1. Introduction
Uterine leiomyomas, termed uterine fibroids, are
benign smooth muscle tumors, which are enriched in the
extracellular matrix (ECM). They are associated with the highest
morbidity rate in female reproductive tract tumors (1), and females currently tend to present
with larger leiomyomas and are younger at diagnosis (2). Myomas are the primary indication for
performance of a hysterectomy, accounting for >200,000
hysterectomies annually in the USA (1). The most common symptoms of leiomyomas
are heavy bleeding and pelvic pain, which are associated with
infertility and adverse birth outcomes, including fetal mortality
(3). Heavy menstrual bleeding may
be severe enough to lead to anemia, which requires blood
transfusions. Furthermore, females that are diagnosed with uterine
leiomyomas account for >2.5-fold higher healthcare expenses
compared with females without a leiomyoma diagnosis and additional
work disability costs (2). Notably,
fibroids are the leading indication for performance of a
hysterectomy in the USA, however, little is known regarding their
etiology or pathogenesis despite their particularly high prevalence
and serious impact on the lives of females.
Tumorigenesis is a multistep process, which is
considered to be analogous with Darwinian evolution, whereby
genetic changes result in a growth advantage in a subset of cells
and their subsequent progression from a normal to a malignant state
(4). However, a tumor mass is not
defined by tumor cells alone, it is defined as a tissue in which a
tumor microenvironment (TME) prevails. Thus, in the past decade,
the TME and its constituent ‘stromal’ cells have collectively
gained prominence, and are currently being widely investigated
(5,6). Previously, tumor-associated
fibroblasts (TAF) were presumed to be passive structural elements,
however, currently there is a growing awareness that TAF and the
complex cellular TME may be involved in early tumor development
(4,7,8). The
phenomenon of tumor-associated desmoplasia, characterized by
enhanced fibroblast accumulation and a modified, collagenized ECM,
has been comprehensively reviewed in tumors exhibiting a marked
desmoplastic reaction, including tumors of the pancreas, breast and
gastrointestinal tract (7,8).
Myomas are firm, circumscribed masses. They possess
a smooth muscle component and a significant ECM, which principally
consists of fibroblasts, often termed myofibroblasts, which
predominantly produce collagens type I and III (9). Myomas have been found to mimic the
fibrotic process, and have been shown to specifically upregulate
collagen types I and III (9–11). In
addition, it has been proposed that the pathogenesis of myomas is
comparable to an injury response (analogous to keloid development)
following surgery (12).
Mladenović-Mihailović et al (13) investigated the immunocytochemical
characteristics of smooth muscle cells (SMCs) and connective tissue
components of uterine submucosal myomas. They were found to consist
of SMCs of the highly differentiated contractile and proliferate
phenotypes [α-smooth muscle actin (SMA)-, desmin- and proliferating
cell nuclear antigen-immunoreactivity], as well as connective
tissue as a result of the synthetic activity of the fibroblasts.
The two components markedly differ in their immunocytochemical
characteristics from SMCs of the synthetic phenotype (13). Furthermore, Moore et al
(9) revealed that human uterine
leiomyoma-derived fibroblasts stimulate uterine leiomyoma cell
proliferation and collagen type I production, as well as activate
receptor tyrosine kinases and transforming growth factor
(TGF)-β-receptor signaling in co-cultures. These findings indicate
the importance of the interactions between fibroid tumor cells and
ECM fibroblasts in vivo, as well as the role of growth
factors and ECM proteins in the pathogenesis of uterine fibroids.
Thus, it may be hypothesized that carcinoma-associated fibroblasts
are important in the pathogenesis of myomas. The present review
focuses predominantly on the overall activation of TAFs in the
tumorigenesis of uterine fibroids.
TAFs
Fibroblasts are the predominant source of the ECM in
normal and tumor tissues, and are capable of producing several
ECM-modulating factors (8). Due to
the abundance of ECM often observed in fibroids, Moore et al
(9) concluded that the interactions
between leiomyoma SMCs and fibroblasts are important for the growth
of such tumors as a result of their impact on the production of
growth factors and ECM proteins. Tumor-associated ECM is an
aberrant and complex meshwork of collagens, fibrillar glycoproteins
and proteoglycans that determine abnormal tumor architecture.
Furthermore, perturbations in the production, deposition and
degradation of matrix components have been observed in numerous
human tumors, including leiomyomas (9).
Quiescent fibroblasts, an arrested phenotype of TAF,
are unable to promote the desmoplastic reaction of tumors during
wound healing, tissue repair and scar-like pathogenesis unless they
are activated or have differentiated into myofibroblasts. The
present review focuses on the fibroblast activation pathway.
Activated fibroblasts and myofibroblast cells that exhibit the
appearance of fibroblasts, but express myocyte markers, including
the unique marker fibroblast activation protein and α-SMA (the most
reliable markers for the maturation of fibrocytes) are critical in
the genesis of uterine tumor fibrosis during genital tract
inflammation (4,14). One consistent phenotype of TAF, the
myofibroblast, exhibits a muscle-like morphology and marked
microfilamentous apparatus, resulting in a contractile profile.
Once the fibroblasts are activated, TGF-β promotes mitogenesis and
upregulates the synthesis of numerous components of ECM, leading to
fibrosis.
2. TGF-β stimulate stromal fibroblasts
TGF-β is a multifunctional cytokine, which is
important in embryonic development, and the regulation of repair
and regeneration processes following tissue injury (15). This large superfamily of soluble
factors includes three isoforms, TGF-β1, -β2 and -β3, which are
encoded by three separate genes, but bind to the same high affinity
receptor (16,17). Powell et al (18) reported that TGF-β1 is the isoform
that is commonly upregulated in the presence of a tissue injury. It
is secreted in a latent form following cleavage from a large
pro-molecule. It binds non-covalently to the membrane-associated
latency-associated peptide, which is formed from the cleavage
fragments of the TGF-β1 precursor. This latent TGF-β1 is then
stored on the cell surface or in the ECM, awaiting the conversion
to active TGF-β1, via an unknown mechanism (19).
Feghali et al (16) reported that TGF-β is primarily
produced by active T cells, platelets and monocytes in an
anti-infection immunity milieu. At the site of injury, TGF-β, which
is stored in platelets is released upon degranulation. Sarkar et
al (20) also demonstrated that
T cells, however, not tumor cells are a critical source of TGF-β1,
which inhibits antitumor T cell responses and, thus, fosters tumor
growth, which promotes tumor development. However, which cells are
actually responsible for the chronicity of inflammation remains
unclear. Immune cells may be activated by an unknown primary
antigen or by the products of surrounding non-immune or mesenchymal
cells activated by immune cells or self-derived cytokines. It is
known that TGF-β attracts monocytes and other leukocytes to the
inflammation site, thus participating in the initial step of
chronic inflammation. Recently, a seventh hallmark,
cancer-associated inflammation, was proposed by Colotta et
al (21), two years following
the hypothesis proposed by Wegienka et al (2) that leiomyomas are caused in part by a
systemic immune milieu that is chronically inflammatory (22). Inflammation may be problematic if it
is not well regulated, and thus a proper treatment for inflammation
would substantially reduce the mortality and the therapy costs
associated with these tumors.
