Introduction
In advanced malignant diseases, cachexia appears to
be one of the most common systemic manifestations. The presence of
cachexia always indicates a poor prognosis, having a marked impact
on the patients’ quality of life and survival (1). Several important molecular mechanisms
have been shown to be involved in the increased muscle catabolism
observed in cancer-induced cachexia, such as greater
ubiquitin-proteasome-dependent proteolysis, apoptosis, and
activation of uncoupling proteins (2–5).
Interaction of these mechanisms leads to muscle-mass loss by
promoting protein and DNA breakdown and energy inefficiency.
Myostatin, also known as growth and differentiation
factor-8 (GDF factor-8), which was cloned in 1997, is a member of
the transforming growth factor-β (TGF-β) superfamily of secreted
growth factors. It is a negative regulator of skeletal muscle
development (6–8). During the embryogenetic process,
myostatin expression is only observed in developing skeletal
muscles, but the protein continues to be expressed and secreted by
skeletal muscles during adult life (9,10).
Mice and cattle with genetic deficiencies in myostatin exhibit
marked increases in skeletal muscle mass, thus supporting the role
of myostatin in supressing muscle growth (11). Myostatin acts systemically (it is
produced in muscle and adipose tissue and released in the
circulation) and binds to cell-surface receptors, causing muscle
loss. The myostatin protein can be detected in the circulation as a
full-length precursor that is cleaved into an amino-terminal
pro-peptide and a carboxy-terminal mature region, which is the
active form of the peptide. In skeletal muscle and circulation,
myostatin is detected in inactive complexes of differing
composition with other proteins, including its own pro-peptide,
follistatin-like 3 (Fstl3, also known as follistatin-related gene),
and latent TGFβ binding protein (12–15).
The mechanism of activation of these inactive complexes (or whether
they can be activated at all) is unknown. For complexes containing
the pro-peptide, activation likely requires proteolysis of the
pro-peptide, possibly by specific target cells (16,17).
Once activated, myostatin has a high affinity for the activin IIB
receptor (Acvr2b, also known as ActRIIB) and a weak affinity for
Acvr2a (also known as ActRII and ActRIIA), both of which, as with
other receptors for TGF-β family members, bind multiple ligands
(18).
Skeletal muscle possesses the ability to respond and
adapt to changing environmental stimuli, leading to a set of
metabolic and morphological adaptations, which allow it to better
meet the energy demands of sustained physical activity. Active
myostatin binds to its receptor ActRIIB with high affinity and
regulates the expression of target genes through a TGF-β signaling
pathway (10). Myostatin signaling
acts through this receptor on skeletal muscle by activating an
intracellular cascade of events. First, there is presumed
recruitment of a type I co-receptor. Thus, activin receptor-like
kinases 4 and/or 5 (ALK-4, ALK-5) are candidate coreceptors that
are phosphorylated by ActRIIB. This activates phosphorylation of
TGF-α-specific Smads 2 and 3, which form a complex with Smad 4. The
Smad 2/3/4 complex is translocated to the nucleus to regulate the
expression of targeted genes such as MyoD and other myogenic
regulatory factors (19). Myostatin
also activates the p38 MAPK, Erk1/2 and Wnt pathways (10,19–22).
Inhibition of myostatin by injection of neutralizing antibodies or
antagonists causes an increase in skeletal muscle mass, thus
myostatin inhibitors exhibit great potential as candidates for
treatment of muscle-wasting diseases (15,23).
Conversely, the ectopic expression of myostatin induces atrophy in
adult skeletal muscle by decreasing muscle structural genes such as
those of myofibrillar proteins (myosin heavy-chain IIb, troponin I
and desmin) and myogenic transcription factors (MyoD and myogenin)
(24). Myostatin induces muscle
wasting by activating the ubiquitin proteolytic system. The
ubiquitin-associated genes atrogin-1, MuRF-1 and E214k are
upregulated following myostatin treatment (25). Of note, myostatin signaling is able
to inhibit Akt phosphorylation, thus downregulating the
IGF-1/PI3K/AKT hypertrophy pathway. In addition, myostatin
increases the levels of the active form of the transcription factor
FoxO1, allowing for increased expression of atrophy-related genes
(atrogenes). Notably, myostatin does not exert its action through
NF-κB (25).
