Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1) is a DNA nick
sensor nuclear enzyme involved in the surveillance and maintenance
of genomic integrity. PARP-1 functions in the repair of DNA
single-strand breaks (SSBs) via the base excision repair (BER)
pathway (1,2). The inactivation of SSB repair by
PARP-1 inhibition during the S phase impedes replication fork
progression. This leads to replication-associated DNA double-strand
breaks (DSBs), which are the most toxic DNA lesions. Therefore,
pharmacological inhibitors of PARP-1 may be able to enhance the
cytotoxicity of DNA damage agents (3,4). There
are currently at least six PARP inhibitors in clinical trial that
are being used as chemo/radiotherapy sensitizers (5).
PARP inhibitors were first recognized to be
potentially therapeutic by the discovery that PARP inhibition is
toxic to cancer cell lines and human tumors with deficient function
of homologous recombination (HR), the most important DSB repair
pathway. This effect was termed synthetic lethality; when two
components act in a co-operating and semi-redundant manner in cell
survival, targeting one while the other is defective in a cancer
will selectively eliminate the tumor cells, but not be toxic to the
normal cells (6). This creates a
large therapeutic window.
Olaparib is a well-known PARP inhibitor and has been
used clinically in combination therapy for the treatment of
multiple cancers (5). Olaparib has
advanced into a phase III program as a single treatment for ovarian
cancer patients with BRCA gene mutations, which confer HR repair
dysfunction in tumor cells (7).
Although heterozygous germ-line mutations in the BRCA1/2 genes
render a risk of up to 85% for the development of breast cancer,
and 10–40% for ovarian cancer, only a small fraction of tumors are
BRCA-deficient, accounting for 3–5% of all breast cancers and 15%
of ovarian cancers (8,9). This therefore limits the therapeutic
utility of olaparib monotherapy. The phase III trial of olaparib
has emphasized the requirement for identifying those candidates who
are most likely to respond to treatment with the drug (7).
To provide further evidence for clinical
application, the sensitivity to olaparib of a range of cancer cell
lines, other than the commonly used breast or ovary cell lines,
were compared in the present study. Furthermore, the cellular
mechanism of the sensitive cell line was preliminarily
investigated.
Materials and methods
Reagents
6(5H)-phenanthridinone (PHE; Sigma-Aldrich, St.
Louis, MO, USA) and olaparib (Selleck, Burlington, USA) were
dissolved in dimethylsulfoxide (DMSO) to produce a stock solution
of 10 mM, and stored at −20°C for the in vitro studies. For
the in vivo experiment, olaparib was dissolved in
phosphate-buffered solution (PBS)/DMSO at 1 mg/ml.
Cell lines
JF-305 cells were obtained from the Tumor Research
Institute of China Medical University (Shenyang, China).
MDA-MB-436, Capan-1 and T47D cells were purchased from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China). Rin5f,
B16, Acc-3, Patu8988, Bel7402, HNE2, HepG2, DU145, SGC7901 and A549
cells were preserved in the lab. The cells, unless stated
otherwise, were maintained in RPMI 1640 medium containing 10% (v/v)
fetal bovine serum (FBS). The T47D cells were maintained in the
same manner, but supplemented with 0.2 U/ml insulin (Hisun
Pharmaceutical Co., Ltd., Taizhou, China). The Capan-1 cells were
maintained in Iscove’s modified Dulbecco’s medium containing 20%
FBS. The MDA-MB-436 cells were cultured in Leibovitz L-15 medium
supplemented with 10% FBS and 0.2 U/ml insulin. The cells were
maintained at 37°C in a humidified atmosphere of 5% CO2
and 95% air, except for MDA-MB-436, which was cultured at 37°C and
in 100% air.
Clonogenic assay for cell
proliferation
Exponentially proliferating cells were plated into
six-well plates at a density of 300 cells per well. The following
day, the cells were incubated with a series of concentrations of
PHE for five days or olaparib for seven days. The cells were fixed
and stained with 0.1% crystal violet in methanol/PBS (1:4) and
colonies consisting of >10 cells (PHE test) or >50 cells
(olaparib test) were subsequently manually counted. The results
were calculated as the percentage of colonies in the olaparib
treatment group compared with that in the PHE control group.
CCK-8 assay for cell viability
The cells were seeded into 96-well plates at
1,000–4,000 cells per well depending on the growth rate and left to
attach overnight. Olaparib at a concentration of 1 nM-10 μM was
added, and the cells were continually incubated for four days
(10). The cell viability was
measured using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan).
