Introduction
Aberrant activities of interrelated signaling
pathways contribute to squamous cell carcinoma (SCC) development
and are the focus of several molecular targeted therapy approaches,
such as epidermal growth factor pathway and tyrosine kinase
inhibitors. In this regard, particularly the mechanisms of inherent
radioresistance in this entity remain to be defined. We recently
showed that the MAPK pathway, which can be upregulated by ionizing
radiation (IR), seems to play a pivotal role in irradiation
resistance development (1). The
cascade is involved in cell growth, thereby conferring a survival
advantage (2,3). ERK activation induced by IR is assumed
to lead to an increase of vascular endothelial growth factor (VEGF)
expression levels and, therefore, to a feedback loop of
radioresistance (4).
Our present study focuses on the role of the tumor
microenvironment in radioresistance development. A benefit of IR as
a therapeutic tool is the locoregional application, which prevents
systemic toxicity. However, besides targeting cancer cells, IR
affects surrounding normal cells, which is a dose-limiting factor.
Unknown effects may occur particularly from the response of
tumor-infiltrating fibroblasts.
There is increasing knowledge that solid tumors do
not only contain neoplastic cells, but are rather assembled of a
mixture of cells and extracellular matrix constituting the tumor
stroma (5). Cancer-associated
fibroblasts (CAFs) are known to promote growth and invasion of
cancer cells by various mechanisms (6), maintaining a permanent crosstalk with
tumor cells. They are therefore considered a key player in cancer
progression and, similarly, a promising target in therapy regimes.
In this regard, it has already been shown that exposure to lower
doses of IR (<20 Gy) enhances the capacity of senescent
fibroblasts to promote the survival of co-cultured cancer cells
(7).
In the present study we continued to explore
cellular responses to a combination therapy regimen. Tumor and
fibroblast cell lines were used as models for cancer and stroma
cells and were treated with the specific MEK/ERK inhibitor U0126
before irradiation in an attempt to mimic the therapeutical
situation. CAFs were analyzed in order to depict potential
interference mechanisms of this cellular subgroup in response to
treatment. Our aim was to investigate the impact of irradiation and
kinase inhibition on tumor and normal cells, including CAFs,
focusing on their colony forming and proliferative properties. Our
data contribute to a better understanding of the responses of CAFs
to avoid unwanted responses when developing therapeutic
strategies.
Materials and methods
Tumor cell lines
The oral squamous cell carcinoma (OSCC) cell line
HNSCCUM-02T was previously established and characterized in our
laboratory (8). Non-small cell lung
cancer (NSCLC) cell line A549 and mouse embryonic fibroblast cell
line NIH3T3 were purchased from ATCC (Manassas, VA, USA).
Fibroblast cell lines derived from OSCC and normal tissues were
established and cultured in our laboratory. Experiments were
approved by the relevant ethics committee. Cells were maintained in
DMEM (Gibco® Invitrogen, Auckland, NZ) supplemented with
5% FCS (PAA Laboratories, Cölbe, Germany) and antibiotics
(penicillin 100 U/ml and streptomycin 100 μg/ml (Gibco Invitrogen)
at 37°C in 5% CO2.
Inhibitor treatment and irradiation of
cell cultures
Tumor cell lines were isolated by tryptic digestion
(PAA Laboratories), stained with trypan blue (Sigma, Taufkirchen,
Germany) and counted. Cells (7×105) were seeded per T25
flask and cultured in DMEM with 5% FCS and antibiotics. The cells
were serum-starved in DMEM containing antibiotics for 24 h. U0126
(Cell Signaling Technology Inc., Danvers, MA, USA) was added in a
final concentration of 10 μM. After incubation for 2 h, cells were
irradiated with intensities of 1, 8 and 30 Gy, using a γ-source
(Cs137). After 5 min, cells were harvested. Controls
were not irradiated.
Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and western blotting
A volume of homogenate containing 20 μg of total
protein was loaded onto 12.5% acrylamide gels and subjected to
SDS-PAGE. Gels were transferred to nitrocellulose and were western
blotted using an anti-phospho-ERK1/2 antibody (R&D,
Minneapolis, MN, USA) and an anti-β-actin antibody (Sigma) as a
loading control. Blots were developed using Western Lightning
Chemiluminescence Reagent Plus (Perkin-Elmer Life Sciences, Boston,
MA, USA) and AGFA Curix HT 1.000 plus X-ray film. Densitometric
analysis for ECL blots was performed using the Corel Photo-Paint X3
Image system and the respective software and band densities were
normalized to that of β-actin in the same sample. Blots were
digitally scanned using Epson Smart Panel (Epson, Munich, Germany).
Each experiment was performed in triplicate.
Clonogenic assay and data analysis
Monolayers (80% confluence) of cell line NIH3T3 were
dispersed by tryptic digestion (PAA Laboratories) and re-suspended
in 10 ml (240 and 720 cells/ml for doses of 6 and 8 Gy,
respectively) of standard media supplemented with FCS and
antibiotics. U0126 (Cell Signaling Technology Inc.) was added at a
final concentration of 10 μM. Following incubation for 2 h, cells
were irradiated with the following doses: 0.5, 1, 2, 4, 6 and 8 Gy,
using a γ-source (Cs137). Mock-irradiated cultures were
processed in parallel. Cells were seeded into T25 flasks and
cultured for 10 days. After fixation with ethanol/acetone (50%,
v/v), cells were stained with 10% Giemsa (Sigma, St. Louis, MO,
USA). Colony formation was defined as a colony of >50 cells.
Each experiment was performed in duplicate and repeated three
times. Survival curves were then created. We examined the
proportion of cells at 0 Gy that were still present at a given
dose. The purpose of the statistical analysis was to model the
dependence of the proportion of surviving tumor cells on the type
of experiment (DMSO or U0126) and on the radiation dose. We assumed
a linear-quadratic relationship between log (proportion) of
surviving cells and radiation dose.
We took repeated measurements on assays into account
by first fitting a linear mixed model with SAS PROC Mixed. We
found, however, that the assay variance components were estimated
zero. Therefore, we assumed independence of measurements and fitted
a linear quadratic model using SAS PROC GLM. We used SAS PROC
GLMSelect to determine what degree of polynomial was suitable for
the data and whether common constant, linear or quadratic terms
might be assumed. The statistical analysis was performed using SAS
9.3 (SAS Institute Inc., Cary, NC, USA).
Alamar blue proliferation assays
In addition to the HNSCCUM-02T, A549 and NIH3T3 cell
lines, we also used CAFs and fibroblasts derived from normal tissue
for this experiment.
Cell proliferation under the influence of IR and
U0126 application was assessed with the oxidation-reduction
sensitive dye Alamar Blue (Biozol, Eching, Germany), a
fluorometric/colorimetric indicator of metabolic activity. Briefly,
cells were seeded into T25 flasks (7×105) and incubated
at 37°C in a 5% CO2 humidified incubator. After 48 h,
cells were treated with 10 μM of the U0126 MEK inhibitor and DMSO,
respectively, for 2 h. Subsequently, cells were harvested and
counted. Cells (750,000) were transferred into falcon tubes in a
total volume of 4.5 ml medium and were then irradiated with a
dosage of 30 Gy. DMSO-treated cells as control and cells treated
with U0126 were processed in parallel.
Following treatment, cells were plated onto a
96-well-plate in triplicate (25,000 cells/150 μl). Cells treated
with 100% ethanol for 5 min were used as a death control. Cells
were left to attach at 37°C. After 24 or 72 h, respectively, 10%
Alamar Blue was added and the cells were incubated for 4 h before
recording the fluorescence on a Fluorescence Microplate Reader
(Fluoroskan Ascent, Thermo Electron Corp., Dreieich, Germany).
Results were given as fluorescence units using a 538-nm excitation
filter and a 600-nm emission filter. Each experiment was performed
6-fold and repeated three times.
