKO cells were plated into XF24 cell culture
microplates (Seahorse Bioscience, North Billerica, MA, USA) at a
density of 20,000 cells/well. The cells were then treated with 10
mM at 37°C in an atmosphere containing 5% CO2. Following
24 h of treatment, the cell culture growth medium in the microplate
was replaced with warmed Seahorse XF base medium (catalog no.
102353-100; Seahorse Bioscience) using a multichannel pipette.
Oligomycin (2 µM), FCCP (0.1 µM), antimycin A (1 µM) and rotenone
(1 µM), from the Agilent Seahorse XF Cell Mito Stress Test kit
(Agilent Technologies, Inc., Santa Clara, CA, USA; catalog no.
103015-100) were then consecutively added to the each well. The
cells were then incubated at 37°C in an incubator with 0%
CO2 for 45 min to 1 h prior to the assay. OCR was
measured using an XF24 Analyzer (Seahorse Bioscience). Measured
values were normalized by cell number, which was assessed by BrdU
assay.
Immunofluorescence
Cells were seeded onto cover glasses in a 12-well
plate. After 16 h the cells were treated with 10 mM metformin at
37°C in an atmosphere containing 5% CO2. After 24 h, the
cells were fixed with 4% paraformaldehyde for 10 min and
permeabilized in 0.1% Triton X-100 for 20 min at room temperature.
Cells were then blocked with 2% bovine serum albumin for 1 h at
room temperature and incubated at 4°C overnight with primary
antibodies against AMPK and β-catenin. Following washing, cells
were incubated with fluorochrome-conjugated anti-mouse (1:200;
catalog no. TI-2000) and anti-rabbit IgG (1:100; catalog no.
FI-1000; both from Vector Laboratories, Inc., Burlingame, CA, USA)
at room temperature for 1 h. Subsequently, nuclei were stained with
10 µM Hoechst 33342 (Invitrogen; Thermo Fisher Scientific, Inc.;
catalog no. H1399) for 10 min at room temperature, and visualized
using a fluorescence microscope (magnification, ×400) and a digital
camera.
Immunoprecipitation
Cells were cultured in a 100 mm culture dish for 24
h and then treated with 10 mM metformin for 24 h at 37°C in an
atmosphere containing 5% CO2. Cells were harvested by
centrifugation at 1,000 × g at 4°C for 10 min, washed with PBS, and
lysed with protein lysis buffer. Cell extracts were then incubated
with Protein G agarose beads at 4°C for 3 h. Following washing with
lysis buffer three times, extracts were incubated with an
anti-AMPKα1/2 antibody at 4°C overnight. Following washing with
lysis buffer, SDS-loading buffer containing β-mercaptoethanol was
added and the samples were boiled for 5 min. The proteins [whole
cell lysate (WCL), 10 µg; and immunoprecipitation (IP), 10 µl] were
loaded on 9% SDS-PAGE gels and then electrophoretically transferred
to polyvinylidene fluoride membranes. Subsequently, the membranes
were blocked with 10% skim milk at room temperature for 1 h. The
proteins were then subjected to immunoblot analysis using AMPK1/2α,
β-catenin and β-actin antibodies. Primary antibodies were added for
16 h at 4°C followed by horseradish peroxidase-conjugated
goat-anti-mouse IgG secondary antibodies (Bio-Rad Laboratories,
Inc.; 1:3,000; catalog no. 172-1011) at room temperature for 1 h.
Proteins were visualized using enhanced chemiluminescent reagent
(PerkinElmer, Inc; catalog no. NEL120001EA).
Statistical analysis
Data are presented as the mean ± standard deviation.
All data were analyzed using SPSS 20.0 software (IBM Corp., Armonk,
NY, USA). Comparisons between groups were performed using Student's
t-test or one-way analysis of variance followed by Duncan's
multiple-range test. P<0.05 was considered to indicate a
statistically significant difference. All experiments were
conducted in triplicate.