The theory that injury or reproductive tract
infections may trigger fibroid development was introduced many
decades ago (22), however, it has
not been adequately analyzed. In addition, Laughlin et al
(23) indicated that certain
pathogens do not remain latent in fibroid tissue and hypothesized
that they may exhibit an acute ‘hit and run’ effect on tumor
initiation or tumor growth, whereby having infected the tissue
once, they may induce macrophage activity and immunocyte lethality
(24). Innate immune responses
initiate an anti-inflammatory process, starting with the
recognition of mucopeptides and the activation of alternative
complement pathways. Certain types of protein in the cell wall
stimulate CD4+ T cells and produce large quantities of
cytokines, including TGF-β. As a result of immunogenic variation
and other forms of immune invasion, innate and adaptive immunity
may fail to clear pathogens, which contribute to chronic
inflammation and subsequent persistent and repeated infections.
The extracellular concentration of active TGF-β is
primarily regulated by the conversion of latent TGF-β to active
TGF-β. However, numerous studies have overlooked the activation
process, possibly due to the complex biological nature of TGF-β.
Mammalian TGF-β is secreted in a latent form that is composed of
three proteins derived from two genes. One of the genes encodes for
TGF-β and latency-associated peptide (LAP) (25). The mechanism of latent TGF-β
activation is a topic of intense investigation and various details
require investigation. Latent TGF-β binding protein is primarily
involved in TGF-β localization by interacting with the local matrix
during activation, whereby TGF-β is liberated from LAP and becomes
activated (25). As soon as the
repair is complete, TGF-β and ECM production is subsequently shut
down by an unknown mechanism. The two functions are critical for
maintaining homeostasis (15).
Mechanism of fibroblast activation
TGF-β appears to be the most important cytokine that
activates the fibroblasts (25,26).
Recently, it has been demonstrated that the activation of the
myofibroblast requires the presence of matrix molecules, in
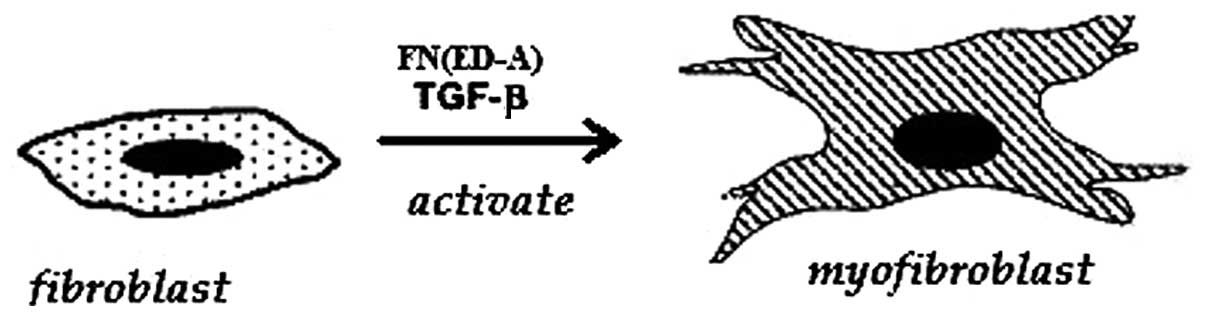
particular, the ED-A (EIIIA) domain of fibronectin. Tissue injury
results in the production of this specific ED-A domain splice
variant of fibronectin. ED-A is the binding site for cell membranes
and for other matrix molecules. Furthermore, it has been shown, in
skin granulation tissue and hepatic models, that the fibronectin
ED-A domain is necessary for TGF-β to trigger α-SMA expression and
collagen secretion in the stellate transformation of myofibroblasts
(18)(Fig. 1).
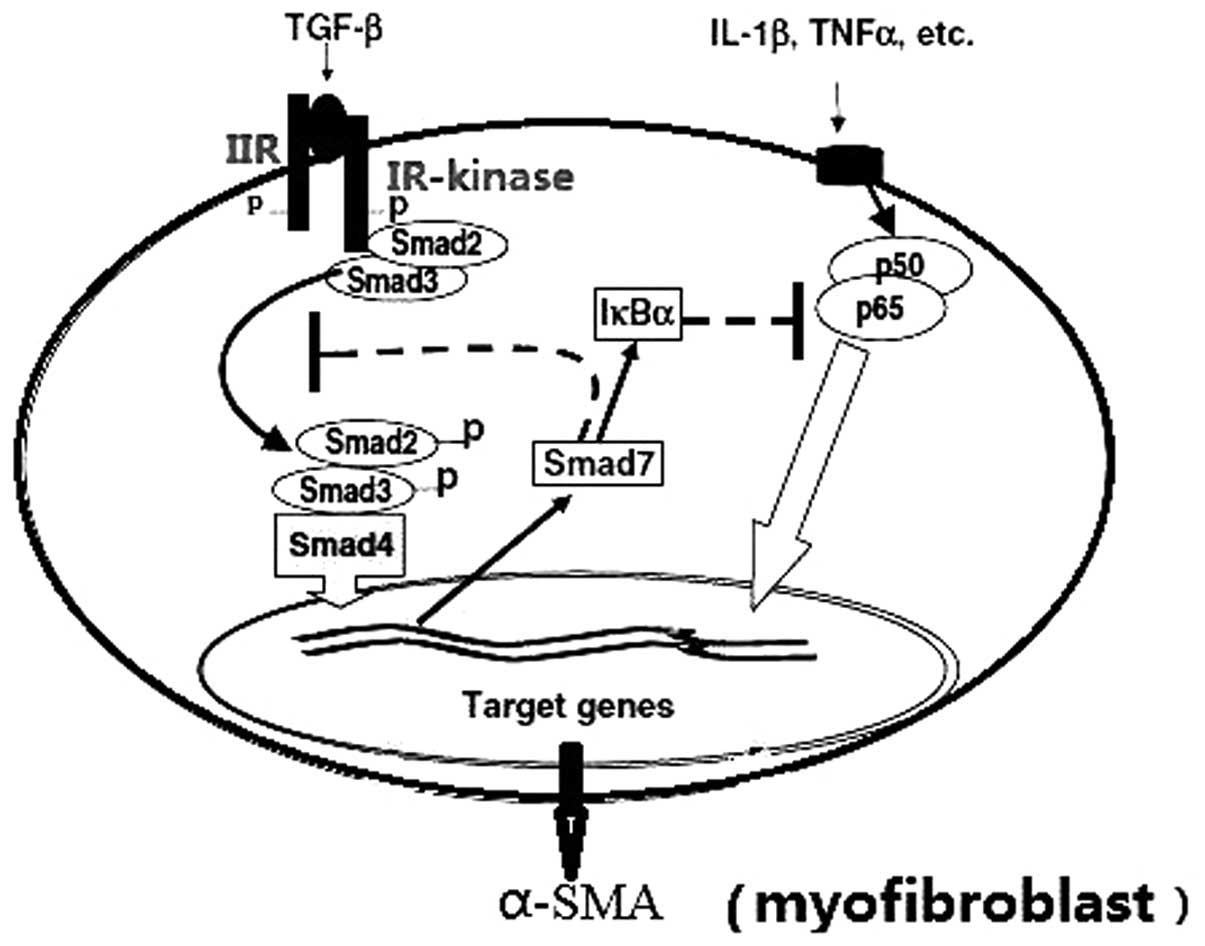
Briefly, TGF-β initiates the cellular response by
binding to its distinct TGF-β II receptor. The ligand binding
cascade activates the TGF-β-RI kinase, which phosphorylates the
receptor regulated Smads (R-Smads). The activated R-Smads form
oligomeric complexes with the common Smad (Co-Smad; Fig. 2).