Results
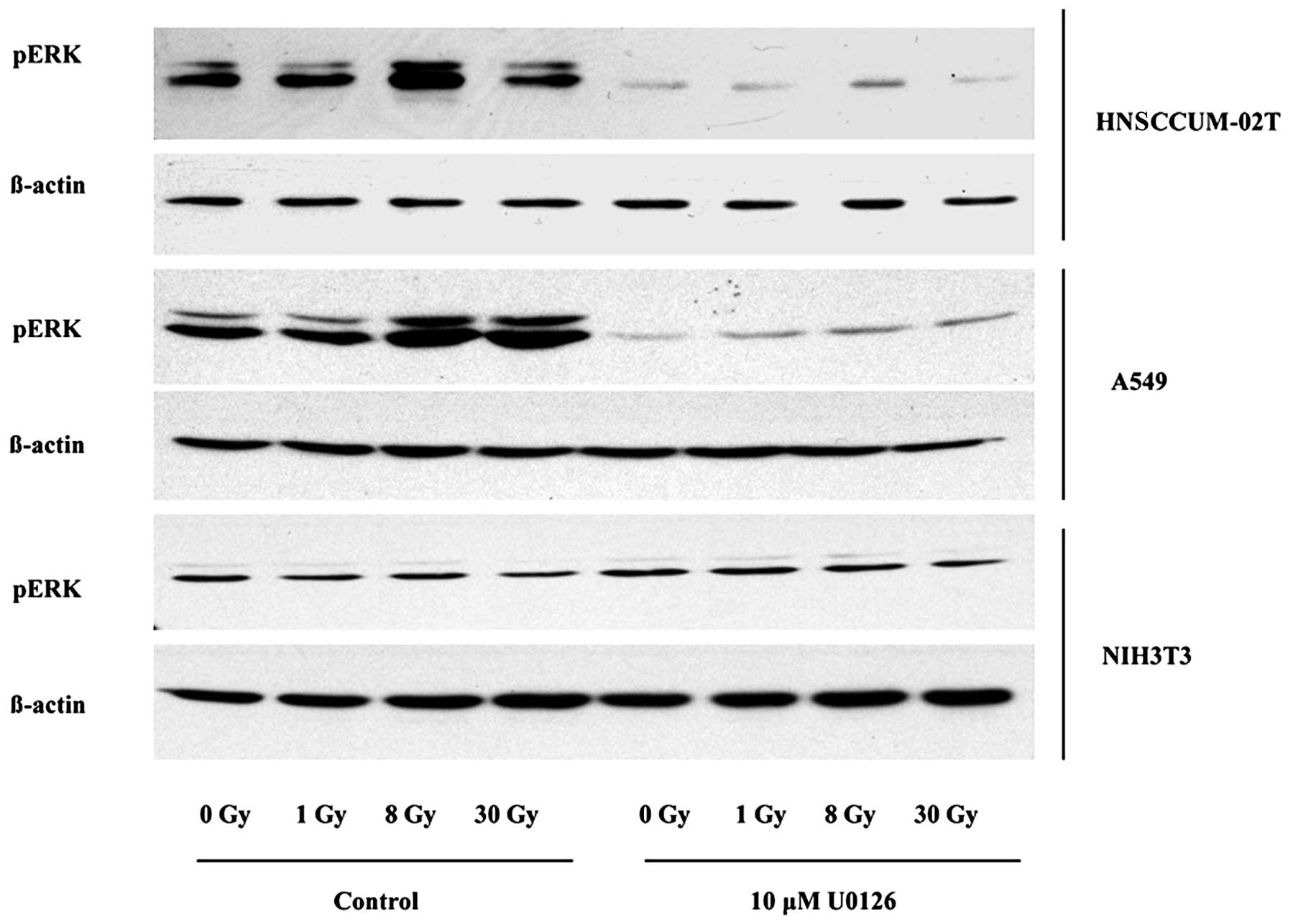
MEK/ERK inhibitor U0126 reduces pERK
expression in irradiated tumor cells, but not in fibroblasts
We previously reported that radiation-induced
phosphorylation of ERK was distinctly diminished after U0126
application in HNSCCUM-02T cells when performing dose course
experiments with different IR intensities and treatment with 10 μM
U0126 followed by immunoblotting (4). In order to verify whether the other
cell lines might express post-radiogenically elevated pERK
(activated ERK) levels as well, we irradiated with dosages of 0, 1,
8 and 30 Gy. The oral cancer cell line HNSCCUM-02T displayed
elevated pERK levels after irradiation compared to mock-irradiated
cells with maximum levels when treated with 8 Gy. HNSCCUM-02T cells
showed a 117% increase of pERK after an intensity of 8 Gy in
comparison to mock-irradiated cells. Phospho-ERK levels were
markedly suppressed after treatment with U0126 throughout all the
applied intensities in HNSCCUM-02T. Cells showed a decrease of 86%
in their pERK expression when applying the inhibitor without
irradiation, in comparison with untreated cells, and a decrease of
96% after utilization of 30 Gy in combination with U0126. Similar
to the OSCC cells, the A549 NSCLC cell line displayed maximum
