Introduction
Extraskeletal Ewing’s sarcoma (EES)/primitive
neuroectodermal tumor (PNET) is a rare entity, accounting for 15%
of all Ewing’s sarcomas (1). The
most common clinical manifestation of this disease is a rapidly
growing, painful lump (2).
Pathological, cytogenetic, immunohistochemical and molecular
genetic analysis contribute to an accurate diagnosis (3). Multidisciplinary treatment modalities
comprising extended resection, aggressive chemotherapy and local
irradiation are recommended (4). At
present, EES/PNET is viewed as a potentially curable disease
(5).
Primary mediastinal EES is extremely rare. The
present study describes the case of a middle-aged female with
mediastinal EES who received sequential chemotherapy and
radiotherapy and achieved a marked response. Written informed
consent was obtained from the patient. Additionally, the associated
studies on EES are also reviewed. Further studies are required to
establish the standard treatment strategy for EES. Written informed
consent was obtained from the patient.
Case report
On December 25, 2012, a 51-year-old female presented
with intermittent chest pain that had been apparent for one year.
The physical examination was unremarkable. The patient’s
performance status was 1 according to an Eastern Cooperative
Oncology Group (ECOG) evaluation (6).
Laboratory investigations revealed a normal complete
blood cell count, coagulation routine and serum biochemical
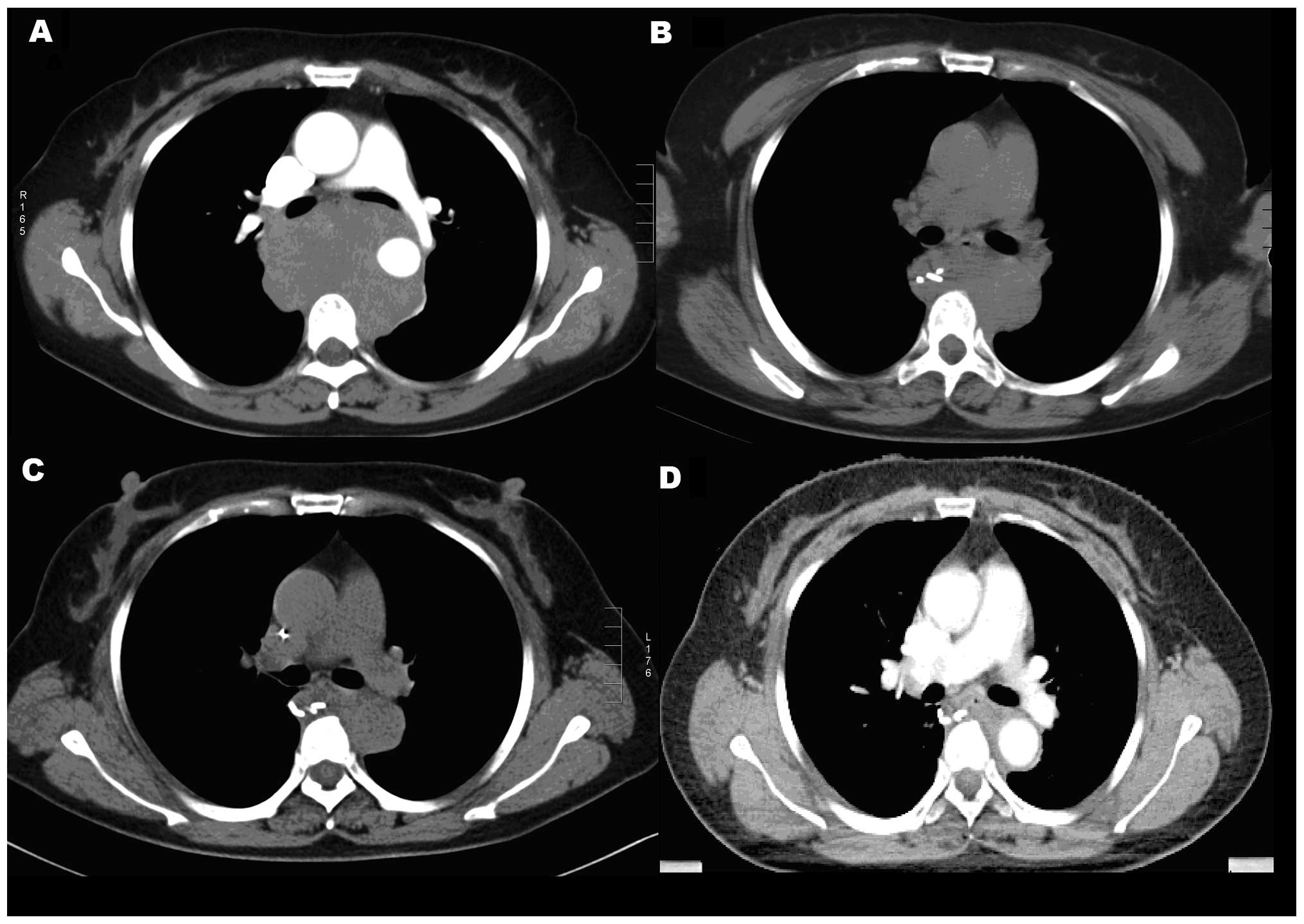
profile. The thoracic computed tomography (CT) scan revealed a huge
mass in the posterior mediastinum, with invasion of the esophagus,
descending aorta and right pulmonary artery (Fig. 1A). On December 31, 2012, a biopsy of
the mediastinal mass was performed by thoracoscopy, during which an
irregular posterior mediastinal mass measuring ~8×8 cm was located
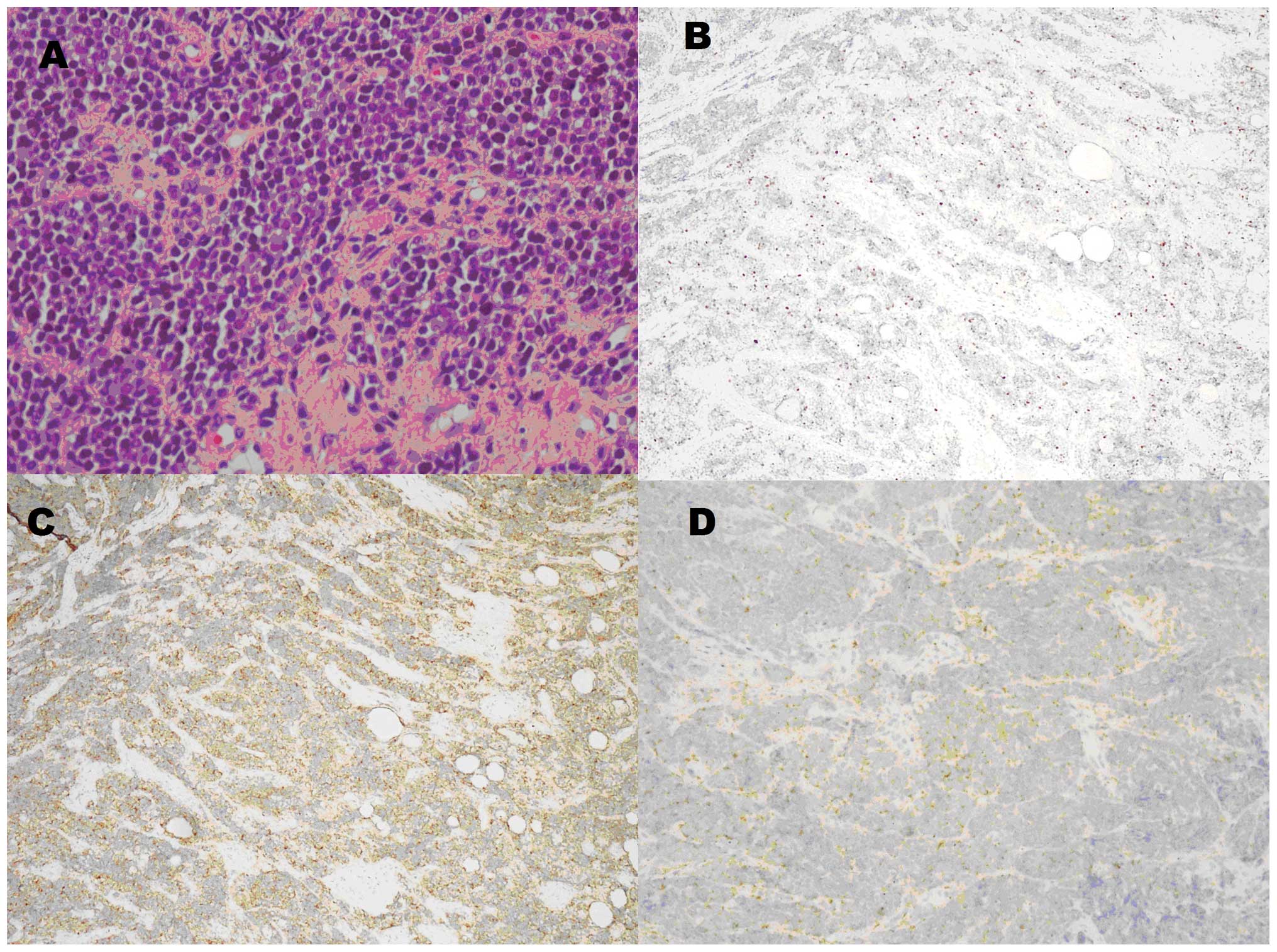
under the arch of the azygos vein. The final pathology showed a
malignancy of small round cells, which was consistent with an
Ewing’s sarcoma/PNET (Fig. 2A). The
immunohistochemical results demonstrated a Ki-67 of 20% (Fig. 2B), positivity for cluster of
differentiation (CD)99 (Fig. 2C)
and synaptophysin (Fig. 2D),
partial positivity for CD56, neuron-specific enolase and S-100, and
negativity for CD1a, Wilms tumor 1 protein, octamer-binding
transcription factor 3/4, vimentin, epithelial membrane antigen,
cytokeratin (CK)5/6, desmin, thyroid transcription factor-1,
inhibin A, CD34, terminal deoxynucleotidyl transferase, CK and
leukocyte common antigen. The following abdominal CT, brain
magnetic resonance imaging and bone scan excluded the possibility
of metastasis.