The oligomeric complexes then translocate into the
nucleus, where they regulate the transcription of target genes by
binding to DNA directly or indirectly via interaction with various
cofactors (Fig. 2). TGF-β may also
stimulate inhibitory Smads, which negatively regulate TGF-β
signaling transduction (27).
R-Smads, including Smad2, -3 and the Co-Smad
(Smad4), contain conserved amino- and carboxyl-terminal
mad-homologies (MH) 1 and 2, respectively, which flank a more
divergent middle linker region (26,27).
The MH1 domain is the functional unit that binds DNA directly to
regulate gene transcription, whereas the MH2 domain contains the
SSXS phosphorylation site (Ser-465/Ser-467), which is typically
phosphorylated by the TGF-β receptor I serine kinase (28). TGF-β induced accumulation of ECM
predominantly occurs via the Smad3-associated downregulation of
matrix metalloproteinase-1 and positive regulation of tissue
inhibitor of matrix metalloproteinases-1. Smad3 binds directly to
DNA, whereas Smad2 binds to coactivators or repressors to regulate
its target gene activities. As a result of Smad signals that
promote the expression α-SMA, the fibroblasts are activated and
differentiated.
3. Mechanical forces activate
fibroblasts
A study by Petersen et al (29) presented a novel insight with regards
to the effects of mechanical loading on the production and
remodeling of ECM components, as well as the impact of the altered
mechanical cell environment on these processes. The theory of
cellular mechanotransduction has been proposed in recent years,
which indicates that mechanical and chemical signals may interact
to control cell growth, differentiation, movement and death. Ingber
(30) reported that cytoskeletal
tension affects the integrity of the shape and function of cells,
analogous to the tent model (31).
The association between cell mechanics and biochemistry is
dependent on integrins, discrete focal adhesions, ECM substrates
and the cytoskeleton; therefore, controlling cell shape is
important in managing the structural and informational complexity
of living cells.
Connective tissues do not passively bear the stress
resulting from gravity, compression and muscle-generated forces.
They interplay with these factors dynamically by modifying their
composition and mechanical properties. At the cellular level,
mechanical signals influence cell morphology, cytoskeletal
reorganization, cell survival, cell differentiation and gene
expression (32). Similarly, cells
contain a set of specific structures, the cytoskeleton, which is
capable of generating forces and bearing elastic deformation
(33).
Mechanical forces include fluid flow, direct
compression and tensile stress. They are essential regulators of
tissue homeostasis and are essential for the correct functioning of
connective tissues, since these are subjected to the greatest
levels of stress in an organism (34). All adherent cells, including
endothelial cells, fibroblasts and myofibroblasts sense tension,
which originates from the environment. Tension is transmitted via
cell-ECM contact, which leads to the reorganization of the
cytoskeleton and the elicitation of specific signals that modulate
gene expression. Cells are continuously recognizing alterations in
mechanical forces and their functions are adapted according to the
biological requirements. When mechanical tension is removed
(30), tissues undergo atrophy,
which demonstrates the importance of mechanical signals in
maintaining the proper functioning of the organism. Malik et
al (36) investigated the
altered mechanical homeostasis in uterine leiomyomas, which had
been exposed to increased mechanical stress. Structural and
biochemical features were observed to be consistent with the
activation of solid-state signaling. Thus, stress may be a
contributing factor to leiomyoma growth.
As previously stated, cells firmly attach to ECM
structures via matrix adhesions. These include focal complexes, and
focal and fibrillar adhesions. The major structures that are
required to form such matrix contacts are the integrin receptors,
which directly connect the ECM structures to the intracellular
cytoskeleton network (36).
Mechanical forces act on focal adhesions, resulting in further
structural maturation. The mechanisms by which fibroblasts transmit
mechanical signals remain unclear, however, they may involve
stretch-activated ion channels, direct interactions between
structural and signaling components or the activation of small
guanosine triphosphatases (GTPases).
As previously described, numerous cooperative
interactions exist between integrins and growth factor signaling.
Specifically, fibroblast to myofibroblast conversion and α-SMA
expression depend on a combination of mechanical tension and TGF-β
activity. Thus, in scarring, the generated tensions may induce
myofibroblast formation, resulting in a self-perpetuating loop
(37). A similar autocrine loop is
observed in the induction of collagen synthesis in fibroblasts by
mechanical tension, whereby TGF-β is induced by tension, which in
turn activates collagen synthesis via the usual signaling
pathways.
The formation of stress fibers and the
neo-expression of α-SMA is a hallmark of fibroblast to
myofibroblast differentiation. This change is a significant event
in the development of fibro-contractive diseases and in wound
granulation tissue contraction. The incorporation of the SMA
isoform into stress fibers confers a high contractile activity to
myofibroblasts. This is subsequently transmitted to the ECM at
sites of specialized adhesions, termed ‘fibronexus’ in tissue and
‘supermature focal adhesions’ in two-dimensional cell cultures
(38). In addition, Hinz (39) proposed that myofibroblast
differentiation requires a mechanically restrained environment in
conjunction with the action of growth factors (TGF-β) and
specialized matrix molecules (ED-A splice variant of fibronectin).