Foci formation of γ-H2AX and RAD51 by
co-immunostaining
The cells were seeded onto sterile confocal dishes
and exposed to a medium containing 3 μM olaparib, or PBS, for 24 h.
The cells were fixed in pre-chilled methanol/acetone (7:3) at −20°C
for 10 min. Subsequent to air-drying, the dishes were washed three
times with PBS and blocked using 5% skimmed dry milk and 0.1%
Triton X-100 in PBS at room temperature for 1 h. Samples were then
incubated overnight at 4°C with mouse anti-phospho-Histone H2AX
(Ser139) monoclonal antibody (Millipore, Billerica, MA, USA:
dilution, 1:50) and rabbit anti-RAD51 polyclonal antibody (Santa
Cruz, Dallas, TX, USA: dilution, 1:50) (11). Subsequent to being washed, the cells
were incubated with secondary Cy3-labeled goat anti-mouse
immunoglobulin G (IgG), and Alexa Fluor 488-labeled goat
anti-rabbit IgG (H+L) antibodies (Beyotime, Suzhou, China), for 1 h
at room temperature and protected from light. Subsequent to being
washed again, the nuclei were stained with 1 μg/ml DAPI (Beyotime)
for 10 min. Images were obtained with a confocal laser scanning
microscope (Leica TCS SP8; Leica, Wetzlar, Germany).
Cell cycle analysis
The cells were plated in six-well plates at
concentrations determined to reach 70–80% confluence when analyzed.
Following attachment, the cells were incubated with 0, 0.3 or 3 μM
olaparib for 48 h, then washed twice with PBS, treated with trypsin
and centrifuged at 800 × g for 5 min. The cells were fixed with 70%
ethanol for 2 h at 4°C and the pellet was then removed from the
ethanol and washed twice in ice-cold PBS. The cell pellet was
resuspended in PBS with 50 μg/ml RNase at 37°C for 30 min, followed
by 50 μg/ml propidium iodide in the dark at 4°C for 30 min. Samples
were analyzed using a flow cytometer (BD FACSCalibur; BD
Biosciences, San Diego, CA, USA) and data was analyzed with ModFit
software (Verity Software House, Inc., Topsham, ME, USA).
Nucleus staining and
photomicrography
The cells were exposed to olaparib for four days,
washed and fixed, and then stained with 1 μg/ml DAPI for 10 min.
Images were captured by a video camera (Nikon Coolpix 54, Nikon,
Tokyo, Japan) mounted on a Leica CME microscope.
Xenograft tumor studies
CByJ-Cg-Foxn1nu/Nju mice (male, aged 3–4 weeks) from
Nanjing Biochemical Research Institute of Nanjing University
(Nanjing, China) were used in the xenograft experiments. The
protocol was approved by the Animal Ethics Committee of Jiangnan
University (Wuxi, Jiangsu, China). The mice were maintained and
handled in isolators under specific pathogen-free conditions and
were inoculated to the right axillary cavity with 5×106
cells in 0.1 ml of medium without serum. Tumor volumes were
estimated using the formula: Tumor volume = (length / 2) ×
(width2) (10). When the
mean tumor volume reached 150 mm3, the tumor-bearing
mice were randomly split into two groups, with six animals in each
group. Mice in the test group received 10 mg/kg olaparib once daily
for 22 consecutive days, whilst those in the vehicle group received
PBS as a vehicle containing the same concentration of DMSO. The
tumor volumes were measured every three days and the established
tumors in each animal were individually normalized to their size at
the start of the treatment administration. The relative tumor
volume (RTV) was calculated according to the formula (12): RTV = TVx / TV0, where TVx is the
tumor volume on any given day and TV0 is the tumor volume at the
initiation of dosing (i.e., day 0).
Statistical analysis
Results are presented as the mean ± standard
deviation. All statistical analyses were performed using GraphPad
Prism version 5.0 for Windows Software (GraphPad Software, La
Jolla, CA, USA). Statistical differences were determined by
two-tailed Student’s t-test unless stated otherwise. P<0.05
denotes a statistically significant difference.
Results
JF-305 cells are hypersensitive to PARP
inhibitors
In the present study, the effects of the PARP
inhibitor, PHE (IC50, 350 nM) (13), on the colony formation efficiency of
various cell lines was investigated. The pancreatic cancer JF-305
cell line was identified to be the most sensitive to PHE, as its
colony formation efficiency decreased to <10% when treated with
10 μM PHE, a concentration at which other cell lines retained at
least 60% colony formation efficiency (P<0.05) (Fig. 1).