Results
Metformin suppresses cell
proliferation and ATP production, resulting in AMPK activation
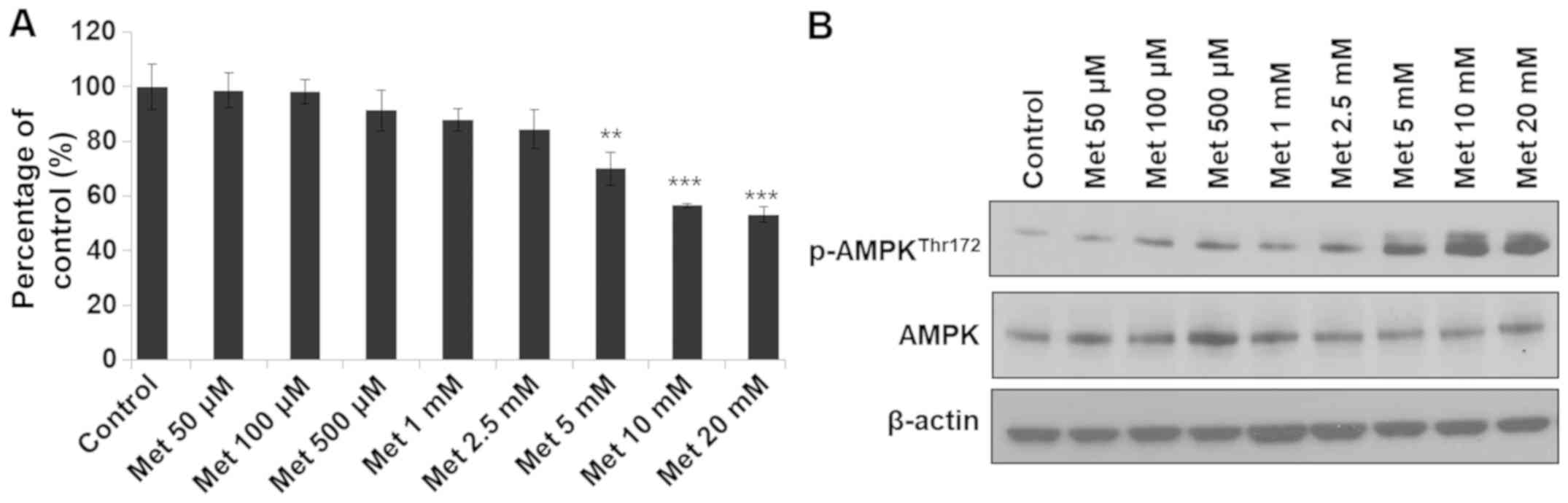
The effects of metformin on cell proliferation and
energy production in colon cancer RKO cells were examined. RKO
cells were treated with metformin at low to high concentrations for
24 h. AMPK activation was observed in the groups treated with
metformin at 100 µM to 20 mM and cell growth inhibition was
observed in groups treated with 5–20 mM metformin (Fig. 1). Therefore, the concentrations of
metformin that effectively activated AMPK and inhibited cell growth
were selected for further experiments. Treatment with metformin at
different concentrations (5–20 mM) for 24 h significantly inhibited
RKO cell viability (Fig. 2A and B)
and reduced ATP production (Fig. 2C).
Subsequent analysis demonstrated that metformin increased the
expression level of pAMPK, but decreased the expression level of
β-catenin (Fig. 2D). AMPK is a master
regulator of cellular energy homeostasis (6); therefore, AMPK may be activated by the
reduction of cellular ATP. Several types of cancer cell have
demonstrated increased energy metabolism through glycolysis and/or
oxidative phosphorylation with increased production of ATP
(14). In metformin-treated RKO cells