β2-adrenergic agonists are potent muscle growth
promoters in many animal species (26,27),
resulting in skeletal muscle hypertrophy (28–31).
Formoterol is one of these compounds with important anti-cachectic
effects in animal models. The model of action of this drug is based
on its ability to prevent muscle-wasting by inhibiting proteolysis
in skeletal muscle. Thus, this β2-agonist decreases the activation
of the ubiquitin-dependent proteolytic system, the main mechanism
activated in muscle-wasting conditions (32). In addition to its anti-proteolytic
effects, formoterol decreases muscle apoptosis in muscle-wasting
animals (32).
Bearing the above in mind, the aim of the present
study was, in addition to observing the myostatin gene expression
in a catabolic condition characterised by profound muscle-wasting
implantation of the Yoshida AH-130 ascites hepatoma, to test the
hypothesis that certain anabolic actions of formoterol on skeletal
muscle (32) involve the myostatin
system.
Materials and methods
Animals
Male Wistar rats (Interfauna, Barcelona, Spain) of 5
weeks of age were used in the experiments. The animals were
maintained at 22±2°C with a regular light-dark cycle (light on from
08:00 a.m. to 08:00 p.m.) and had free access to food and water.
The food intake was measured daily. All animal manipulations were
performed in accordance with the European Community guidelines for
the use of laboratory animals.
Tumour inoculation and formoterol
treatment
Rats were divided into control and tumour host
groups. The latter group received an intraperitoneal inoculum of
108 AH-130 Yoshida ascites hepatoma cells obtained from
exponential tumours (33). The two
groups were further divided into treated and untreated sub-groups,
the former being administered a daily subcutaneous dose of
formoterol [0.3 mg/kg body weight (bw), dissolved in physiological
solution], and the latter the corresponding volume of solvent. On
day 7 after tumour transplantation, the animals were weighed and
anaesthetised with an intra-peritoneal injection of
ketamine/xylazine mixture (3:1) (Imalgene® and
Rompun®, respectively). The tumour was harvested from
the peritoneal cavity and its volume and cellularity were
evaluated. Tissues were rapidly excised, weighed, and frozen in
liquid nitrogen.
Real-time PCR
Total RNA from the gastrocnemius muscle was
extracted by TriPure™ kit (Roche, Barcelona, Spain), a commercial
modification of the acid guanidinium
isothiocyanate/phenol/chloroform method (34).
First-strand cDNA was synthesised from total RNA
with oligo dT15 primers and random primers pdN6 using a cDNA
synthesis kit (Transcriptor Reverse Transcriptase, Roche,
Barcelona, Spain). Analysis of mRNA levels for myostatin, ActIIB,
FLST288, FLST315, FLST-L3 and Ub was performed with primers
designed to detect these products: myostatin (Forward, 5′ CTA CCA
CGG AAA CAA TCA TTA CCA 3′; Reverse, 5′ AGC AAC ATT TGG GCT TTC CAT
3′), ActIIB (Forward, 5′ TTG GCT GCG TCT GGA AGG CT 3′; Reverse, 5′
TGC CAC GAC TGC TTG TCC TGA 3′), FLST288 (Forward, 5′ AGG GAA AGT
GTA TCA AAG CAA AGT C 3′; Reverse, 5′ AAC CTT GAA ATC CCA TAG GCA
TT 3′); FLST315 (Forward, 5′ TGC TCC TCC GGC GTA CTG 3′; Reverse,
5′ CCG AGA TGG AGT TGC AAG ATC 3′); FLST-L3 (Forward, 5′ CCA ACC
CTG GCC AAG AAC T 3′; Reverse, 5′ AAT GGA ACG GCC CAG AAA G 3′),
Ubiquitin (Forward, 5′ GAT CCA GGA CAA GGA GGG C 3′; Reverse, 5′
CAT CTT CCA GCT GCT TGC CT3′) and hydroxymethylbilane synthase
(hmbs) (Forward, 5′ TGC CAG AGA AAA GTG CCG TGG G 3′; Reverse, 5′
TGC AGC TCA TCC AGC TTC CGT 3′), 18S (Forward, 5′-GCG AAT GGC TCA
TTA AAT CAG TTA-3′; Reverse, 5′-TGG TTT TGA TCT GAT AAA TGC
ACG-3′), p0 (Forward, 5′ GAG GTC CTC CTT GGT GAA CA 3′; Reverse, 5′
CCT CAT TGT GGG AGC AGA CA 3′). To avoid possible contamination by
genomic DNA, primers were designed in different exons. The
real-time PCR was performed using a commercial kit (LightCycler™
480 SYBR-Green I Master, Roche, Barcelona, Spain). The relative
amount of all mRNA was calculated using a comparative Ct method.