levels of activated ERK after an irradiation dosage of 8 Gy.
Compared to the mock-irradiated cells, we observed a 55% increase
in pERK levels. A549 displayed an average decrease of 89% in pERK
levels after U0126 treatment without irradiation and a pERK
reduction of 89% when applying a 30-Gy intensity. However, NIH3T3
mouse fibroblasts did not respond to lower dosages of 1 and 8 Gy
with or without U0126 application. Only after U0126 application at
a maximum radiation dosage of 30 Gy and U0126 was there a minor
decrease of pERK levels of 65% compared to untreated cells
(Fig. 1). Table I shows the mean values and SEM of
densitometric measurements.
 | Table IWestern blots (mean values and SEM of
densitometric analyses). |
Table I
Western blots (mean values and SEM of
densitometric analyses).
| A, Effect of IR and
U0126 treatment on expression levels of pERK in HNSCCUM-02T |
|---|
|
|---|
| HNSCCUM-02T | Mean value | SEM |
|---|
| 0 Gy | 194,211 | 44,162 |
| 1 Gy | 259,657 | 89,303 |
| 8 Gy | 421,151 | 91,338 |
| 30 Gy | 327,781 | 166,389 |
| 0 Gy + U0126 | 26,846 | 22,973 |
| 1 Gy + U0126 | 13,112 | 6,189 |
| 8 Gy + U0126 | 20,797 | 7,146 |
| 30 Gy + U0126 | 7,533 | 4,294 |
|
| B, Effect of IR and
U0126 treatment on expression levels of pERK in A549 |
|