in the present study, mitochondrial oxidative phosphorylation and
glycolysis were reduced compared with that in the controls
(Fig. 2E and F).
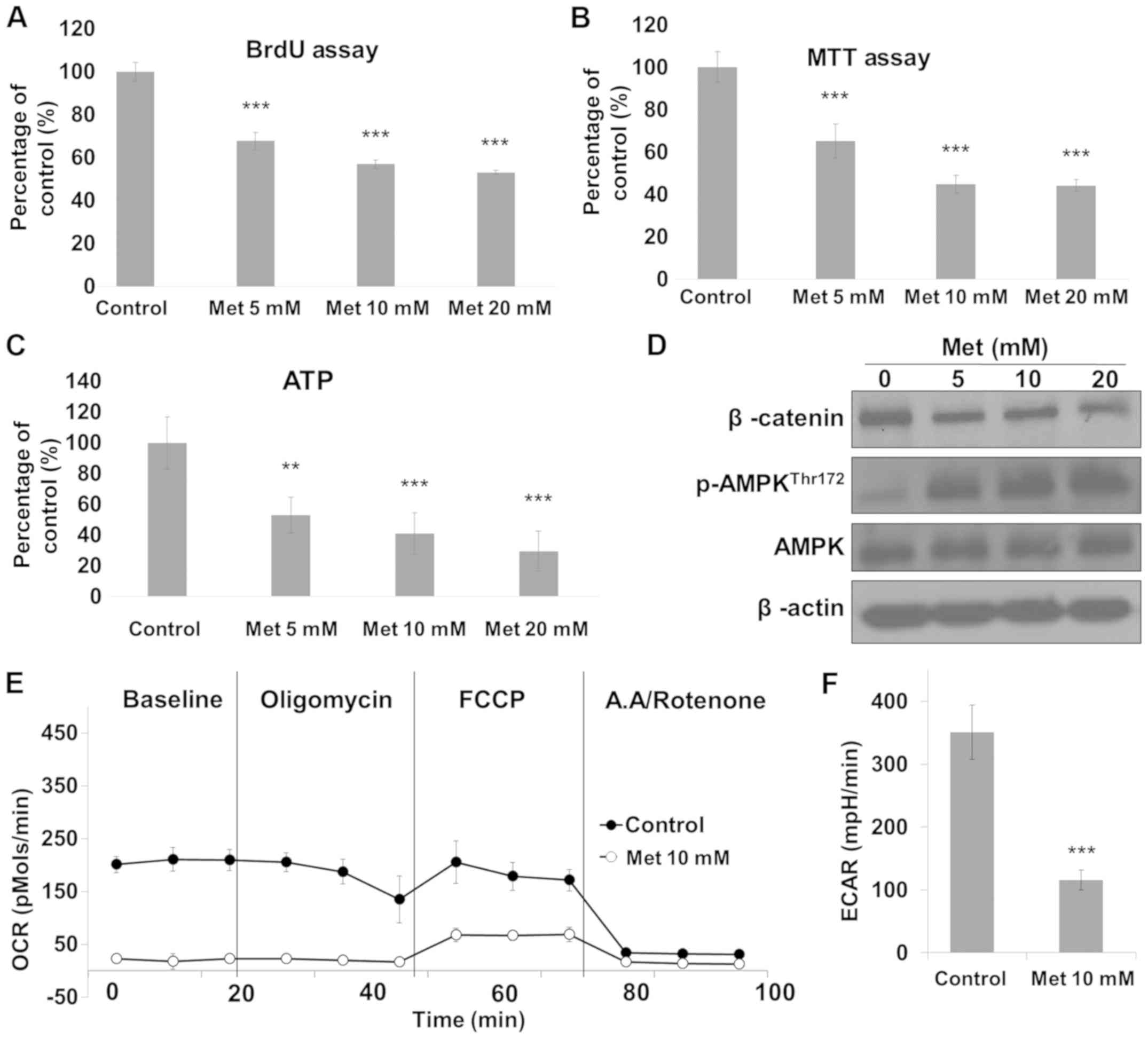 | Figure 2.Metformin inhibits cell proliferation
and ATP production in RKO cells. Cells were treated with metformin
at different concentrations (5–20 mM) for 24 h and cell viability
was measured by (A) BrdU and (B) MTT assay. (C) Cells were
subjected to cellular ATP measurement. (D) Cell lysates were
subjected to immunoblot analysis for β-catenin, p-AMPK, AMPK and
β-actin. Cells were plated into XF24 culture plates and incubated
for 24 h using a medium containing glucose, glutamine and pyruvate.
(E) OCR responses to oligomycin (2 µM), FCCP (0.1 µM), antimycin A
(1 µM) and rotenone (1 µM) were measured and (F) the baseline of
ECAR was measured. **P<0.01 and ***P<0.001 vs. control. p,
phosphorylated; AMPK, 5′-adenosine monophosphate-activated protein
kinase; ATP, adenosine 5′-triphosphate; OCR, oxygen consumption
rate; ECAR, extracellular acidification rate; Met, metformin. |
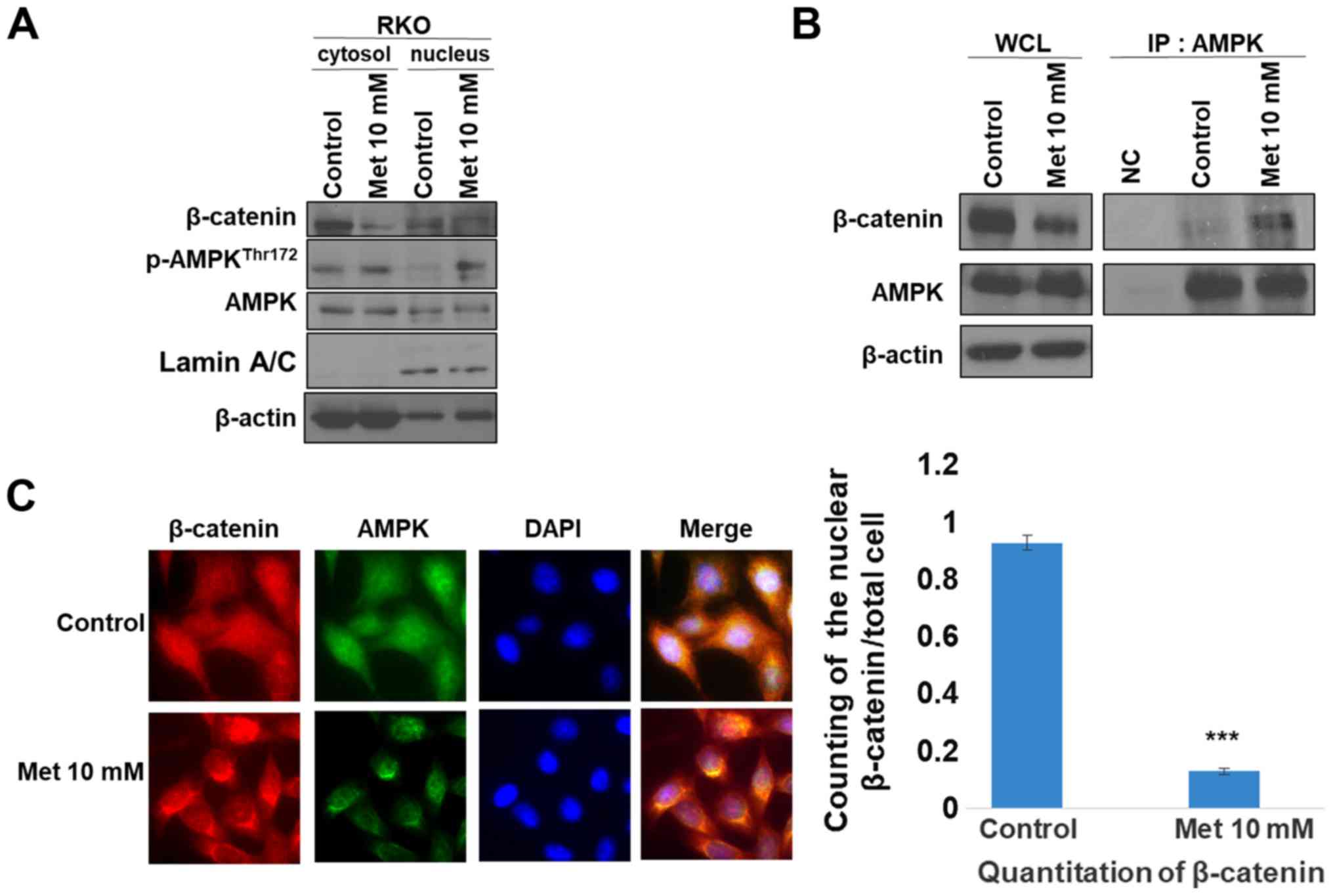
Metformin suppresses β-catenin nuclear
translocation involving binding of AMPK with β-catenin
β-catenin is a major effector of Wnt signaling as it