The average value of 18S, hmbs and acidic ribosomal phosphoprotein
p0 mRNA was used as the invariant control for all studies.
Western blotting
Gastrocnemius muscle was homogenised in 10 mmol/l
HEPES (pH 7.5), 10 mmol/l MgCl2, 5 mmol/l KCl, 0.1
mmol/l EDTA, 0.1% Triton X-100, 1 mmol/l DTT, and 5 μl/ml of buffer
of a protease inhibitor cocktail (Sigma, St. Louis, MO, USA).
Tissue homogenates were then centrifuged at 7,000 rpm for 5 min at
4°C, and the supernatants were collected. Protein concentrations
were determined according to the method of bicinchoninic acid
(Pierce Technology, Rockford, IL, USA). Equal amounts of protein
(50 or 100 μg) were heat-denatured in sample loading buffer [50
mmol/l Tris-HCl (pH 6.8), 100 mmol/l DTT, 2% SDS, 0.1% bromophenol
blue, 10% glycerol], resolved by SDS-PAGE (10% polyacrylamide, 0.1%
SDS), and transferred to Immobilon membranes (Immobilon
polyvinylidene difluoride, Millipore, Billerica, MA, USA). The
filters were blocked with 5% PBS non-fat dry milk and then
incubated with polyclonal antibody anti-myostatin (Millipore).
Mouse antibody anti-Na+, K+-ATPase α subunit
(John Hopkins University, Baltimore, MD, USA) was used as the
invariant control. Goat anti-rabbit horseradish peroxidase
conjugate (BioRad, Hercules, CA, USA) and HRP-conjugated donkey
anti-mouse immunoglobulin-specific monoclonal antibody (Jackson
Immuno Research, West Grove, PA, USA) were used as secondary
antibodies. The membrane-bound immune complexes were detected by an
enhanced chemiluminescence system (EZ-ECL, Amersham Biosciences,
Piscataway, NJ, USA).
Statistical analysis
Statistical analysis of the data was performed by
means of the Student’s t-test.