Myofibroblast adhesions sense matrix stress and transmit
contractile force to the extracellular environment, in addition to
producing the high intracellular tension that is required for
myofibroblast development (39).
This clearly demonstrates that mechanical tension,
which is generated during wound contraction or scar formation, may
modulate the gene expression of fibroblasts and myofibroblasts
embedded into this tissue at different molecular levels. Tension
directly modifies gene transcription via the induction of integrin
signaling, which affects small GTPases or induces/inhibits growth
factor signaling, which subsequently indirectly affects ECM protein
synthesis in the fibroblasts/myofibroblasts (36). Via a combination of these
mechanisms, mechanical tension induces an activated, contractile
fibroblast phenotype, which is characterized by high levels of ECM
protein synthesis and fibrogenic cytokine production, as well as
low protease activity.
Signaling mechanisms potentially involved
in the regulation of actin genes by mechanical stress
Previous data (36–40)
indicates that mechanical signals specifically regulate the
synthesis and degradation of various ECM components. The forces
exerted by the cells themselves are generated by the cytoskeleton
and are measurable. In electrically excitable cells,
stretch-sensitive cation channels are important for sensing strain
(41). Therefore, it is likely that
in connective tissue cells, such as fibroblasts, cell-matrix
adhesions are the functional strain gauges that sense the
mechanical properties of the ECM as well as the environmental
changes. Focal adhesions, evolving from focal complexes (small
dot-like adhesion sites), undergo further structural maturation
depending on externally applied or cytoskeletal forces (33). Furthermore, integrin activation
triggers intracellular signaling events. Mechanical stress applied
directly to integrin ligands elicits chemical responses inside the
cell cascade, including the assembly and growth of focal
contacts.
The earliest responses to mechanical stimulation are
recorded at the cell-ECM adhesion level. These include the opening
of stretch-activated ion channels, release of soluble mediators,
phosphorylation of focal adhesion-associated kinases (for example,
focal adhesion kinase, Src and integrin-linked kinase), activation
of small GTPases (including RhoA), increased phosphatidyl inositol
metabolism and generation of reactive oxygen species (42,43).
Multiple intracellular signaling pathways are subsequently
triggered, including those involving mitogen-activated protein
kinase (MAPK), protein kinase C and nuclear factor κB (44). Overall, the cascades lead to the
regulation of the target gene of α-SMA at the gene transcription
level. There are at least three regulatory mechanisms that use the
abovementioned pathways, subsequently leading to the activation of
fibroblasts (Fig. 3).
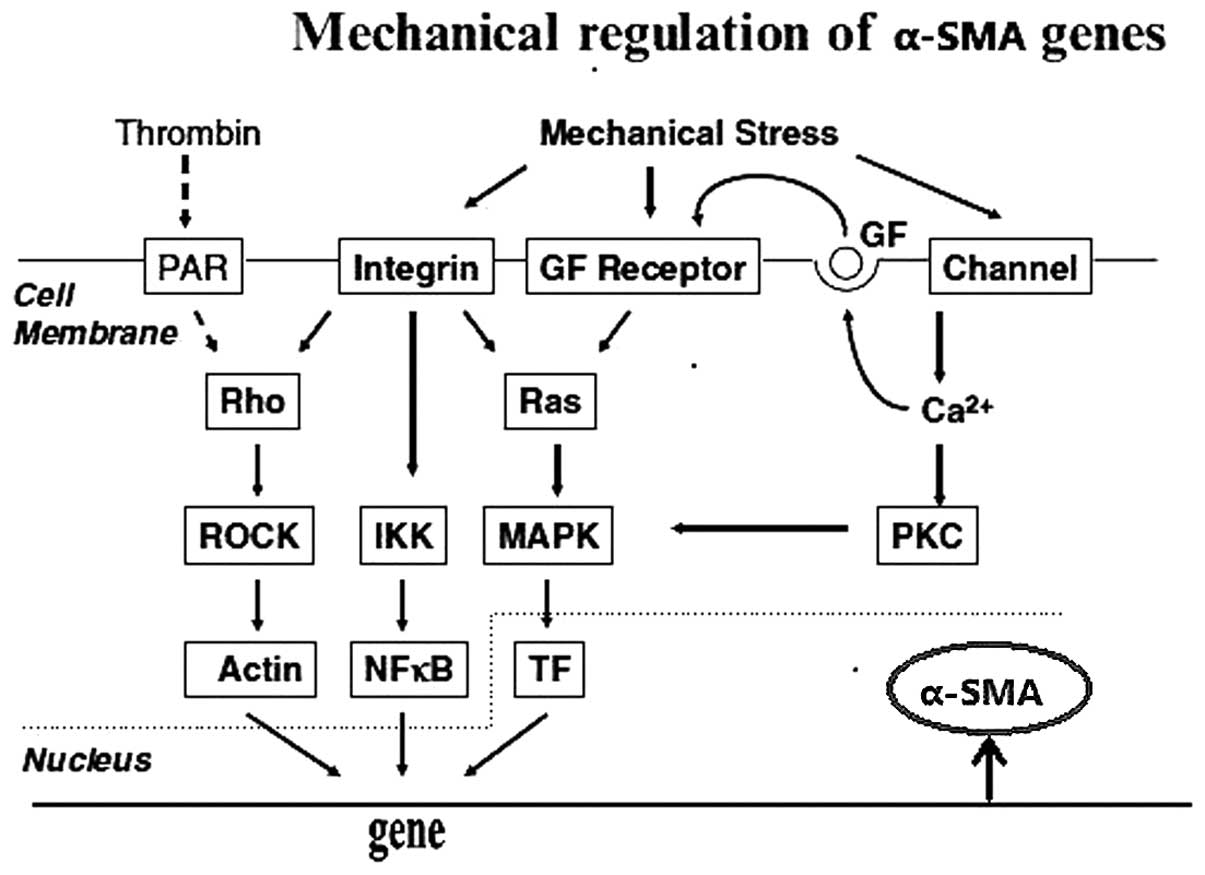 | Figure 3Mechanical forces activate
fibroblasts. Intergrins and stretch-activated ion channels act as
receptors and mechanical forces activate the Rho family, IKK,
mitogen-activated protein kinase p38 and PKC via downstream
elements, which regulates α-SMA. Dotted lines represent inhibitory
interactions (31). α-SMA, α-smooth muscle actin; PAR, protease
activated receptor; GF, growth factor; ROCK, Rho kinase; IKK, IκB
kinase; MAPK, mitogen-activated protein kinase; NF-κβ, nuclear
factor-κβ; TF, transcription factor; PKC, protein kinase C. |
4. Hypoxia
Kawaguchi et al (45) demonstrated that myocardial
ischemia/reperfusion injury triggers the activation of the
inflammasome in fibroblasts. These observations revealed that
chronic and sustained hypoxia, loss of stromal fibroblast,
caveolin-1 (as a biomarker for chronic hypoxia), oxidative stress
and autophagy (46) induce a
proinflammatory and profibrotic microenvironment in rat pulmonary
arteries (47). In addition,
hypoxia-induced proteomic changes in neoplastic and stromal cells
influence tumor propagation (44).