transcriptionally regulates the expression of c-myc, c-Jun and
cyclin D1 to increase cell proliferation and oncogenesis (15). Upon activation of Wnt signaling,
β-catenin binds to Tcf and translocates into the nucleus (16). The suppression of β-catenin
translocation by metformin was evaluated according to the status of
AMPK activity (Fig. 3). Treatment
with metformin decreased nuclear β-catenin expression, according to
immunoblotting and immunofluorescence assays (Fig. 3A and C), which suggests that a
decrease of cytosolic β-catenin resulted in β-catenin nuclear
translocation. A subsequent immunoprecipitation assay demonstrated
the association between AMPK and β-catenin following metformin
treatment. Co-localization of AMPK with β-catenin was confirmed
(Fig. 3B and C). These results
suggest that AMPK can bind to β-catenin in the cytosol and such
binding may suppress translocation of β-catenin into the
nucleus.
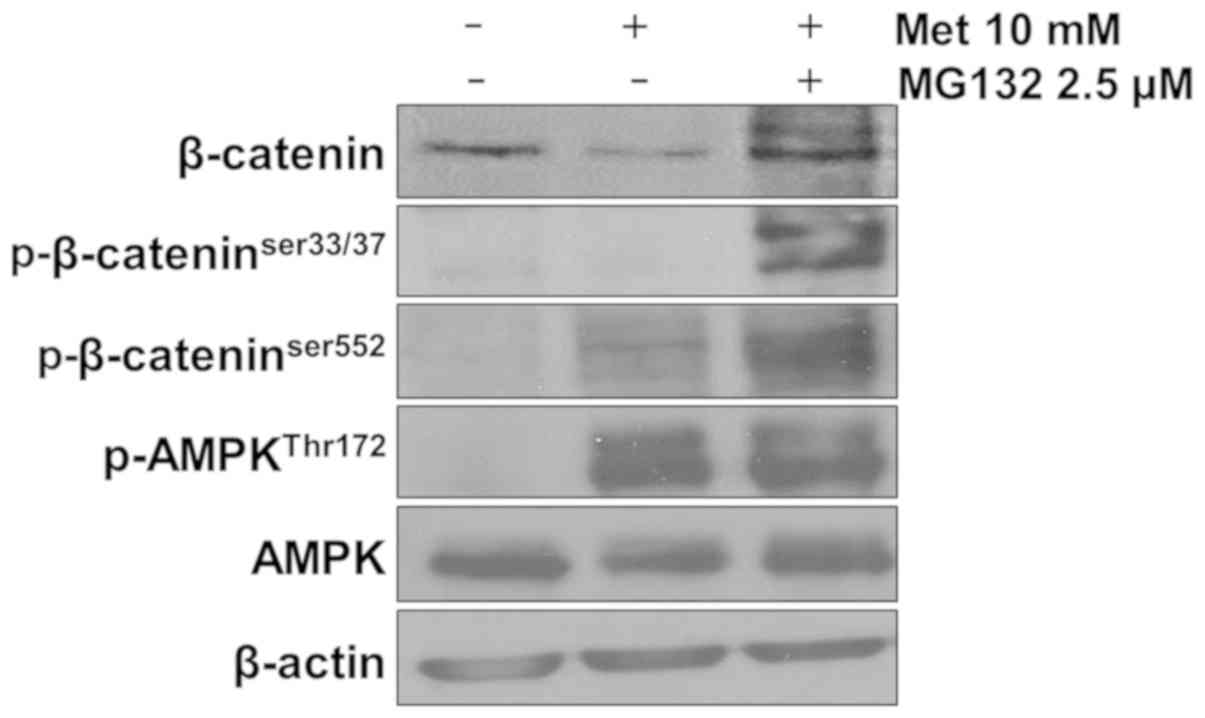
Metformin-induced AMPK activation
leads to β-catenin degradation
Cytoplasmic β-catenin is complexed with adenomatous
polyposis coli, axin and glycogen synthase kinase (GSK), and
subjected to proteasomal degradation following GSK-mediated
phosphorylation of β-catenin (17).
Serine 33/37 phosphorylation of β-catenin is mediated by GSK3β.