The patient was administered four cycles of
dacarbazine and pirarubicin chemotherapy. One cycle consisted of
300 mg dacarbazine on days 1–5 and 40 mg pirarubicin on days 1–2,
for 21 days (patient’s body surface area, ~1.6 m2).
Following one cycle of chemotherapy, the tumor rapidly decreased in
size (Fig. 1B) and the patient’s
discomfort disappeared. The thoracic CT revealed that the tumor
became even smaller after three cycles compared with one cycle
(Fig. 1C). Minimal residual tumor
remained subsequent to four cycles of chemotherapy (Fig. 1D). Radiotherapy was then
administered to the tumor bed at a total dose of 54 Gy/30 fractions
over 42 days (between May 10, 2013, and June 21, 2013) and the
recorded side-effects, such as esophagitis and leukopenia, were
mild.
Discussion
EES is a rare entity with high-grade malignancy
commonly involving the soft tissues of the trunk and extremities.
The thoracic sites of EES include the chest wall, trachea, spinal
epidural space, paraspinal area and mediastinum (7–11).
Primary mediastinal EES/PNET is extremely rare (1,12); to
the best of our knowledge, only five such cases have previously
been reported (1,11–14).
The present case will therefore aid in expanding our understanding
of this distinct neoplasm.
EES often exhibits translocation of (11;22)(q24;q12)
(3). CD99-positive expression plays
a crucial role in the diagnosis of EES (15). Tural et al reported
indicators of poor overall survival, namely a primary tumor of
>8 cm, a high level of lactic dehydrogenase, metastasis at the
time of the first hospital visit, a poor response to chemotherapy,
radiotherapy as the single method to improve local control and
positive margins (16).
For a long period of time, EES had been regarded as
exhibiting no significant differences to osseous Ewing’s sarcoma. A
study by Applebaum et al (17) was the first to reveal that EES
exhibited a different therapeutic response and clinical
characteristics. The mean age of onset is older in EES, with a
biphasic distribution to its peak age of onset: >35 and <5
years. Furthermore, the preponderance of males is less marked in
EES compared with Ewing’s sarcoma of the bone. Axial locations are
more likely than the pelvic cavity. Additionally, more patients
with EES receive radiotherapy.
EES is highly aggressive. Local relapse and distant
metastases are frequent. Multidisciplinary measures are important
for improvements in survival. A retrospective study of 24 cases of
EES showed a 61% five-year overall survival rate following
multimodality therapies (18). In a
study by Lee et al (7), two
cases of EES of the chest achieved at least 30 and 22 months
progression free survival, respectively, following comprehensive
treatment. The two cases utilized extended resections and a
post-operative alternate chemotherapy regimen of vincristine,
Adriamycin and cyclophosphamide, and ifosfamide and etoposide (IE).
One patient also received 54 Gy/30 fractions of radiotherapy to the
tumor bed (7). Extended resection
combined with multidrug chemotherapy often results in a clinical
benefit (11). However, from a
review of the literature, it can be observed that no consensus has
yet been reached with regard to a standard chemotherapy regimen.
Tural et al recommended an intense strategy of administering
vincristine, Adriamycin, cyclophosphamide and actinomycin D
alternately with IE (16). Tao
et al held the view that the use of platinum-based
chemotherapy should be considered (4). In the present case, dacarbazine and
pirarubicin achieved a good response and the side-effects were
mild.