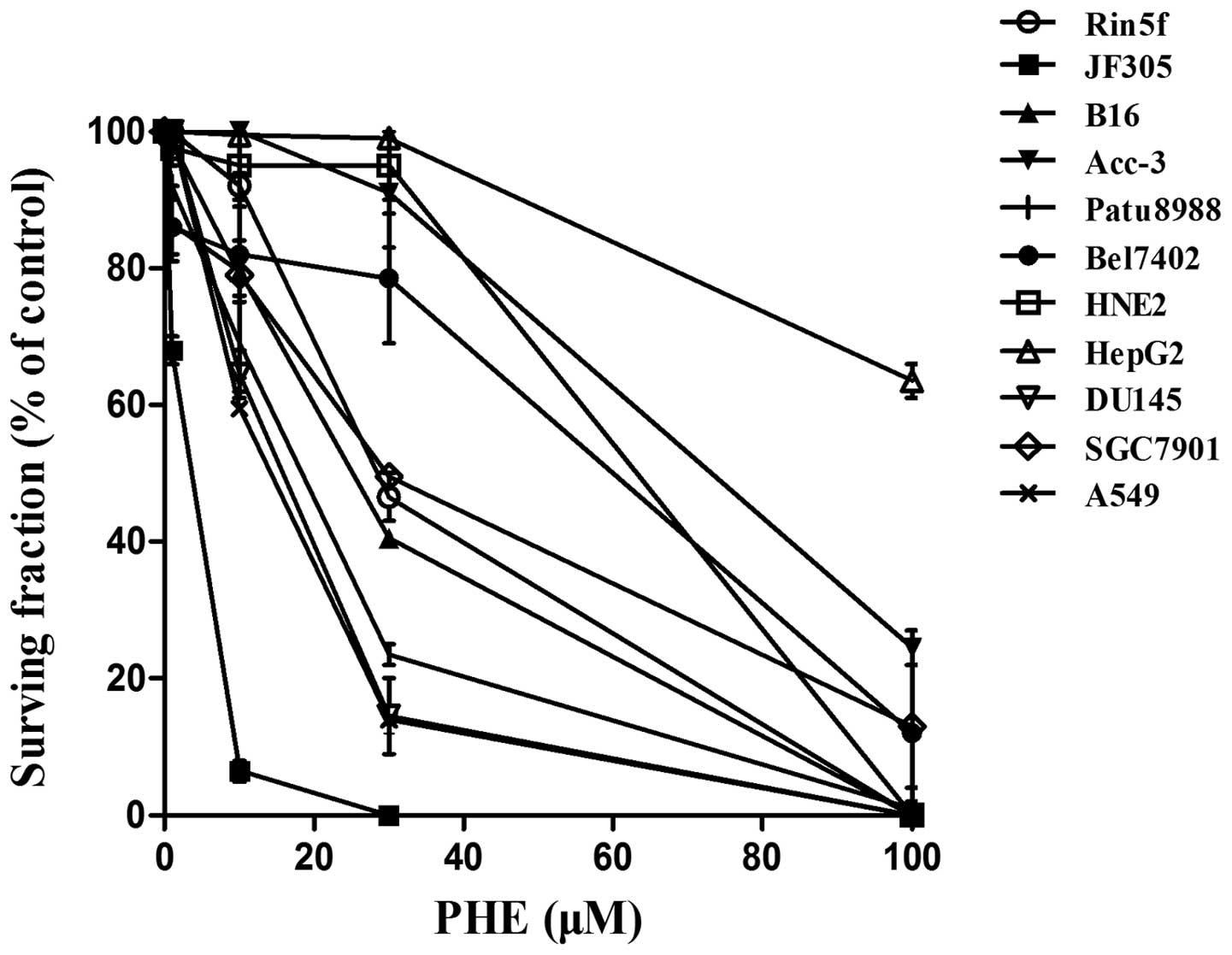 | Figure 1Compared with other cell types, JF-305
cells demonstrate relatively high sensitivity of their colony
formation efficiency to the PARP inhibitor PHE. Rin5f, islet tumor
cell; B16, skin melanoma cell; Acc-3, salivary gland adenoid cystic
carcinoma cell; Patu8988, pancreatic cancer cell; Bel7402, hematoma
cell; HNE2, nasopharyngeal carcinoma cell; HepG2, hepatocellular
carcinoma cell; DU145, prostate cancer cell; SGC7901, gastric
cancer cell; A549, lung adenocarcinoma cell; PHE,
6(5H)-phenanthridinone. Data are expressed as the mean ± standard
deviation (n=3). |
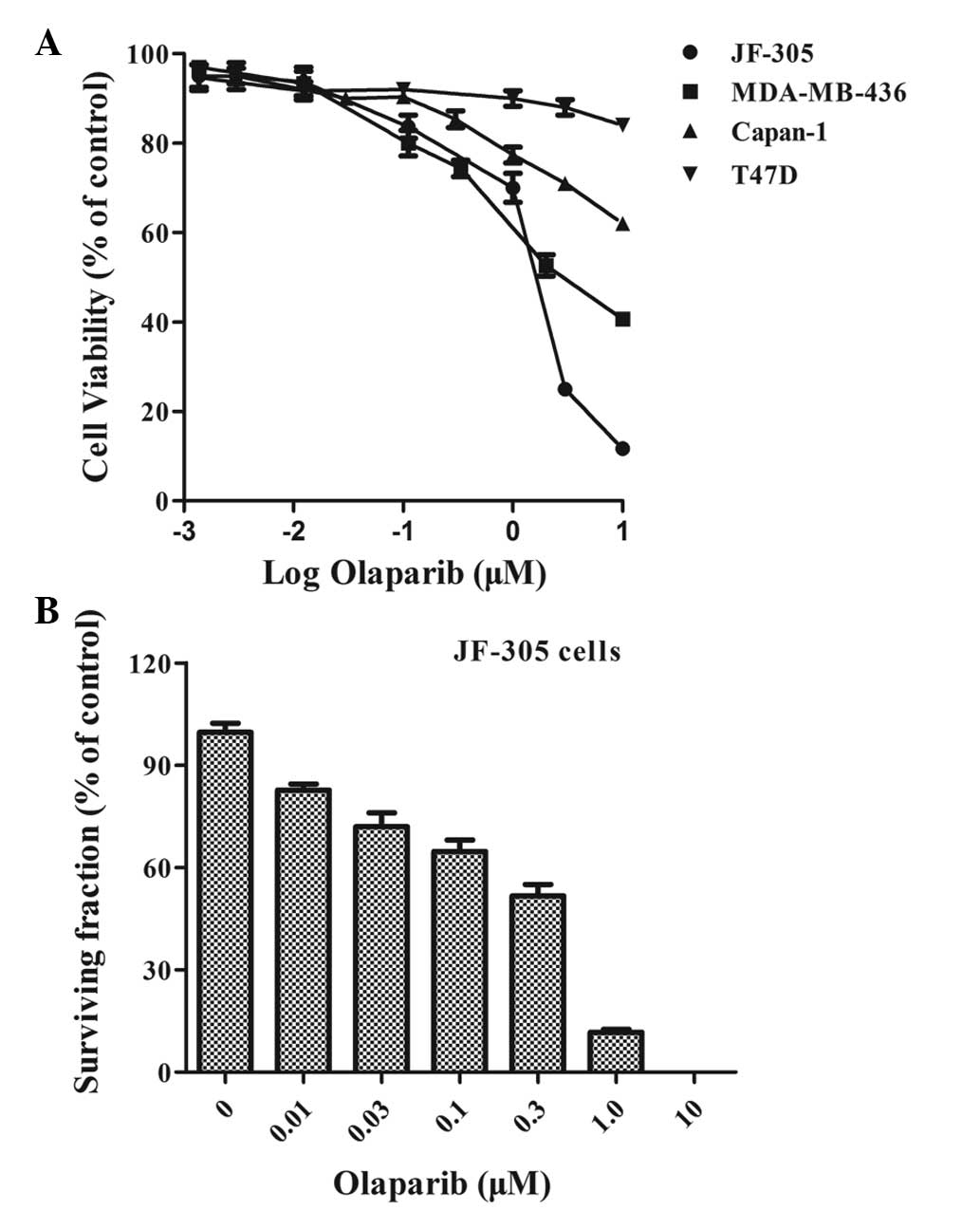
A more potent PARP inhibitor, olaparib
(IC50, 5 nM) (10), was
then used to confirm this result. For the in vitro cell
viability assay, the T47D cells (BRCA-1- and BRCA-2-proficient),
MDA-MB-436 cells [BRCA1 (5382insC) mutated] and Capan-1 cells
[BRCA1 (6174delT) mutated] (10,13)
were used as controls. The JF-305 cells exhibited hypersensitivity
to olaparib, with a percentage viability of ~25% at 3 μM and 11% at
10 μM, compared with 50% and 41%, respectively, in the MDA-MB-436
cells, and 71% and 62%, respectively, in the Capan-1 cells. The
T47D cells, however, demonstrated very little response (P<0.05)
(Fig. 2A). To validate the
sensitivity of the JF-305 cells to olaparib, a clonogenic assay was
performed as a ‘gold standard’ to assess cell proliferation. The
colony formation efficiency of the JF-305 cells was significantly
reduced upon treatment with an increasing concentration of olaparib
(P<0.05). The dosage at which 50% of the cells survived was 0.4
μM (Fig. 2B).