Phosphorylation at this site leads to ubiquitination of β-catenin,
which acts as a bridge for proteasomal degradation (18). In addition, it has been reported that
serine 552 phosphorylation can facilitate β-catenin translocation
from the cytoplasm to the nucleus (18). The present study examined whether the
AMPK-mediated decrease in β-catenin expression may be attributable
to proteasomal degradation. In the presence of MG132, metformin
still activated AMPK; however, it failed to reduce β-catenin
expression. Instead, a ubiquitination laddering pattern was
produced (Fig. 4). Since β-catenin
phosphorylation on residues serine 33/37 was observed in
metformin-treated cells, GSK-mediated β-catenin phosphorylation and
subsequent proteosomal degradation may have adequately occurred. In
addition, β-catenin phosphorylation on serine 552 was observed in
metformin-treated cells (Fig. 4).
These results suggest that an association of β-catenin with AMPK
may sequester β-catenin in the cytoplasm, even when serine 552 is
phosphorylated. This cytoplasmic sequestering of β-catenin may
subject β-catenin to degradation.
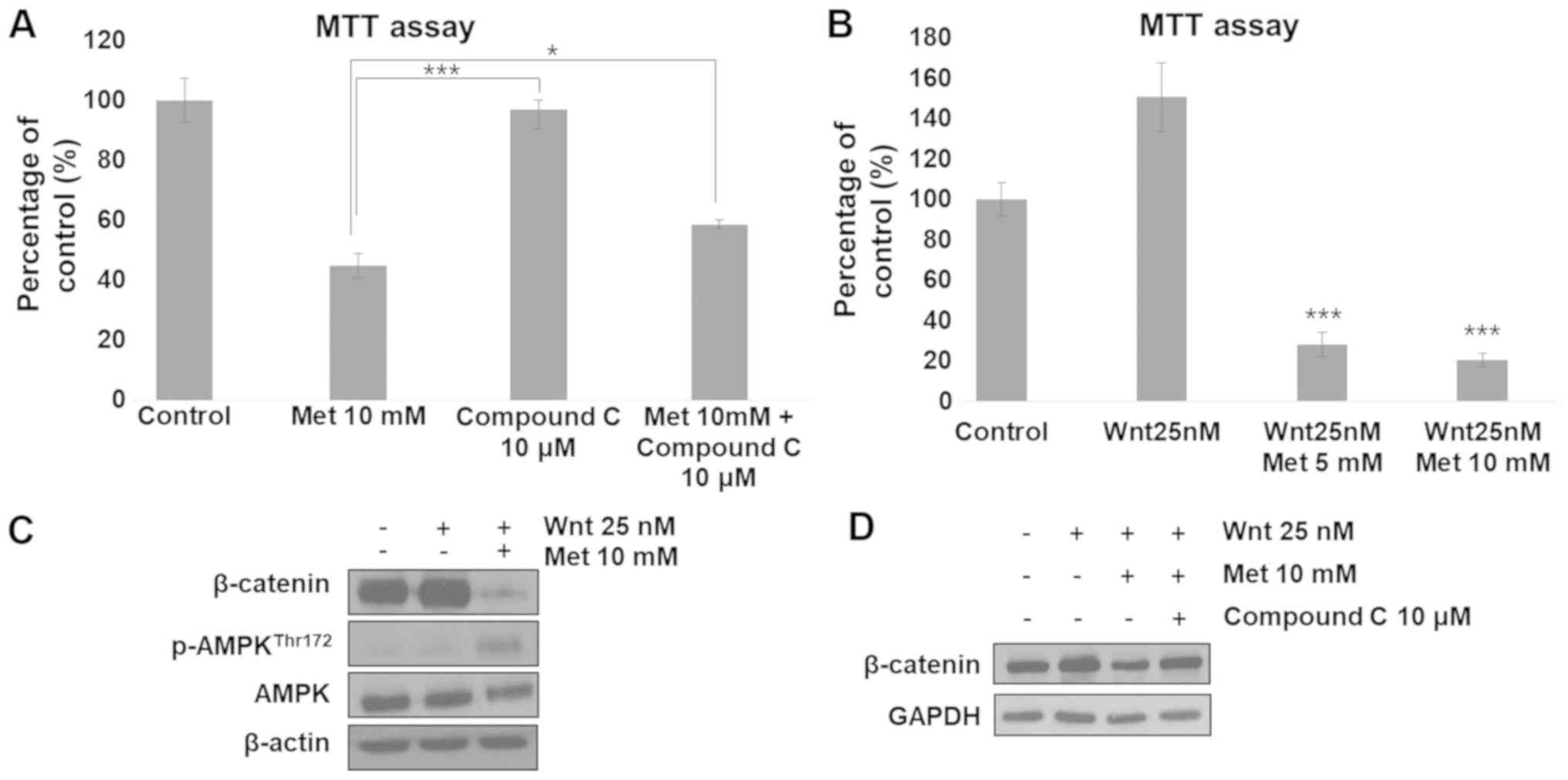
Metformin suppresses
β-catenin-dependent Wnt signaling through AMPK activation
Metformin-induced suppression of cell proliferation
was partially restored by inhibition of AMPK using compound C
(Fig. 5A). Subsequently, the current
study examined whether metformin could suppress RKO cell
proliferation and inhibit β-catenin expression when Wnt signaling
was hyperactivated. It was identified that Wnt-treatment increased
cell proliferation. This increase in cell proliferation was
significantly inhibited by co-treatment with metformin (Fig. 5B). β-catenin expression was markedly
lower in cells co-treated with Wnt and metformin compared with that
in cells treated with Wnt alone (Fig.