| A549 | Mean value | SEM |
|
| 0 Gy | 674,832 | 199,646 |
| 1 Gy | 654,505 | 141,277 |
| 8 Gy | 1,046.941 | 178,202 |
| 30 Gy | 1,027.477 | 216,754 |
| 0 Gy + U0126 | 72,807 | 34,964 |
| 1 Gy + U0126 | 83,556 | 20,153 |
| 8 Gy + U0126 | 99,122 | 31,535 |
| 30 Gy + U0126 | 77,410 | 32,684 |
|
| C, Effect of IR and
U0126 treatment on expression levels of pERK in NIH3T3 |
|
| NIH3T3 | Mean value | SEM |
|
| 0 Gy | 570,900 | 487,118 |
| 1 Gy | 841,258 | 907,611 |
| 8 Gy | 449,025 | 276,554 |
| 30 Gy | 542,448 | 641,098 |
| 0 Gy + U0126 | 359,449 | 163,519 |
| 1 Gy + U0126 | 355,130 | 83,697 |
| 8 Gy + U0126 | 260,587 | 43,929 |
| 30 Gy + U0126 | 200,813 | 44,367 |
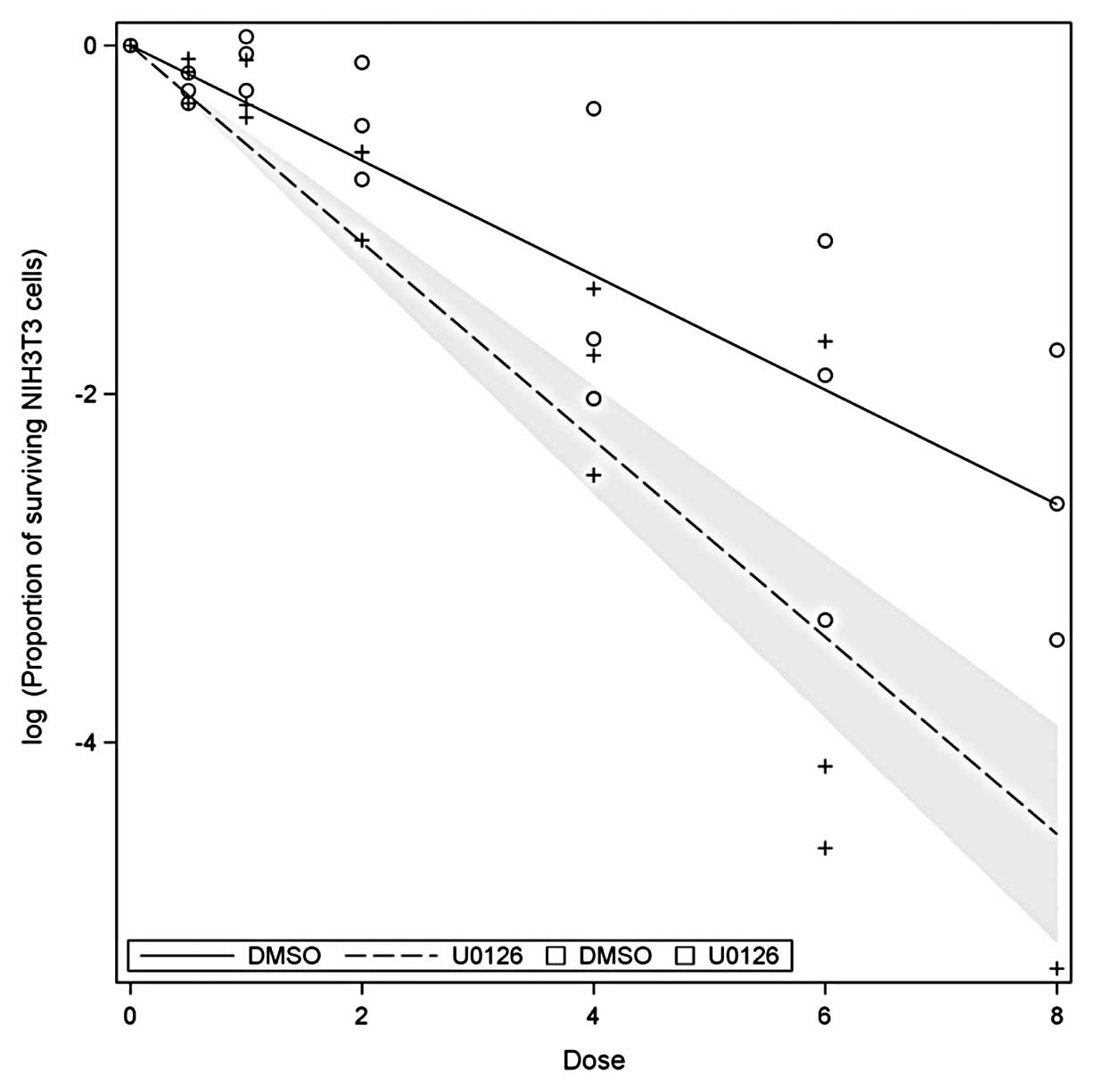
Impact of irradiation and U0126
application on clonogenic survival rates
Clonogenic assays of NIH3T3 cells were performed
with intensities ranging from 0.5 to 8 Gy and compared to
non-irradiated cultures. U0126 was added and compared to
unstimulated samples. When fitting a linear quadratic model to the
log (proportion) of the surviving cells, the corresponding p-values
are all >0.18. Using a simpler model involving only linear
terms, we found R2=0.82. The coefficients for DMSO and
U0126 can clearly be distinguished (p<0.0001). The resulting
functions are displayed in Fig.