Results and Discussion
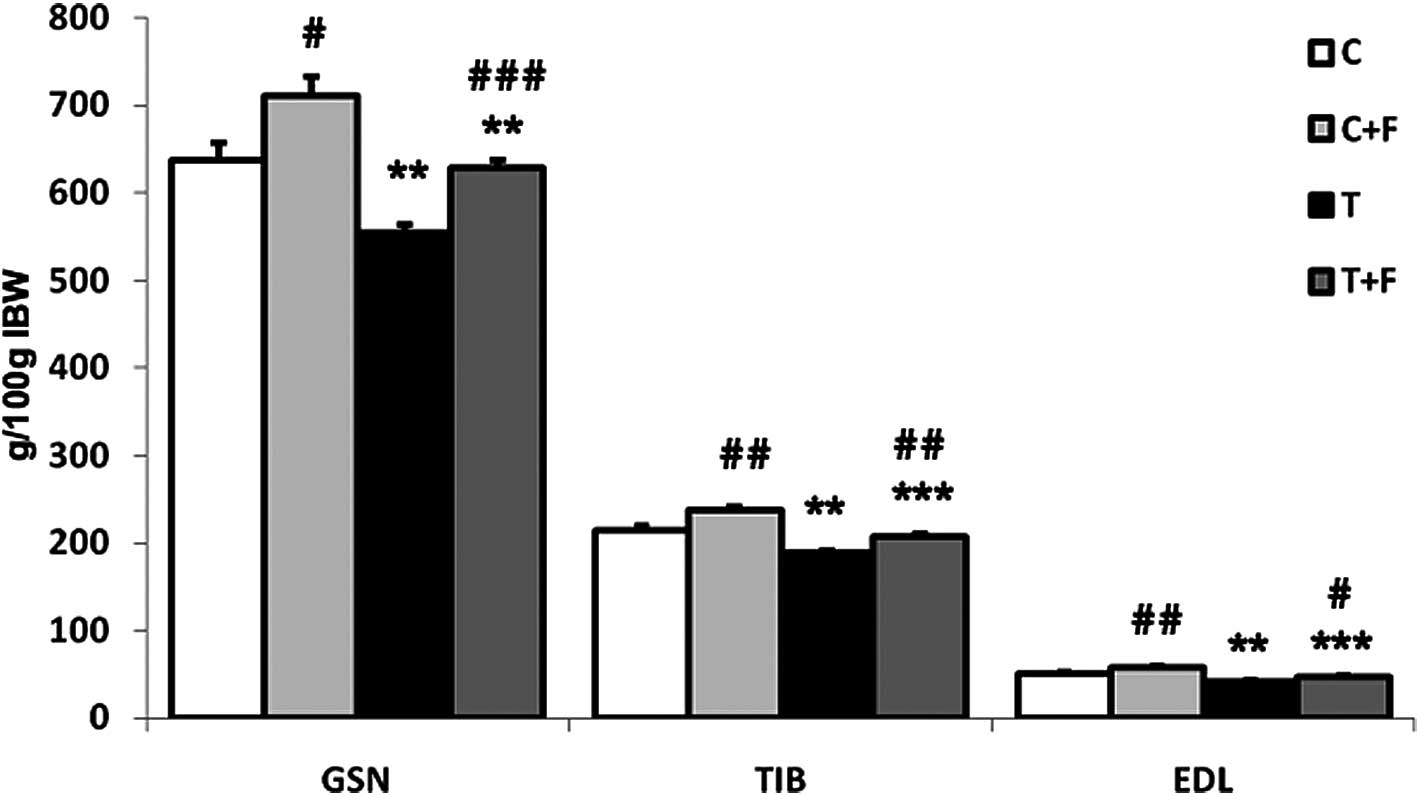
Fig. 1 shows the
implantation of the Yoshida AH-130 ascites hepatoma caused
significant decreases in the weight of the gastrocnemius (13%), EDL
(18%) and tibialis (12%) muscles. Formoterol treatment
significantly increased the weight of the above-mentioned muscles
(Fig. 1). These data are in
agreement with previous results from our own laboratory (32).
Myostatin has been described as a negative regulator
of muscle growth and its development, and is involved in several
forms of muscle wasting, including the severe cachexia observed as
a result of diseases such as AIDS and cancer (35,36).
McFarlane et al have demonstrated that myostatin induces
cachexia through a NF-κB-independent mechanism by antagonizing
hypertrophy signaling through the regulation of the AKT-FOXO1
pathway (25). As with other TGF-β
superfamily members, myostatin interacts with a serine/threonine
transmembrane receptor known as activin IIB (ActIIB) (22,37,38).
Binding of the ligand to ActRII leads to phosphorylation and
activation of the activin type I receptor, which in turn initiates
intracellular signaling mediated by SMAD proteins (22,37,38).
Although the levels of gene expression for myostatin were not
affected by tumour burden (Table
I), the cachectic animals presented a significant increase
(113%) in the gastrocnemius muscle mRNA levels of the myostatin
receptor (ActIIB). This increase indicates that muscle wasting in
this experimental tumour model involved the myostatin system.