As the aforementioned data demonstrated (Figs. 1 and 2), the JF-305 cells were sensitive to PARP
inhibitors in vitro.
Olaparib results in DSBs and cell cycle
arrest with activated HR repair in JF-305 cells
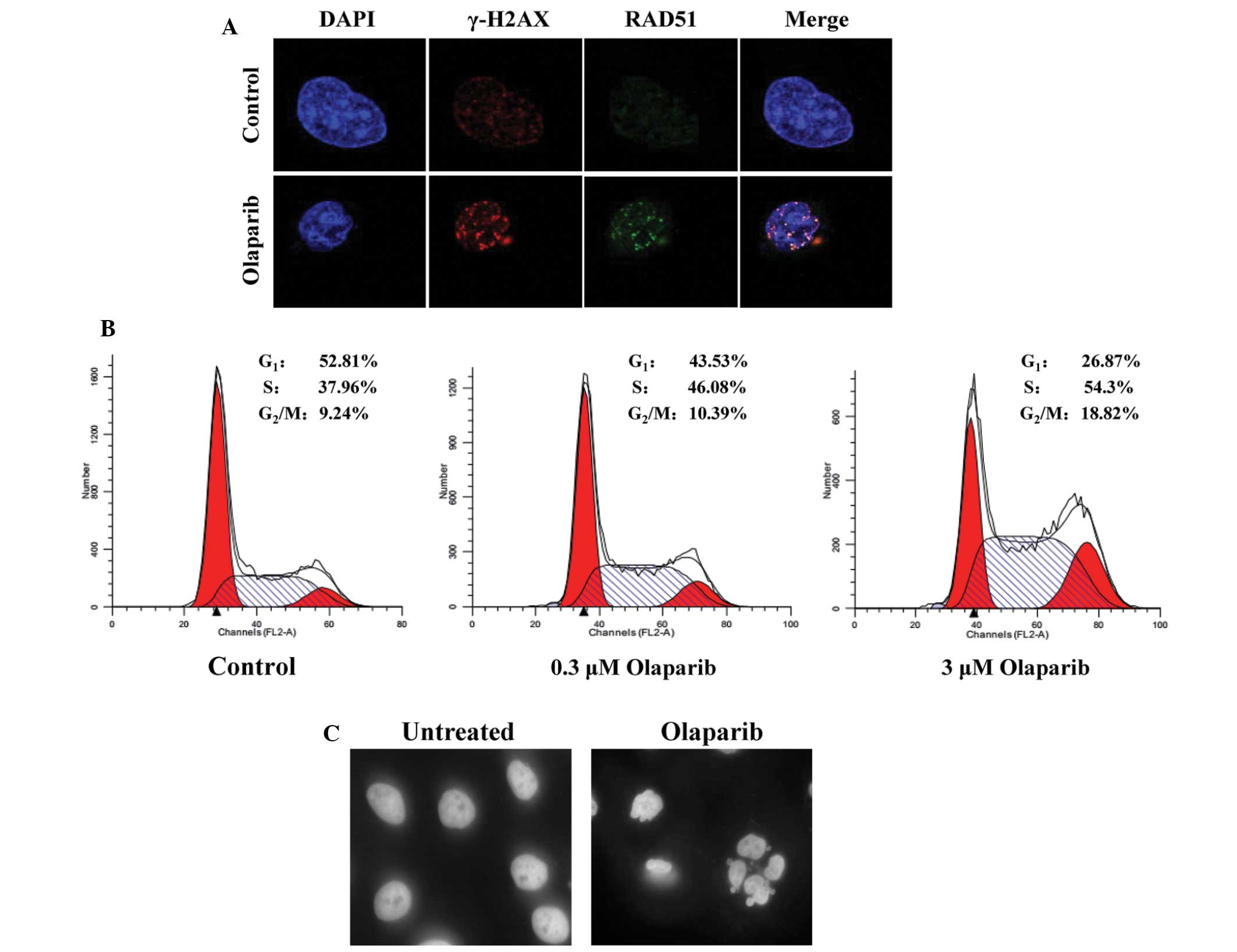
Olaparib targets PARP-1, a component of the BER
pathway. Blockage of the BER pathway will induce a large number of
potentially lethal DSBs when encountered by replication forks. The
nuclear γ-H2AX foci occur at the sites of DSBs (14), therefore, the present study
identified γ-H2AX foci in the JF-305 cells to reveal the existence
of DNA damage. Treatment of JF-305 cells with olaparib increased
the formation of γ-H2AX foci in the nucleus compared with the
control (Fig. 3A), which indicated
the interaction of olaparib with a functional DNA sensor. In
addition, the formation of RAD51 foci, which play a key role in DNA
HR during DSB repair, were investigated in the present study. The
co-immunostaining analysis revealed that the increased RAD51 foci
overlapped at the sites of DSBs (Fig.