In terms of local control, no random trials have yet
been initiated to demonstrate whether surgery combined with
radiotherapy is superior to surgery alone. However, complete
resection plus radiotherapy is known to have an advantage over
radiotherapy. For unresectable cases or tumors that cannot be
removed completely, radiotherapy with a radical dose of 50–60 Gy
should be performed (1).
In conclusion, the present study describes a
relatively rare case of primary mediastinal EES/PNET that could not
be resected. Sequential chemotherapy and radiotherapy achieved a
favorable response with mild side effects. We propose that
dacarbazine and pirarubicin chemotherapy should be considered for
unresectable EES/PNET cases for its beneficial effect, and
chemoradiotherapy is a good treatment option for such cases.
Successful published protocols using collaborative clinical trials
will aid in the development of a standard treatment strategy.
References
|
1
|
Reali A, Mortellaro G, Allis S, et al: A
case of primary mediastinal Ewing’s sarcoma/primitive
neuroectodermal tumor presenting with initial compression of
superior vena cava. Ann Thorac Med. 8:121–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rud NP, Reiman HM, Pritchard DJ, Frassica
FJ and Smithson WA: Extraosseous Ewing’s sarcoma. A study of 42
cases. Cancer. 64:1548–1553. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Delattre O, Zucman J, Melot T, et al: The
Ewing family of tumors - a subgroup of small-round-cell tumors
defined by specific chimeric transcripts. N Eng J Med. 331:294–299.
1994. View Article : Google Scholar
|
|
4
|
Tao HT, Hu Y, Wang JL, et al:
Extraskeletal Ewing sarcomas in late adolescence and adults: a
study of 37 patients. Asian Pac J Cancer Prev. 14:2967–2971. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Halliday J, Soon SY, Monaghan H, Walker WS
and Zamvar V: Extraskeletal Ewing’s sarcoma presenting as a
mediastinal mass. Ann Thorac Surg. 90:1016–1017. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oken MM, Creech RH, Tormey DC, et al:
Toxicity and response criteria of the Eastern Cooperative Oncology
Group. Am J Clin Oncol. 5:649–655. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee WS, Kim YH, Chee HK, et al: Multimodal
treatment of primary extraskeletal Ewing’s sarcoma of the chest
wall: report of 2 cases. Cancer Res Treat. 41:108–112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elmi M, Ko MA, Gupta A, et al: Primary
tracheal Ewing’s sarcoma. Ann Thorac Surg. 90:1349–1352. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ozturk E, Mutlu H, Sonmez G, et al: Spinal
epidural extraskeletal Ewing sarcoma. J Neuroradiol. 34:63–67.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Perouli E, Chrysikopoulos H, Vlachos A, et
al: Imaging findings in paraspinal extra osseous Ewing sarcoma.
JBR-BTR. 89:310–312. 2006.
|
|
11
|
Manduch M, Dexter DF, Ellis PM, et al:
Extraskeletal Ewing’s sarcoma/primitive neuroectodermal tumor of
the posterior mediastinum with t(11;22)(q24;q12). Tumori.
94:888–891. 2008.
|
|
12
|
Halefoglu AM: Extraskeletal Ewing’s
sarcoma presenting as a posterior mediastinal mass. Arch
Bronconeumol. 49:82–84. 2013. View Article : Google Scholar
|
|
13
|
El-Essawy MT: Extraskeletal Ewing’s
sarcoma. Saudi Med J. 30:840–843. 2009.PubMed/NCBI
|
|
14
|
Kuzucu A, Erkal HS, Soysal O and Serin M:
Extraskeletal Ewing’s sarcoma presenting with multifocal
intrathoracic mass lesions associated with mediastinal shift. Ann
Thorac Surg. 81:1487–1488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guiter GE, Gamboni MM and Zakowski MF: The
cytology of extraskeletal Ewing sarcoma. Cancer. 87:141–148. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tural D, Molinas Mandel N, Dervisoglu S,
et al: Extraskeletal Ewing’s sarcoma family of tumors in adults:
prognostic factors and clinical outcome. Jpn J Clin Oncol.
42:420–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Applebaum MA, Worch J, Matthay KK, et al:
Clinical features and outcomes in patients with extraskeletal Ewing
sarcoma. Cancer. 117:3027–3032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahmad R, Mayol BR, Davis M and Rougraff
BT: Extraskeletal Ewing’s sarcoma. Cancer. 85:725–731. 1999.
View Article : Google Scholar : PubMed/NCBI
|