2.
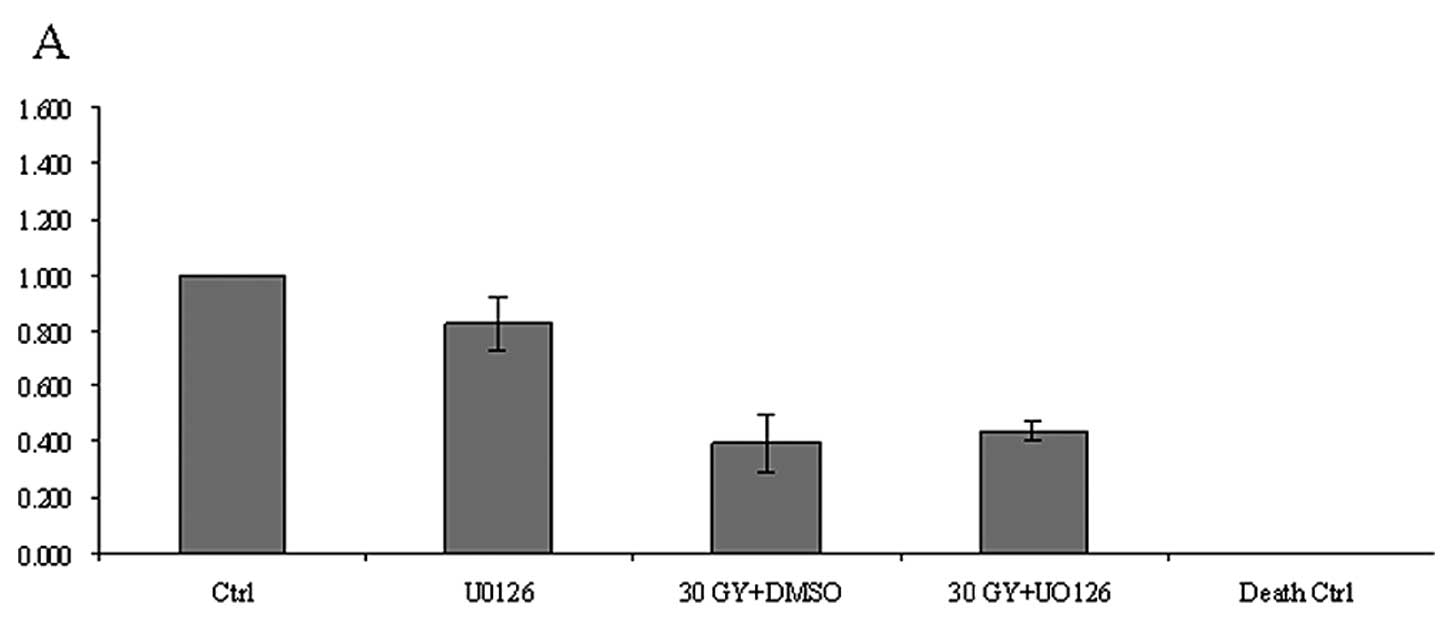
Irradiation and U0126 application impede
the proliferation of cancer cells and normal fibroblasts but not of
CAFs
There was no effect of IR or inhibitor treatment
seen after 24 h of incubation in proliferation (MTT) assays.