3A), which identified activated HR repair in JF-305 cells
treated with olaparib.
To determine how olaparib leads to the decrease of
cell viability and colony formation efficiency, the cell cycle of
the JF-305 cells was analyzed in the present study. Following 48 h
of exposure, 3 μM olaparib elicited a 2-fold accumulation of
tetraploid DNA content in the JF-305 cells, indicating an arrest in
the G2/M phase of the cell cycle. The number of cells in
the S phase also increased by 43% compared with the untreated group
(Fig. 3B). Following 96 h of
treatment with olaparib, the DAPI-stained JF-305 cells demonstrated
an increase in apoptosis, which usually manifests with chromatin
condensation and nuclear fragmentation (15) (Fig.
3C).
Together, this data suggested that despite activated
HR, olaparib induced DNA DSBs by PARP inhibition, initiated S and
G2/M cell cycle arrest and ultimately induced the cells
to undergo apoptosis.
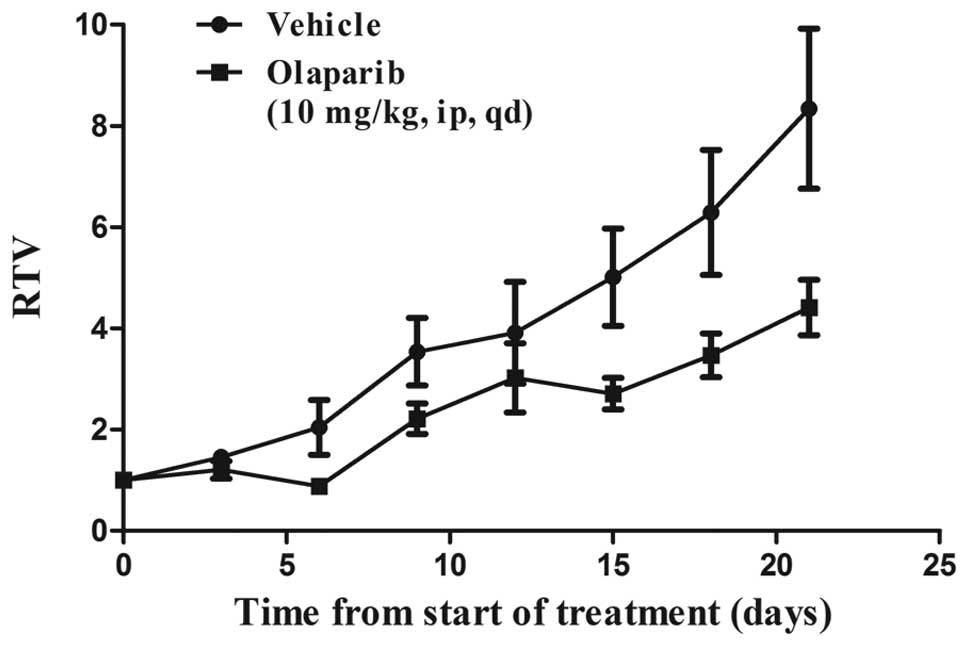
JF-305 tumor growth is delayed by
olaparib in vivo
The results from the present study demonstrated that
the JF-305 cells were sensitive to olaparib in vitro. In
addition, JF-305 cells have also previously been reported to
exhibit tumorigenicity when transplanted into nude mice (16). In the present study, upon assessment
of the response of JF-305 tumors to olaparib in vivo, tumor
formation was detected after two weeks of inoculation. The mean
tumor volume in the control group increased to 1,368 mm3
by the 5th week, and to 687 mm3 in the olaparib-treated
group (P<0.05), a 49.8% reduction following 22 consecutive days
of administration (Fig. 4).
The comparable activity of olaparib upon the JF-305
cells in vitro and the JF-305 tumors in vivo supports
a therapeutic model for the preclinical study of pancreatic cancer
and the potential applications of olaparib in an Asian
population.