Notably, the expression of the different isoforms of follistatin
[FLST288 and FLST315 (39,40)], a protein with the opposite effects
to those of myostatin, were significantly reduced as a result of
the implantation of the tumour (46 and 58% for FLST288 and FLST315,
respectively, Table I). This
observation further supports a role for the myostatin system in
muscle wasting in the Yoshida AH-130 ascites hepatoma model.
 | Table IEffects of formoterol treatment on
gastrocnemius muscle mRNA content of ubiquitin, myostatin,
follistatin isoforms and ActIIB receptor and muscle protein content
of myostatin in rats bearing the Yoshida AH-130 ascites
hepatoma. |
Table I
Effects of formoterol treatment on
gastrocnemius muscle mRNA content of ubiquitin, myostatin,
follistatin isoforms and ActIIB receptor and muscle protein content
of myostatin in rats bearing the Yoshida AH-130 ascites
hepatoma.
| C | C + F | T | T + F |
|---|
| Gastrocnemius
muscle |
| mRNA content |
| Myostatin | 100±21 | 140±19 | 102±23 | 160±60 |
| ActIIB | 100±12 | 133±13 |
213±31b |
106±27d |
| FLSTL3 | 100±12 | 94±16 | 172±38 |
291±48b |
| FLST315 | 100±21 | 108±28 |
42±14a |
129±29d |
| FLST288 | 100±13 |
55±10d |
54±13a |
105±10b,d |
| Ubiquitin | 100±36 | 90±16 |
439±83b |
103±25e |
| Protein
content |
| Myostatin 40
kDa | 100±19 | 115±14 | 98±14 |
53±12b,d |
| Myostatin 26
kDa | 100±16 | 108±13 | 106±10 |
72±7a,d |
| Heart |
| mRNA content |
| Myostatin | 100±24 | 85±10 | 67±7 | 87±11 |
| ActIIB | 100±15 | 125±13 | 126±17 | 140±23 |
| FLSTL3 | 100±12 | 83±13 | 82±8 | 94±13 |
| FLST315 | 100±7 | 118±21 | 98±15 | 116±31 |
| FLST288 | 100±33 | 111±21 | 74±13 | 94±15 |
| Ubiquitin | 100±5 | 93±9 |
143±4c |
119±9d |
Follistatin is a secreted protein capable of binding
and neutralising the actions of any member of the TGF-β family of
proteins such as myostatin. In addition to our finding that
follistatin is decreased in tumour-bearing animals, two independent
approaches provide clear evidence that downregulation of this gene
contributes to the atrophic phenotype in cachectic muscles. One
approach is based on the observation that follistatin is required
to increase the muscle capillary density and improve endothelial
cell function in ischemic mice and that its expression is
associated with AKT1 signaling (41). This result indicates that
follistatin downregulation in cachectic muscles leads to a decrease
in AKT1 expression resulting in a decreased protein synthesis
(41). The other approach, using
transgenic mice and C2C12 cells overexpressing follistatin,
demonstrated that follistatin over-expression in mouse skeletal
muscle promotes hypertrophy and an oxidative fibre-type switch,
leading to increased whole-body strength and fatigue resistance;
follistatin overexpression enhances myoblast fusion, resulting in
hypertrophic myotubes in C2C12 cells, acting via regulation of the
CnA-NFAT pathway (42). Our results
are in agreement with those of Costelli et al who observed
similar changes in both myostatin and follistatin mRNA levels in
gastrocnemius muscles of tumour-bearing rats at day 4 of tumour
growth (36). However, at later
stages of tumour growth (day 7, as used in the present study) the
same authors observed increased myostatin and follistatin mRNA
levels. The reasons for this discrepancy are not known.
No changes in myostatin, myostatin receptor or
follistatin mRNA were observed in the heart of the tumour-bearing
animals (Table I). This indicates
that the myostatin system does not appear to be responsible for
cardiac muscle wasting. Similar observations have been made in
patients with chronic heart failure (43).