5C). Wnt treatment did not affect the phosphorylation status of
AMPK (Fig. 5C). To determine whether
these effects were attributable to AMPK, cells were treated with
compound C, an AMPK inhibitor. The results revealed that the
metformin-induced decrease in β-catenin expression level was
restored following treatment with compound C (Fig. 5D). These results suggest that
metformin-induced AMPK activation can regulate β-catenin
expression.
Discussion
The Wnt pathway is one of the most important
signaling pathways with regard to cell survival and
differentiation. Overexpression and mutations of proteins
associated with the Wnt signaling pathway have been identified in
several types of cancer (19,20). Aberrant expression of β-catenin has
been demonstrated in various types of human cancer, including colon
cancer. Furthermore, a number of studies have demonstrated that
β-catenin, a transcription factor involved in the Wnt signaling
pathway, is frequently upregulated in various types of cancer cells
(21,22). β-catenin exhibits two functions in
cells. Firstly, it is responsible for cell-cell adhesion at the
cell membrane by binding to E-cadherin. Secondly, it is a
transcription factor in the nucleus and serves as the final
messenger of the Wnt signaling pathway (23). Therefore, the intracellular β-catenin
expression level is maintained at a certain level. β-catenin forms
a cytoplasmic complex with APC, axin and GSK to maintain these
proteins at appropriate levels, which causes ubiquitination and
proteasomal degradation of β-catenin (17).
Numerous studies have reported that AMPK is
frequently downregulated in several types of cancer cells (24). AMPK is one of the most important
targets of cancer therapy. Evidence suggests that several AMPK
activators can suppress cancer cell growth and induce apoptosis
(25,26). Metformin is known to be an AMPK
activator. AMPK is a crucial cellular energy sensor that can
increase intracellular AMP level (13). It has been reported that AMPK
activation can suppress cancer cell proliferation in various types
of carcinoma by inhibiting signaling molecules, including protein
kinase B and mTOR, and regulating tumor suppressors, including
cellular tumor antigen p53, which induces cancer cell apoptosis.
The present study demonstrated that the activation of AMPK was
associated with a reduction in the nuclear β-catenin expression
level, and cell growth was significantly reduced in
metformin-treated RKO cells.
The importance of inhibiting the nuclear
translocation of β-catenin has been recognized for suppressing
cancer cell proliferation and metastasis (27). A number of reports have suggested that
AMPK-regulated genes are associated with Wnt signaling, including
GSK3β and Dishevelled (28,29). As chemical activators of AMPK can
suppress Wnt3a-induced TCF-dependent transcriptional activity, we
hypothesized that AMPK activity may participate in the regulation
of Wnt signaling. However, a limited number of studies have
investigated the direct association between AMPK and β-catenin. The
current study performed immunoprecipitation and immunofluorescence
analysis to identify whether activated AMPK could bind to β-catenin
in metformin-treated RKO cells. The results demonstrated that
binding between AMPK and β-catenin occurred in the cytoplasm near
the nuclear membrane. In addition, phosphorylation of the serine
552 residue of β-catenin was maintained; however, the translocation
of β-catenin to the nucleus was not observed. These results suggest
that AMPK-inhibited β-catenin nuclear translocation may be due to
cytoplasmic sequestering of β-catenin through the association with
AMPK. Sequestered β-catenin remains targeted to proteosomal
degradation since serine 33/37 residues of β-catenin are
phosphorylated and a ubiquitination laddering pattern was produced
when cells were treated with proteasome inhibitor MG132 and
metformin. Furthermore, metformin-induced AMPK activation reduced
Wnt-mediated cell viability and β-catenin degradation, suggesting
that AMPK activation may regulate Wnt activity. In primary
hepatocytes, a lower concentration (<100 nM) of metformin
activated AMPK, resulting in suppression of glucose production
(30,31). In ovarian cancer cells, >1 mM
metformin demonstrated a growth inhibitory effect, as well as AMPK
activation (32). In the present
study, with a low dose of metformin, AMPK was weakly activated
without an inhibitory effect on cell proliferation, suggesting that
a higher level of AMPK activity may be required for the suppression
of cell proliferation. In human clinical trials for patients with
diabetes, the safety of metformin has been proven and the
dose-limiting toxicity of metformin was reported as 2,500 mg/day
(33).
A number of studies have described the ability of
metformin to reduce the risk of cancer among patients with
metformin-treated diabetes (34–36).
Therefore, metformin may be considered as a candidate drug to be
used for the prevention of cancer; however, the exact mechanism
remains to be elucidated. The current results, which demonstrated
that metformin-activated AMPK may be due to mitochondrial
dysfunction, suggest that AMPK activity is an important factor for
the effects of metformin; however, inhibition of AMPK only
partially restored metformin-induced cell death, suggesting that
unidentified additional mechanisms may be involved. Although the
safety of metformin has been demonstrated in diabetes treatment,
further studies investigating the complete mechanism of metformin
are required, particularly with regard to cancer, prior to clinical
trials using the drug.