Hypoxia-mediated malignant progression has been debated as a
leading factor that leads to multidrug resistance. In previous
animal models (41), the earliest
and most evident structural changes following hypoxic exposure were
identified in the adventitial compartment of the vascular walls.
Furthermore, resident adventitial fibroblasts have been shown to
exhibit early and sustained increases in proliferation that exceed
those observed in endothelial or SMCs.
Hypoxia-induced proliferation is
dependent on MAPKs
The increased expression of α-SMA-positive cells
(myofibroblasts) has also been observed in neonatal calves
following acute hypoxic exposure (48). Hypoxia has been reported to activate
MAPK signaling pathways in numerous cell types, although very few
of those cells demonstrate a proliferative response under hypoxic
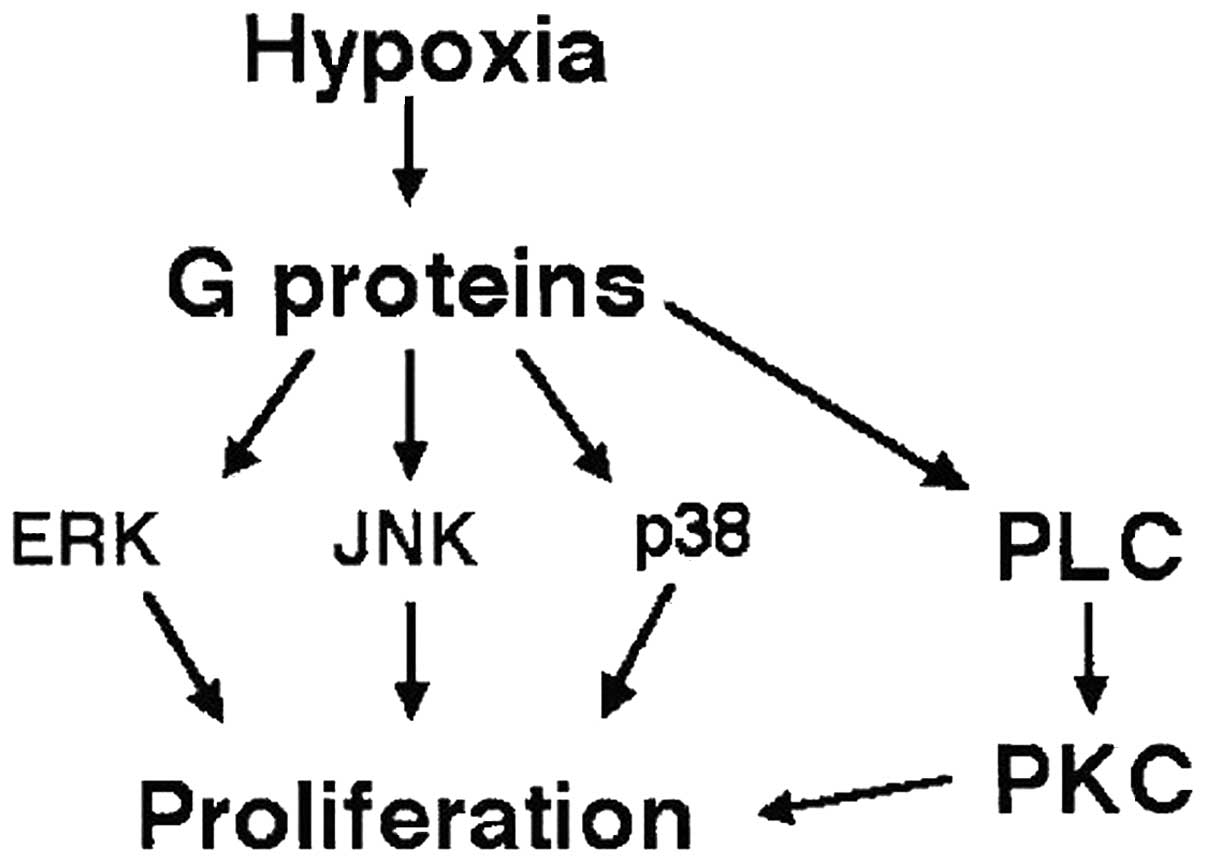
conditions. In fibroblasts, a hypoxia-induced transient activation
of extracellular signal-regulated kinases 1/2 and c-Jun N-terminal
kinase and a biphasic activation of p38 MAPK was observed (Fig. 4).
Activation of fibroblasts induced by
hypoxia
Based on observations demonstrating that stimuli,
including sheer stress, pH and osmolality, may activate Gi-proteins
with subsequent activation of MAPK signaling, Gerasimovskaya et
al (49) proposed that hypoxia
in the absence of exogenous ligands directly activates
Gi/o-mediated signaling. In addition, hypoxia itself may act as a
growth-promoting stimulus for bovine neonatal adventitial
fibroblasts via Gi/o (and possibly Gq)-mediated activation of a
complex network of MAPKs (48).
Furthermore, hypoxia has been shown to activate
G-protein-coupled receptor signaling pathways. Stenmark et
al (48) hypothesized that
hypoxia may act as a stimulus for the induction of the
differentiation of fibroblasts into myofibroblasts. Hypoxia was
found to induce a marked increase in α-actin protein in fibroblast
subpopulations of neonatal bovine PA adventitial fibroblasts
(50). To investigate the
underlying molecular mechanisms, fibroblasts were transiently
transfected with a luciferase-tagged α-SMA promoter and
subsequently exposed to hypoxia. Hypoxia induced an increase in
α-SMA promoter activity and this induction of α-SMA promoter
activity was observed to be largely independent of TGF-β activity.
Thus, these results indicate that hypoxia-induced α-SMA expression
in fibroblasts is mediated by Gi-proteins (Fig. 4).
Synergy between hypoxia and adenosine
receptors
Chronic inflammatory diseases are commonly
associated with hypoxia, which has been shown to be a powerful
stimulus for gene expression and cell differentiation (51,52).
Hypoxia and the activation of A2B adenosine receptors
act synergistically to promote the release of interleukin (IL)-6.