When the animals were treated with formoterol, a
β-agonist with anti-cachectic potential (32), in addition to an anabolic action of
skeletal muscle that was clearly reflected on increases in muscle
weights [gastrocnemius (13%), tibialis (10%) and EDL (12%)
(Fig. 1)], the β-agonist
significantly decreased ubiquitin gene expression (Table I). This is a clear marker of protein
degradation, a process that has extensively been described during
muscle wasting (44–49). Formoterol treatment significantly
increased the two follistatin isoforms (94% for FLST288 and 201%
for FLST315) as well as the FLSTL3 (69%), termed FLST-like 3.
FLSTL3 is a protein that has recently been discovered to share
significant structural and functional homology with follistatin
(50). Moreover, treatment
significantly decreased the levels of expression of the myostatin
receptor (50%) (Table I). In
addition, formoterol treatment resulted in a significant decrease
in the myostatin protein content [both active (32%) and inactive
myostatin (46%)] of gastrocnemius muscle (Table I). It is clear that the antiwasting
effects of formoterol involve the myostatin system.
In conclusion, the results presented indicate that
certain anabolic actions of formoterol on the skeletal muscle of
cachectic animals may be mediated via the myostatin system. This
treatment modality should therefore be regarded as a potential
therapeutic target in cancer cachexia.
Acknowledgements
This study was supported by a grant from the
Ministerio de Ciencia y Tecnología (SAF-02284-2008). The authors
would like to thank Industriale Chimica s.r.l. (Saronno, Italy),
who kindly provided micronised formoterol fumarate.
References
|
1
|
Harvey KB, Bothe A Jr and Blackburn GL:
Nutritional assessment and patient outcome during oncological
therapy. Cancer. 43:2065–2069. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Argiles JM, Alvarez B and Lopez-Soriano
FJ: The metabolic basis of cancer cachexia. Med Res Rev.
17:477–498. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Argiles JM and Lopez-Soriano FJ: The
ubiquitin-dependent proteolytic pathway in skeletal muscle: its
role in pathological states. Trends Pharmacol Sci. 17:223–226.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sanchis D, Busquets S, Alvarez B, Ricquier
D, Lopez-Soriano FJ and Argiles JM: Skeletal muscle UCP2 and UCP3
gene expression in a rat cancer cachexia model. FEBS Lett.
436:415–418. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Royen M, Carbo N, Busquets S, et al:
DNA fragmentation occurs in skeletal muscle during tumor growth: A
link with cancer cachexia? Biochem Biophys Res Commun. 270:533–537.
2000.PubMed/NCBI
|
|
6
|
Lee SJ and McPherron AC: Myostatin and the
control of skeletal muscle mass. Curr Opin Genet Dev. 9:604–607.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma M, Langley B, Bass J and Kambadur
R: Myostatin in muscle growth and repair. Exerc Sport Sci Rev.
29:155–158. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsuchida K: Targeting myostatin for
therapies against muscle-wasting disorders. Curr Opin Drug Discov
Devel. 11:487–494. 2008.PubMed/NCBI
|
|
9
|
McPherron AC, Lawler AM and Lee SJ:
Regulation of skeletal muscle mass in mice by a new TGF-beta
superfamily member. Nature. 387:83–90. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Elkasrawy MN and Hamrick MW: Myostatin
(GDF-8) as a key factor linking muscle mass and bone structure. J
Musculoskelet Neuronal Interact. 10:56–63. 2010.PubMed/NCBI
|
|
11
|
Lee SJ: Sprinting without myostatin: a
genetic determinant of athletic prowess. Trends Genet. 23:475–477.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee SJ: Regulation of muscle mass by
myostatin. Annu Rev Cell Dev Biol. 20:61–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anderson SB, Goldberg AL and Whitman M:
Identification of a novel pool of extracellular pro-myostatin in
skeletal muscle. J Biol Chem. 283:7027–7035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hill JJ, Davies MV, Pearson AA, et al: The
myostatin propeptide and the follistatin-related gene are
inhibitory binding proteins of myostatin in normal serum. J Biol
Chem. 277:40735–40741. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo T, Jou W, Chanturiya T, Portas J,
Gavrilova O and McPherron AC: Myostatin inhibition in muscle, but
not adipose tissue, decreases fat mass and improves insulin
sensitivity. PLoS One. 4:e49372009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wolfman NM, McPherron AC, Pappano WN, et
al: Activation of latent myostatin by the BMP-1/tolloid family of
metalloproteinases. Proc Natl Acad Sci USA. 100:15842–15846. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SJ: Genetic analysis of the role of
proteolysis in the activation of latent myostatin. PLoS One.