Metformin can suppress cellular energy production by
inhibiting mitochondrial complex I (12). This reduction of cellular energy may
promote AMPK activation. In the present results, the baseline OCR
of metformin-treated RKO cells was lower compared with that of
control RKO cells treated with oligomycin, an inhibitor of
mitochondrial complex V, but similar to that of RKO cells treated
with rotenone, an inhibitor of mitochondrial complex I. This result
is consistent with that of a previous study, which demonstrated
that cell proliferation is suppressed in metformin-treated H1299
and HCT116 cells by inhibition of mitochondrial complex I (37). Therefore, mitochondrial dysfunction
and reduction of energy production are required for
metformin-induced AMPK activation. In addition, Wnt signaling
activity is also associated with energy metabolism. Wnt signaling
is understood to serve an important role in the survival of cancer
cells (38). Wnt signaling increases
aerobic glycolysis by transcribing c-myc, which upregulates the
expression of glycolytic genes, including glucose transporter-1 and
lactate dehydrogenase in cancer cells (39). Axin, a negative regulator for Wnt
signaling, can reduce mitochondrial energy metabolism by
suppressing mitochondrial complex IV function (40). Considering how AMPK activation
inhibits Wnt signaling, reduced Wnt signaling activity may also
participate in the regulation of metabolic energy production. This
reduced Wnt signaling activity may be an additional mechanism to
reduce energy production further in metformin-treated
AMPK-activated cells.
In summary, the current study suggests sequential
mechanisms are involved in metformin-induced suppression of Wnt
signaling and cell proliferation. Metformin treatment suppresses
mitochondrial energy metabolism, which activates AMPK. Activated
AMPK can sequester β-catenin in the cytoplasm, which increases
β-catenin proteosomal degradation rather than β-catenin nuclear
translocation. Overall, these events can reduce Wnt signaling
activity. Therefore, AMPK is an important regulator of the
translocation of β-catenin from the cytosol into the nucleus to
exert anti-proliferative activities with metformin.
Acknowledgements
Not applicable.
Funding
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education, Science and Technology (grant
no. NRF-2015R1D1A1A09056775).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SYP and SHK designed the experiments. SYP and DK
performed the experiments. SYP, DK and SHK analyzed the data. SYP
wrote the paper. SHK edited the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nusse R: Wnt signaling in disease and in
development. Cell Res. 15:28–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DiMeo TA, Anderson K, Phadke P, Fan C,
Feng C, Perou CM, Naber S and Kuperwasser C: A novel lung
metastasis signature links Wnt signaling with cancer cell
self-renewal and epithelial-mesenchymal transition in basal-like
breast cancer. Cancer Res. 69:5364–5373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Komiya Y and Habas R: Wnt signal
transduction pathways. Organogenesis. 4:68–75. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Drees F, Pokutta S, Yamada S, Nelson WJ
and Weis WI: Alpha-catenin is a molecular switch that binds
E-cadherin- beta-catenin and regulates actin-filament assembly.
Cell. 123:903–915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial- mesenchymal transition in cancer: Parallels between
normal development and tumor progression. J Mammary Gland Biol
Neoplasia. 15:117–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patel VA, Massenburg D, Vujicic S, Feng L,
Tang M, Litbarg N, Antoni A, Rauch J, Lieberthal W and Levine JS:
Apoptotic cells activate AMP-activated protein kinase (AMPK) and
inhibit epithelial cell growth without change in intracellular
energy stores. J Biol Chem. 290:22352–22369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaw RJ: LKB1 and AMP-activated protein
kinase control of mTOR signalling and growth. Acta Physiol (Oxf).
196:65–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Corradetti MN, Inoki K, Bardeesy N,
DePinho RA and Guan KL: Regulation of the TSC pathway by LKB1:
Evidence of a molecular link between tuberous sclerosis complex and
Peutz-Jeghers syndrome. Genes Dev. 18:1533–1538. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zulato E, Bergamo F, De Paoli A, Griguolo
G, Esposito G, De Salvo GL, Mescoli C, Rugge M, Nardin M, Di Grazia
L, et al: Prognostic significance of AMPK activation in advanced
stage colorectal cancer treated with chemotherapy plus bevacizumab.
Br J Cancer. 111:25–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Madiraju AK, Erion DM, Rahimi Y, Zhang XM,
Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald
MJ, et al: Metformin suppresses gluconeogenesis by inhibiting
mitochondrial glycerophosphate dehydrogenase. Nature. 510:542–546.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Viollet B, Guigas B, Sanz Garcia N,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yue W, Yang CS, DiPaola RS and Tan XL:
Repurposing of metformin and aspirin by targeting AMPK-mTOR and
inflammation for pancreatic cancer prevention and treatment. Cancer
Prev Res (Phila). 7:388–397. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liberti MV and Locasale JW: The warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shang S, Hua F and Hu ZW: The regulation
of β-catenin activity and function in cancer: Therapeutic
opportunities. Oncotarget. 8:33972–33989. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cadigan KM and Waterman ML: TCF/LEFs and
Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol.