Zhong et al (54)
demonstrated that the activation of A2B adenosine
receptors increased the release of IL-6. This proinflammatory
cytokine, which mediates inflammation, is exhibited at elevated
concentrations in the lung of individuals with asthma and induces
the differentiation of human lung fibroblasts into myofibroblasts.
The induction of α-SMA expression (by adenosine) is an essential
feature during this process (53).
At present, the cellular source of the adenosine is unknown. Under
hypoxia, the effect of adenosine on α-SMA expression is not
completely blocked by anti-IL-6. There may be additional factors,
including platelet-activating factor and platelet-derived growth
factor, which also contribute to the synergistic effect of
adenosine and hypoxia on α-SMA. Notably, IL-6 was demonstrated to
inhibit the proliferation of normal fibroblasts and induce
proliferation of idiopathic pulmonary fibrosis fibroblasts
(54).
The initial evidence regarding the critical effect
of hypoxia on TAFs (55) may lead
to further investigation into the regulatory mechanisms, which are
relevant to fibroblasts in an oxygen-deficient (tumor)
micro-milieu, in order to establish novel fibroblast-based
therapeutic designs.
5. Inactivation of fibroblasts
Novel role of thrombospondin-1
(TSP-1)
Wu et al (56) demonstrated that a downregulation of
TSP-1 during cervical carcinogenesis was accompanied by the
upregulation of stromal markers, α-SMA and desmin. The transfection
of NIH/3T3 cells with TSP-1 and purified TSP-1 did not alter the
protein levels of α-SMA and desmin, however, significantly
inhibited matrix metalloprotease-2 activity.
TSP-1 expression was higher in the tumors or
tumor-associated stroma when compared with the expression in normal
epithelial (57). TSP-1 inhibited
fibroblast invasion regardless of the presence of TGF-β, however, a
higher dose of TSP-1 was required for the complete inhibition of
TGF-β-treated NIH/3T3 cells. The complexity and duality of the
functions of TSP-1 and TGF-β may result from the ability to
suppress tumor cell proliferation at the early stage, whilst
enhancing the host stroma reaction at the later stages (58). Further investigation is required to
elucidate the dynamic interaction that exists between TSP-1 and
TGF-β in the regulation of cervical cancer growth. Notably,
TSP-1-mediated inhibition was only demonstrated in the fibroblasts
with manipulated TSP-1 expression, however, not in the tumor cells.
TSP-1 exerts its effects, including the inhibition of fibroblast
migration, decreasing the recruitment of inflammatory cells,
induction of endothelial cell apoptosis or the activation of SMC
proliferation, in multiple types of stromal cells (59).
The effects of TSP-1 on tumorigenesis differ
markedly from those on stromal cells. This indicates that TSP-1
exerts various biological functions in different cell types. For
example, a switch in angiogenesis phenotype during the transition
from low- to high-grade squamous intraepithelial lesion occurs
partly due to the downregulation of TSP-1 (57). The genetic manipulation of TSP-1
expression levels in cells revealed that TSP-1-mediated inhibition
of stromal reactions is primarily due to the inhibition of
activated fibroblast migration and invasion, rather than a direct
effect on stromal marker expression (60).
Unlike TSP-1, secreted protein, acidic and rich in
cysteine (SPARC), was shown to inhibit fibroblast activation by
blocking α-SMA overexpression (61,62).
Although SPARC and TSP-1 are matricellular proteins, which inhibit
angiogenesis and interfere with ECM organization (63), TSP-1 inhibits stromal reactions via
a mechanism that is distinct from SPARC.
The function of SPARC
SPARC, also termed osteonectin or BM-40, is a
Ca2+-binding matricellular glycoprotein involved in
wound healing, neoplasia and the mediation of cell-matrix
interactions. Chlenski et al (62) reported that in addition to stromal
formation enhancement, SPARC prevented fibroblast activation in 293
xenografts, indicating that the anticancer effects of SPARC may be
due to the formation of tumor stroma, which do not support tumor
growth (56).
Interactions between tumor and inflammatory cells
determine tumor progression or regression via numerous mechanisms,
including stromal formation, angiogenesis, adhesion and cell
migration. A cytokine- and chemokine-rich milieu, together with
other factors contributes to tissue remodeling. Emerging evidence
proposes that SPARC produced by host leukocytes, rather than the
tumor, determines the assembly and function of tumor-associated
stroma via collagen type IV organization (64).
The actin cytoskeleton of animal cells maintains the
cellular shape and is significant in cell motility. Rho and Rac
(two members of the Ras-associated superfamily of small GTPases)
and Cdc42 (another member of the Rho family), regulate the
polymerization of actin to produce stress fibers or lamellipodia,
respectively; the Rho family of small GTPases controls stress fiber
formation. In particular, the activation of the Rho-Rac-Cdc42
signaling pathway results in stress fiber assembly via the
activation of actomyosin contractility and suppression of the
actin-severing activity of cofilin. Overexpression of SPARC in DAOY
medulloblastoma cells inhibits Rho-Rac-Cdc42 GTPase activity and
thus contributes to the inactivation of fibroblasts (65).
6. Conclusion
Uterine fibroids, the benign smooth muscle tumors
originating from the myometrium, are responsible for the incidence
of morbidity in a large number of females. Although their exact
pathogenesis remains unknown, there is substantial evidence, which
indicates that myomas consist of large quantities of uterine
leiomyoma cells and fibroblasts. The present review predominantly
focuses on a novel mechanism of fibroblast activation and its
potential association with uterine fibroids. Thus, such novel
insights may be considered useful for further investigation and
future non-surgical treatment of leiomyomas.
Acknowledgements
This review was supported by the Natural Science
Foundation of China (grant no. 81172461).
References
|
1
|
Walker CL and Stewart EA: Uterine
fibroids: the elephant in the room. Science. 308:1589–1592.
2005.
|
|
2
|
Wegienka G: Are uterine leiomyoma a
consequence of a chronically inflammatory immune system? Med
Hypotheses. 79:226–231. 2012.
|
|
3
|
Ciarmela P, Islam MS, Reis FM, et al:
Growth factors and myometrium: biological effects in uterine
fibroid and possible clinical implications. Hum Reprod Update.
17:772–790. 2011.
|
|
4
|
Räsänen K and Vaheri A: Activation of
fibroblasts in cancer stroma. Exp Cell Res. 316:2713–2722.
2010.
|
|
5
|
Hanahan D and Coussens LM: Accessories to
the crime: functions of cells recruited to the tumor
microenvironment. Cancer Cell. 21:309–322. 2012.
|
|
6
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011.
|
|
7
|
Kunz-Schughart LA and Knuechel R:
Tumor-associated fibroblasts (part I): Active stromal participants
in tumor development and progression? Histol Histopathol.