Discussion
Given the potential of olaparib as a therapeutic
approach for the treatment of cancer, certain studies have
demonstrated interest in the concept of synthetic lethality in
cancers with defects in DNA metabolic processes other than HR
repair. Previous studies have confirmed that a loss of function of
RAD51C, XRCC3, PTEN, ATM, CHK1 or CHK2 (17–20)
causes cells and tumors to become sensitive to PARP inhibition.
High-throughput RNA interference analysis also identified DDB1,
XAB2 and CDK12 as novel genetic determinants of PARP inhibitor
sensitivity (21,22). Furthermore, cells deficient in the
aforementioned genes became an ideal model for the preclinical
study of cancers. The present study identified a cell line, JF-305,
which was hypersensitive to olaparib. Although olaparib induced an
increase in DNA DSBs, HR was effectively activated. Furthermore,
the PARP inhibitors, KU0058684 and olaparib, have previously been
shown to arrest the cells in phase G2 of the cell cycle
in wild-type cells, an effect that was enhanced in BRCA1/2- or
RAD51C-deficient cells (23,24).
In addition to G2/M phase arrest, JF-305 cells also
exhibit arrest at the S phase following treatment with olaparib.
These factors may indicate a novel molecular mechanism contributing
to the sensitivity of PARP inhibitors in JF-305 cells. Identifying
the cellular events that occur following the accumulation of RAD51
foci, prior to the cell progressing through the cell cycle
checkpoints, will be the objective of a future study.
Pancreatic adenocarcinoma is currently the fourth
most common cause of cancer-related lethality. The disease
incidence is almost equal to the disease mortality due to a high
resistance to chemo/radiotherapy and a poor prognosis (25). The standard first-line therapy for
pancreatic adenocarcinoma of gemcitabine alone, or in combination
with fluorouracil, demonstrates limited efficacy (26). Attempts have been made to improve
the outcome for BRCA-mutated pancreatic cancer by using the PARP
inhibitor veliparib, or combining gemcitabine with olaparib or
veliparib (6). The results from the
present study revealed that the pancreatic JF-305 cell line was
sensitive to olaparib as a single treatment either in vitro
or in vivo. This finding further confirmed the clinical
potential of olaparib as a combined treatment or monotherapy for
pancreatic cancers without a BRCA/HR deficiency.
Furthermore, as is the case for BRCA mutations, the
biomarkers of PARP inhibitor sensitivity differ between Asian and
Western populations (27,28). It is also unclear how representative
the Western findings on pancreatic cancer are for Asian populations
(29). The JF-305 cells of Chinese
origin from the present study may support an accurate model for
PARP inhibitor sensitivity and pancreatic carcinoma in an Asian
population.
In conclusion, the present study identified that
JF-305, a pancreatic cancer cell line of Chinese origin, was
sensitive to olaparib in vitro and in vivo. Although
HR repair was effectively activated in the cells treated with
olaparib, the cell cycle was arrested in the S and G2/M
phases following numerous DSBs. In addition to the regional
diversity of gene mutations, this may indicate another functional
impairment mutation in JF-305 cells that has the potential to be a
model for the preclinical investigation of pancreatic cancer
chemotherapy with PARPs as a target.
References
|
1
|
Peralta-Leal A, Rodríguez-Vargas JM,
Aguilar-Quesada R, Rodríguez MI, Linares JL, de Almodóvar MR and
Oliver FJ: PARP inhibitors: new partners in the therapy of cancer
and inflammatory diseases. Free Radic Biol Med. 47:13–26. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rouleau M, Patel A, Hendzel MJ, Kaufmann
SH and Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev
Cancer. 10:293–301. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Helleday T, Petermann E, Lundin C, Hodgson
B and Sharma RA: DNA repair pathways as targets for cancer therapy.
Nat Rev Cancer. 8:193–204. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Graziani G and Szabó C: Clinical
perspectives of PARP inhibitors. Pharmacol Res. 52:109–118. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee JM, Ledermann JA and Kohn EC: PARP
Inhibitors for BRCA1/2 mutation-associated and BRCA-like
malignancies. Ann Oncol. 25:32–40. 2014. View Article : Google Scholar :
|
|
6
|
Ashworth A: A synthetic lethal therapeutic
approach: poly(ADP) ribose polymerase inhibitors for the treatment
of cancers deficient in DNA double-strand break repair. J Clin
Oncol. 26:3785–3790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
No authors listed. Olaparib enters phase
III clinical testing. Cancer Discovery. 3:12102013. View Article : Google Scholar
|
|
8
|
Rottenberg S, Jaspers JE, Kersbergen A, et
al: High sensitivity of BRCA1-deficient mammary tumors to the PARP
inhibitor AZD2281 alone and in combination with platinum drugs.