3:e16282008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
De Caestecker M: The transforming growth
factor-beta superfamily of receptors. Cytokine Growth Factor Rev.
15:1–11. 2004.
|
|
19
|
Rodino-Klapac LR, Haidet AM, Kota J, Handy
C, Kaspar BK and Mendell JR: Inhibition of myostatin with emphasis
on follistatin as a therapy for muscle disease. Muscle Nerve.
39:283–296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Allendorph GP, Vale WW and Choe S:
Structure of the ternary signaling complex of a TGF-beta
superfamily member. Proc Natl Acad Sci USA. 103:7643–7648. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Steelman CA, Recknor JC, Nettleton D and
Reecy JM: Transcriptional profiling of myostatin-knockout mice
implicates Wnt signaling in postnatal skeletal muscle growth and
hypertrophy. FASEB J. 20:580–582. 2006.PubMed/NCBI
|
|
22
|
Joulia-Ekaza D and Cabello G: The
myostatin gene: physiology and pharmacological relevance. Curr Opin
Pharmacol. 7:310–315. 2007. View Article : Google Scholar
|
|
23
|
Whittemore LA, Song K, Li X, et al:
Inhibition of myostatin in adult mice increases skeletal muscle
mass and strength. Biochem Biophys Res Commun. 300:965–971. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Durieux AC, Amirouche A, Banzet S, et al:
Ectopic expression of myostatin induces atrophy of adult skeletal
muscle by decreasing muscle gene expression. Endocrinology.
148:3140–3147. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McFarlane C, Plummer E, Thomas M, et al:
Myostatin induces cachexia by activating the ubiquitin proteolytic
system through an NF-κB-independent, FoxO1-dependent mechanism. J
Cell Physiol. 209:501–514. 2006.PubMed/NCBI
|
|
26
|
Stock MJ and Rothwell NJ: Effects of
β-adrenergic agonists on metabolism and body composition. Control
and manipulation of Animal Growth. Buttery PJ, Hayes NB and Lindsay
DB: Butterworths; London: pp. 249–257. 1985
|
|
27
|
Kim YS and Sainz RD: Beta-adrenergic
agonists and hypertrophy of skeletal muscles. Life Sci. 50:397–407.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Agbenyega ET and Wareham AC: Effect of
clenbuterol on skeletal muscle atrophy in mice induced by the
glucocorticoid dexamethasone. Comp Biochem Physiol Comp Physiol.
102:141–145. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rajab P, Fox J, Riaz S, Tomlinson D, Ball
D and Greenhaff PL: Skeletal muscle myosin heavy chain isoforms and
energy metabolism after clenbuterol treatment in the rat. Am J
Physiol Regul Integr Comp Physiol. 279:R1076–R1081. 2000.PubMed/NCBI
|
|
30
|
Hinkle RT, Hodge KM, Cody DB, Sheldon RJ,
Kobilka BK and Isfort RJ: Skeletal muscle hypertrophy and
anti-atrophy effects of clenbuterol are mediated by the
beta2-adrenergic receptor. Muscle Nerve. 25:729–734. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wineski LE, von Deutsch DA, Abukhalaf IK,
Pitts SA, Potter DE and Paulsen DF: Muscle-specific effects of
hindlimb suspension and clenbuterol in mature male rats. Cells
Tissues Organs. 171:188–198. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Busquets S, Figueras MT, Fuster G, et al:
Anticachectic effects of formoterol: a drug for potential treatment
of muscle wasting. Cancer Res. 64:6725–6731. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tessitore L, Costelli P, Bonetti G and
Baccino FM: Cancer cachexia, malnutrition, and tissue protein
turnover in experimental animals. Arch Biochem Biophys. 306:52–58.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chomczynski P and Sacchi N: Single-step
method of RNA isolation by acid guanidinium
thiocyanate-phenol-chloroform extraction. Anal Biochem.