17:599–621. 2002.
|
|
8
|
Kunz-Schughart LA and Knuechel R:
Tumor-associated fibroblasts (part II): Functional impact on tumor
tissue. Histol Histopathol. 17:623–637. 2002.
|
|
9
|
Moore AB, Yu L, Swartz CD, et al: Human
uterine leiomyoma-derived fibroblasts stimulate uterine leiomyoma
cell proliferation and collagen type I production, and activate
RTKs and TGF beta receptor signaling in coculture. Cell Commun
Signal. 8:102010.
|
|
10
|
Stewart EA, Friedman AJ, Peck K and Nowak
RA: Relative overexpression of collagen type I and collagen type
III messenger ribonucleic acids by uterine leiomyomas during the
proliferative phase of the menstrual cycle. J Clin Endocrinol
Metab. 79:900–906. 1994.
|
|
11
|
Stewart EA: Uterine fibroids. Lancet.
357:293–298. 2001.
|
|
12
|
Flake GP, Andersen J and Dixon D: Etiology
and pathogenesis of uterine leiomyomas. Environ Health Perspect.
111:1037–1054. 2003.
|
|
13
|
Mladenović-Mihailović A,
Mladenović-Bogdanović Z, Mitrović P, et al: Immunocytochemical
characteristics of submucosal uterine myomas. Vojnosanit Pregl.
67:977–982. 2010.(In Serbian).
|
|
14
|
Sugimoto H, Mundel TM, Kieran MW and
Kalluri R: Identification of fibroblast heterogeneity in the tumor
microenvironment. Cancer Biol Ther. 5:1640–1646. 2006.
|
|
15
|
Border WA and Ruoslahti E: Transforming
growth factor-beta in disease: the dark side of tissue repair. J
Clin Invest. 90:1–7. 1992.
|
|
16
|
Feghali CA and Wright TM: Cytokines in
acute and chronic inflammation. Front Biosci. 1:12–26. 1997.
|
|
17
|
Gentle ME, Shi S, Daehn I, et al:
Epithelial cell TGFβ signaling induces acute tubular injury and
interstitial inflammation. J Am Soc Nephrol. 24:787–799. 2013.
|
|
18
|
Powell DW, Mifflin RC, Valentich JD, Crowe
SE, Saada JI and West AB: Myofibroblasts. II. Intestinal
subepithelial myofibroblasts. Am J Physiol. 277:C183–C201.
1999.
|
|
19
|
Powell DW, Mifflin RC, Valentich JD, et
al: Myofibroblasts. I Paracrine cells important in health and
disease. Am J Physiol. 277:C1–C9. 1999.
|
|
20
|
Sarkar A, Donkor MK and Li MO: T cell- but
not tumor cell-produced TGF-β1 promotes the development of
spontaneous mammary cancer. Oncotarget. 2:1339–1351. 2011.
|
|
21
|
Colotta F, Allavena P, Sica A, et al:
Cancer-related inflammation, the seventh hallmark of cancer: links
to genetic instability. Carcinogenesis. 30:1073–1081. 2009.
|
|
22
|
Witherspoon JT and Butler VW: The etiology
of uterine fibroids with special reference to the frequency of
their occurrence in the Negro: a hypothesis. Surg Gynecol Obstet.
58:57–61. 1934.
|
|
23
|
Laughlin SK, Schroeder JC and Baird DD:
New directions in the epidemiology of uterine fibroids. Semin
Reprod Med. 28:204–217. 2010.
|
|
24
|
Grande MT and López-Novoa JM: Fibroblast
activation and myofibroblast generation in obstructive nephropathy.
Nat Rev Nephrol. 5:319–328. 2009.
|
|
25
|
Buscemi L, Ramonet D, Klingberg F, et al:
The single-molecule mechanics of the latent TGF-β1 complex. Curr
Biol. 21:2046–2054. 2011.
|
|
26
|
Le Goff C and Cormier-Daire V: From tall
to short: the role of TGFβ signaling in growth and its disorders.
Am J Med Genet C Semin Med Genet. 160C:145–153. 2012.
|
|
27
|
Wang W, Koka V and Lan HY: Transforming
growth factor-beta and Smad signalling in kidney diseases.
Nephrology (Carlton). 10:48–56. 2005.
|
|
28
|
Wang C, Chen L, Wang L and Wu J: Crystal
structure of the MH2 domain of Drosophila Mad. Sci China C Life
Sci. 52:539–544. 2009.
|
|
29
|
Petersen A, Joly P, Bergmann C, Korus G
and Duda GN: The impact of substrate stiffness and mechanical
loading on fibroblast-induced scaffold remodeling. Tissue Eng Part
A. 18:1804–1817. 2012.
|
|
30
|
Ingber DE: From cellular
mechanotransduction to biologically inspired engineering: 2009
Pritzker Award Lecture, BMES Annual Meeting October 10, 2009. Ann
Biomed Eng. 38:1148–1161. 2010.
|
|
31
|
Ingber DE: Cellular tensegrity: defining
new rules of biological design that govern the cytoskeleton. J Cell
Sci. 104:613–627. 1993.
|
|
32
|
Legant WR, Miller JS, Blakely BL, et al:
Measurement of mechanical tractions exerted by cells in
three-dimensional matrices. Nat Methods. 7:969–971. 2010.
|
|
33
|
Sarasa-Renedo A and Chiquet M: Mechanical
signals regulating extracellular matrix gene expression in
fibroblasts. Scand J Med Sci Sports. 15:223–230. 2005.
|
|
34
|
Carmeliet G, Vico L and Bouillon R: Space
flight: a challenge for normal bone homeostasis. Crit Rev Eukaryot
Gene Expr. 11:131–144. 2001.
|
|
35
|
Rogers R, Norian J, Malik M, et al:
Mechanical homeostasis is altered in uterine leiomyoma. Am J Obstet
Gynecol. 198:e1–e11. 2008.
|
|
36
|
Malik M, Segars J and Catherino WH:
Integrin β1 regulates leiomyoma cytoskeletal integrity and growth.
Matrix Biol. 31:389–397. 2012.
|
|
37
|
Gurtner GC, Dauskardt RH, Wong VW, et al:
Improving cutaneous scar formation by controlling the mechanical
environment: large animal and phase I studies. Ann Surg.