4(pii): a0079062012.PubMed/NCBI
|
|
17
|
Stamos JL and Weis WI: The β-catenin
destruction complex. Cold Spring Harb Perspect Biol. 5:a0078982013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fang D, Hawke D, Zheng Y, Xia Y,
Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T and Lu Z:
Phosphorylation of beta-catenin by AKT promotes beta-catenin
transcriptional activity. J Biol Chem. 282:11221–11229. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhan T, Rindtorff N and Boutros M: Wnt
signaling in cancer. Oncogene. 36:1461–1473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park SY, Lee YK, Lee WS, Park OJ and Kim
YM: The involvement of AMPK/GSK3-beta signals in the control of
metastasis and proliferation in hepato-carcinoma cells treated with
anthocyanins extracted from Korea wild berry Meoru. BMC Complement
Altern Med. 14:1092014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Z, He X, Jia M, Liu Y, Qu D, Wu D, Wu
P, Ni C, Zhang Z, Ye J, et al: β-catenin overexpression in the
nucleus predicts progress disease and unfavourable survival in
colorectal cancer: A meta-analysis. PLoS One. 8:e638542013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kraus C, Liehr T, Hülsken J, Behrens J,
Birchmeier W, Grzeschik KH and Ballhausen WG: Localization of the
human beta-catenin gene (CTNNB1) to 3p21: A region implicated in
tumor development. Genomics. 23:272–274. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Motoshima H, Goldstein BJ, Igata M and
Araki E: AMPK and cell proliferation-AMPK as a therapeutic target
for atherosclerosis and cancer. J Physiol. 574:63–71. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, Saud SM, Young MR, Chen G and Hua B:
Targeting AMPK for cancer prevention and treatment. Oncotarget.
6:7365–7378. 2015.PubMed/NCBI
|
|
26
|
Luo Z, Zang M and Guo W: AMPK as a
metabolic tumor suppressor: Control of metabolism and cell growth.
Future Oncol. 6:457–470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jang GB, Kim JY, Cho SD, Park KS, Jung JY,
Lee HY, Hong IS and Nam JS: Blockade of Wnt/β-catenin signaling
suppresses breast cancer metastasis by inhibiting CSC-like
phenotype. Sci Rep. 5:124652015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang H, Xue J, Li M, Zhao X, Wei D and Li
C: Metformin regulates stromal-epithelial cells communication via
Wnt2/β-catenin signaling in endometriosis. Mol Cell Endocrinol.
413:61–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Markowska A, Pawałowska M, Filas V, Korski
K, Gryboś M, Sajdak S, Olejek A, Bednarek W, Spiewankiewicz B,
Lubin J and Markowska J: Does metformin affect ER, PR, IGF-1R,
β-catenin and PAX-2 expression in women with diabetes mellitus and
endometrial cancer? Diabetol Metab Syndr. 5:762013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cao J, Meng S, Chang E, Beckwith-Fickas K,
Xiong L, Cole RN, Radovick S, Wondisford FE and He L: Low
concentrations of metformin suppress glucose production in
hepatocytes through AMP-activated protein kinase (AMPK). J Biol
Chem. 289:20435–20446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Erices R, Bravo ML, Gonzalez P, Oliva B,
Racordon D, Garrido M, Ibañez C, Kato S, Brañes J, Pizarro J, et
al: Metformin, at concentrations corresponding to the treatment of
diabetes, potentiates the cytotoxic effects of carboplatin in
cultures of ovarian cancer cells. Reprod Sci. 20:1433–1446. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chae YK, Arya A, Malecek MK, Shin DS,
Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L
and Giles F: Repurposing metformin for cancer treatment: Current
clinical studies. Oncotarget. 7:40767–40780. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ruiter R, Visser LE, van Herk-Sukel MP,
Coebergh JW, Haak HR, Geelhoed-Duijvestijn PH, Straus SM, Herings
RM and Stricker BH: Lower risk of cancer in patients on metformin
in comparison with those on sulfonylurea derivatives: Results from
a large population-based follow-up study. Diabetes Care.
35:119–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Staa TP, Patel D, Gallagher AM and de
Bruin ML: Glucose-lowering agents and the patterns of risk for
cancer: A study with the General Practice Research Database and
secondary care data. Diabetologia. 55:654–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Griss T, Vincent EE, Egnatchik R, Chen J,
Ma EH, Faubert B, Viollet B, DeBerardinis RJ and Jones RG:
Metformin antagonizes cancer cell proliferation by suppressing
mitochondrial-dependent biosynthesis. PLoS Biol. 13:e10023092015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li N, Ragheb K, Lawler G, Sturgis J, Rajwa
B, Melendez JA and Robinson JP: Mitochondrial complex I inhibitor
rotenone induces apoptosis through enhancing mitochondrial reactive
oxygen species production. J Biol Chem. 278:8516–8525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sherwood V: WNT signaling: An emerging
mediator of cancer cell metabolism? Mol Cell Biol. 35:2–10. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shin JH, Kim HW, Rhyu IJ and Kee SH: Axin
is expressed in mitochondria and suppresses mitochondrial ATP
synthesis in HeLa cells. Exp Cell Res. 340:12–21. 2016. View Article : Google Scholar : PubMed/NCBI
|