Proc Natl Acad Sci USA. 105:17079–17084. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pal T, Permuth-Wey J, Betts JA, et al:
BRCA1 and BRCA2 mutations account for a large proportion of ovarian
carcinoma cases. Cancer. 104:2807–2816. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Menear KA, Adcock C, Boulter R, et al:
4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one:
a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J
Med Chem. 51:6581–6591. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weston VJ, Oldreive CE, Skowronska A, et
al: The PARP inhibitor olaparib induces significant killing of
ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood.
116:4578–4587. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thomas HD, Calabrese CR, Batey MA, et al:
Preclinical selection of a novel poly(ADP-ribose) polymerase
inhibitor for clinical trial. Mol Cancer Ther. 6:945–956. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Drew Y, Mulligan EA, Vong WT, et al:
Therapeutic potential of poly(ADP-ribose) polymerase inhibitor
AG014699 in human cancers with mutated or methylated BRCA1 or
BRCA2. J Natl Cancer Inst. 103:334–346. 2011. View Article : Google Scholar
|
|
14
|
Mah LJ, El-Osta A and Karagiannis TC:
gammaH2AX: a sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kepp O, Galluzzi L, Lipinski M, Yuan J and
Kroemer G: Cell death assays for drug discovery. Nat Rev Drug
Discov. 10:221–237. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Zhang BG and Jia LL: Establishment
of transplantable human pancreatic carcinoma model in nude mice and
study of its biological characteristics. J China Med Univ.
22:161–163. 1993.(In Chinese).
|
|
17
|
Min A, Im SA, Yoon YK, et al:
RAD51C-deficient cancer cells are highly sensitive to the PARP
inhibitor olaparib. Mol Cancer Ther. 12:865–877. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Issaeva N, Thomas HD, Djureinovic T, et
al: 6-thioguanine selectively kills BRCA2-defective tumors and
overcomes PARP inhibitor resistance. Cancer Res. 70:6268–6276.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mendes-Pereira AM, Martin SA, Brough R, et
al: Synthetic lethal targeting of PTEN mutant cells with PARP
inhibitors. EMBO Mol Med. 1:315–322. 2009. View Article : Google Scholar
|
|
20
|
McCabe N, Turner NC, Lord CJ, et al:
Deficiency in the repair of DNA damage by homologous recombination
and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer
Res. 66:8109–8115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lord CJ, McDonald S, Swift S, Turner NC
and Ashworth A: A high-throughput RNA interference screen for DNA
repair determinants of PARP inhibitor sensitivity. DNA Repair
(Amst). 7:2010–2019. 2008. View Article : Google Scholar
|
|
22
|
Bajrami I, Frankum JR, Konde A, et al:
Genome-wide profiling of genetic synthetic lethality identifies
CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity.
Cancer Res. 74:287–297. 2014. View Article : Google Scholar
|
|
23
|
Farmer H, McCabe N, Lord CJ, et al:
Targeting the DNA repair defect in BRCA mutant cells as a
therapeutic strategy. Nature. 434:917–921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Murai J, Huang SY, Das BB, et al: Trapping
of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res.
72:5588–5599. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hermann PC, Huber SL, Herrler T, et al:
Distinct populations of cancer stem cells determine tumor growth
and metastatic activity in human pancreatic cancer. Cell Stem Cell.
1:313–323. 2007. View Article : Google Scholar
|
|
26
|
Güngör C, Hofmann BT, Wolters-Eisfeld G
and Bockhorn M: Pancreatic cancer. Brit J Pharmacol. 171:849–858.
2014. View Article : Google Scholar
|
|
27
|
Kurian AW: BRCA1 and BRCA2 mutations
across race and ethnicity: distribution and clinical implications.
Curr Opin Obstet Gynecol. 22:72–78. 2010. View Article : Google Scholar
|
|
28
|
Thirthagiri E, Lee SY, Kang P, et al:
Evaluation of BRCA1 and BRCA2 mutations and risk-prediction models
in a typical Asian country (Malaysia) with a relatively low
incidence of breast cancer. Breast Cancer Res. 10:R592008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Untawale S, Odegaard AO, Koh WP, Jin AZ,
Yuan JM and Anderson KE: Body mass index and risk of pancreatic
cancer in a Chinese population. PLoS One. 9:e851492014. View Article : Google Scholar : PubMed/NCBI
|