254:217–225. 2011.
|
|
38
|
Hinz B: Masters and servants of the force:
the role of matrix adhesions in myofibroblast force perception and
transmission. Eur J Cell Biol. 85:175–181. 2006.
|
|
39
|
Prager-Khoutorsky M, Lichtenstein A,
Krishnan R, et al: Fibroblast polarization is a
matrix-rigidity-dependent process controlled by focal adhesion
mechanosensing. Nat Cell Biol. 13:1457–1465. 2011.
|
|
40
|
Eckes B, Nischt R and Krieg T: Cell-matrix
interactions in dermal repair and scarring. Fibrogenesis Tissue
Repair. 3:42010.
|
|
41
|
Walker RG, Willingham AT and Zuker CS:
A Drosophila mechanosensory transduction channel. Science.
287:2229–2234. 2000.
|
|
42
|
Sadoshima J and Izumo S: The cellular and
molecular response of cardiac myocytes to mechanical stress. Annu
Rev Physiol. 59:551–571. 1997.
|
|
43
|
Shi-Wen X, Thompson K, Khan K, et al:
Focal adhesion kinase and reactive oxygen species contribute to the
persistent fibrotic phenotype of lesional scleroderma fibroblasts.
Rheumatology. 51:2146–2154. 2012.
|
|
44
|
Nakayama K, Obara K, Tanabe Y, et al:
Interactive role of tyrosine kinase protein kinase C, and Rho/Rho
kinase systems in the mechanotransduction of vascular smooth
muscles. Biorheology. 40:307–314. 2003.
|
|
45
|
Kawaguchi M, Takahashi M, Hata T, Kashima
Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J,
et al: Inflammasome activation of cardiac fibroblasts is essential
for myocardial ischemia/reperfusion injury. Circulation.
123:594–604. 2011.
|
|
46
|
Martinez-Outschoorn UE, Trimmer C, Lin Z,
Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S,
Martinez-Cantarin MP, Capozza F, et al: Autophagy in cancer
associated fibroblasts promotes tumor cell survival: Role of
hypoxia, HIF1 induction and NFκB activation in the tumor stromal
microenvironment. Cell Cycle. 9:3515–3533. 2010.
|
|
47
|
Burke DL, Frid MG, Kunrath CL, Karoor V,
Anwar A, Wagner BD, Strassheim D and Stenmark KR: Sustained hypoxia
promotes the development of a pulmonary artery-specific chronic
inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol.
297:L238–L250. 2009.
|
|
48
|
Stenmark KR, Gerasimovskaya E, Nemenoff RA
and Das M: Hypoxic activation of adventitial fibroblasts: role in
vascular remodeling. Chest. 122(Suppl 6): S326–S334. 2002.
|
|
49
|
Gerasimovskaya EV, Ahmad S, White CW,
Jones PL, et al: Extracellular ATP is an autocrine/paracrine
regulator of hypoxia-induced adventitial fibroblast growth.
Signaling through extracellular signal-regulated kinase-1/2 and the
Egr-1 transcription factor. J Biol Chem. 277:44638–44650. 2002.
|
|
50
|
Short M, Nemenoff RA, Zawada WM, et al:
Hypoxia induces differentiation of pulmonary artery adventitial
fibroblasts into myofibroblasts. Am J Physiol Cell Physiol.
286:C416–C425. 2004.
|
|
51
|
Wright JL: Diseases of the small airways.
Lung. 179:375–396. 2001.
|
|
52
|
Spivak-Kroizman TR, Hostetter G, Posner R,
et al: Hypoxia triggers hedgehog-mediated tumor-stromal
interactions in pancreatic cancer. Cancer Res. 73:3235–3247.
2013.
|
|
53
|
Haskó G, Csóka B, Németh ZH, et al: A(2B)
adenosine receptors in immunity and inflammation. Trends Immunol.
30:263–270. 2009.
|
|
54
|
Zhong H, Belardinelli L, Maa T and Zeng D:
Synergy between A2B adenosine receptors and hypoxia in activating
human lung fibroblasts. Am J Respir Cell Mol Biol. 32:2–8.
2005.
|
|
55
|
Pilch H, Schlenger K, Steiner E, et al:
Hypoxia-stimulated expression of angiogenic growth factors in
cervical cancer cells and cervical cancer-derived fibroblasts. Int
J Gynecol Cancer. 11:137–142. 2001.
|
|
56
|
Wu MP, Young MJ, Tzeng CC, et al: A novel
role of thrombospondin-1 in cervical carcinogenesis: inhibit stroma
reaction by inhibiting activated fibroblasts from invading cancer.
Carcinogenesis. 29:1115–1123. 2008.
|
|
57
|
Bertin N, Clezardin P, Kubiak R and
Frappart L: Thrombospondin-1 and -2 messenger RNA expression in
normal, benign, and neoplastic human breast tissues: correlation
with prognostic factors, tumor angiogenesis, and fibroblastic
desmoplasia. Cancer Res. 57:396–399. 1997.
|
|
58
|
Wu MP, Tzeng CC, Wu LW, Huang KF and Chou
CY: Thrombospondin-1 acts as a fence to inhibit angiogenesis that
occurs during cervical carcinogenesis. Cancer J. 10:27–32.
2004.
|
|
59
|
Kalas W, Yu JL, Milsom C, et al: Oncogenes
and Angiogenesis: down-regulation of thrombospondin-1 in normal
fibroblasts exposed to factors from cancer cells harboring mutant
ras. Cancer Res. 65:8878–8886. 2005.
|
|
60
|
Kuhn C and Mason RJ: Immunolocalization of
SPARC, tenascin, and thrombospondin in pulmonary fibrosis. Am J
Pathol. 147:1759–1769. 1995.
|
|
61
|
Heinemann V, Reni M, Ychou M, et al:
Tumour-stroma interactions in pancreatic ductal adenocarcinoma:
rationale and current evidence for new therapeutic strategies.
Cancer Treat Rev. 40:118–128. 2014.
|
|
62
|
Chlenski A, Guerrero LJ, Yang Q, Tian Y,
Peddinti R, Salwen HR and Cohn SL: SPARC enhances tumor stroma
formation and prevents fibroblast activation. Oncogene.
26:4513–4522. 2007.
|
|
63
|
Mettouchi A, Cabon F, Montreau N, et al:
SPARC and thrombospondin genes are repressed by the c-jun oncogene
in rat embryo fibroblasts. EMBO J. 13:5668–5678. 1994.
|
|
64
|
Sangaletti S, Stoppacciaro A, Guiducci C,
et al: Leukocyte, rather than tumor-produced SPARC, determines
stroma and collagen type IV deposition in mammary carcinoma. J Exp
Med. 198:1475–1485. 2003.
|
|
65
|
Bhoopathi P, Gondi CS, Gujrati M, et al:
SPARC mediates Src-induced disruption of actin cytoskeleton via
inactivation of small GTPases Rho-Rac-Cdc42. Cell Signal.
23:1978–1987. 2011.
|