162:156–159. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gonzalez-Cadavid NF, Taylor WE, Yarasheski
K, et al: Organization of the human myostatin gene and expression
in healthy men and HIV-infected men with muscle wasting. Proc Natl
Acad Sci USA. 95:14938–14943. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Costelli P, Muscaritoli M, Bonetto A, et
al: Muscle myostatin signalling is enhanced in experimental cancer
cachexia. Eur J Clin Invest. 38:531–538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Joulia-Ekaza D and Cabello G: Myostatin
regulation of muscle development: molecular basis, natural
mutations, physiopathological aspects. Exp Cell Res. 312:2401–2414.
2006. View Article : Google Scholar
|
|
38
|
Kollias HD and McDermott JC: Transforming
growth factor-beta and myostatin signaling in skeletal muscle. J
Appl Physiol. 104:579–587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shimasaki S, Koga M, Esch F, et al:
Primary structure of the human follistatin precursor and its
genomic organization. Proc Natl Acad Sci USA. 85:4218–4222. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Aoki MS, Soares AG, Miyabara EH, Baptista
IL and Moriscot AS: Expression of genes related to myostatin
signaling during rat skeletal muscle longitudinal growth. Muscle
Nerve. 40:992–999. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ouchi N, Oshima Y, Ohashi K, et al:
Follistatin-like 1, a secreted muscle protein, promotes endothelial
cell function and revascularization in ischemic tissue through a
nitric-oxide synthase-dependent mechanism. J Biol Chem.
283:32802–32811. 2008. View Article : Google Scholar
|
|
42
|
Cowling BS, McGrath MJ, Nguyen MA, et al:
Identification of FHL1 as a regulator of skeletal muscle mass:
implications for human myopathy. J Cell Biol. 183:1033–1048. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zamora E, Lupon J, Simo R and Galan A:
Myostatin serum levels in heart failure. Eur J Heart Fail.
12:1379author reply 1379–1380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Llovera M, Carbo N, Lopez-Soriano J, et
al: Different cytokines modulate ubiquitin gene expression in rat
skeletal muscle. Cancer Lett. 133:83–87. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Llovera M, Garcia-Martinez C, Agell N, et
al: Ubiquitin and proteasome gene expression is increased in
skeletal muscle of slim AIDS patients. Int J Mol Med. 2:69–73.
1998.PubMed/NCBI
|
|
46
|
Llovera M, Garcia-Martinez C,
Lopez-Soriano J, et al: Role of TNF receptor 1 in protein turnover
during cancer cachexia using gene knockout mice. Mol Cell
Endocrinol. 142:183–189. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Llovera M, Garcia-Martinez C, Agell N,
Lopez-Soriano FJ and Argiles JM: Muscle wasting associated with
cancer cachexia is linked to an important activation of the
ATP-dependent ubiquitin-mediated proteolysis. Int J Cancer.
61:138–141. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Garcia-Martinez C, Llovera M, Agell N,
Lopez-Soriano FJ and Argiles JM: Ubiquitin gene expression in
skeletal muscle is increased by tumour necrosis factor-alpha.
Biochem Biophys Res Commun. 201:682–686. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Llovera M, Garcia-Martinez C, Agell N,
Marzabal M, Lopez-Soriano FJ and Argiles JM: Ubiquitin gene
expression is increased in skeletal muscle of tumour-bearing rats.
FEBS Lett. 338:311–318. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schneyer A, Tortoriello D, Sidis Y,
Keutmann H, Matsuzaki T and Holmes W: Follistatin-related protein
(FSRP): a new member of the follistatin gene family. Mol Cell
Endocrinol. 180:33–38. 2001. View Article : Google Scholar : PubMed/NCBI
|