However, after 72 h, a dosage of 30 Gy caused a decrease in the
disposition of cells to proliferate in both inhibitor-treated and
untreated samples. Tumor cell lines HNSCCUM-02T and A549 both
showed minor decrease in their proliferative activity when treated
with U0126 without IR. When irradiated with 30 Gy, there was a
strong decrease of proliferating cells with and without inhibitor
application in both tumor cell types compared to the control
(HNSCCUM-02T: 30 Gy + DMSO: 61% decrease, 30 Gy + U0126: 56%
decrease; A549: 30 Gy + DMSO: 47% decrease, 30 Gy + U0126: 48%
decrease). The proliferation pattern of mouse embryonic
fibroblasts, NIH3T3, was similar to those of the cancer cell lines
(30 Gy + DMSO: 48% decrease, 30 Gy + U0126: 55% decrease,
respectively, compared to the control). However, fibroblasts
deriving from non-cancerous human tissue were affected to a lesser
extent when treated with 30 Gy ± U0126 (30 Gy + DMSO: 26% decrease,
30 Gy + U0126: 23% decrease, respectively, compared to the
control). However, when looking at the CAFs, we did not detect any
notable suppression of proliferative activity either by IR alone or
in combination with kinase inhibitor U0126 (30 Gy + DMSO: 5%
decrease, 30 Gy + U0126: 6% increase, respectively, compared to the
control) (Fig. 3). Table II shows the mean values and SEM of
densitometric measurements.
| Table IIProliferation assays (mean values and
SEM of densitometric analyses). |
Table II
Proliferation assays (mean values and
SEM of densitometric analyses).
| A, Effect of IR and
U0126 treatment on the proliferative activity of HNSCCUM-02T |
|---|
|
|---|
| HNSCCUM-02T | Mean value | SEM |
|---|
| Ctrl | 1 | 0 |
| U0126 | 0.824 | 0.1 |
| 30 Gy + DMSO | 0.394 | 0.1 |
| 30 Gy + UO126 | 0.437 | 0.03 |
| Death ctrl | −0.002 | 0 |
|
| B, Effect of IR and
U0126 treatment on the proliferative activity of A549 |
|
| A549 | Mean value | SEM |
|
| Ctrl | 1 | 0 |
| U0126 | 0.908 | 0.06 |
| 30 Gy + DMSO | 0.523 | 0.1 |
| 30 Gy + UO126 | 0.532 | 0.02 |
| Death ctrl | −0.003 | 0 |
|
| C, Effect of IR and
U0126 treatment on the proliferative activity of NIH3T3 |
|
| NIH3T3 | Mean value | SEM |
|
| Ctrl | 1 | 0 |
| U0126 | 0.969 | 0.16 |
| 30 Gy + DMSO | 0.525 | 0.1 |
| 30 Gy + UO126 | 0.447 | 0.06 |
| Death ctrl | −0.002 | 0 |
|
| D, Effect of IR and
U0126 treatment on the proliferative activity of normal
fibroblasts |
|
| Normal
fibroblasts | Mean value | SEM |
|
| Ctrl | 1 | 0 |
| U0126 | 1.165 | 0.32 |
| 30 Gy + DMSO | 0.745 | 0.13 |
| 30 Gy + UO126 | 0.775 | 0.12 |
| Death ctrl | −0.010 | 0.01 |
|
| E, Effect of IR and
U0126 treatment on the proliferative activity of cancer-associated
fibroblasts (CAFs) |
|
| CAFs | Mean value | SEM |
|
| Ctrl | 1 | 0 |
| U0126 | 0.934 | 0.18 |
| 30 Gy + DMSO | 0.951 | 0.2 |
| 30 Gy + UO126 | 1.063 | 0.18 |
| Death ctrl | −0.006 | 0.01 |
Discussion
Two main obstacles of radiotherapy are acquired
radioresistance in cancer cells under therapy and injury of
tumor-surrounding tissue. The Ras-MAPK-pathway has proven to be
instrumental in the development of radioresistance (9). As this cascade is considered to
generate a strong survival signal, specific inhibition of its
components is supposed to increase radiosensitivity. Our study
provides new evidence on the biological response of
tumor-surrounding cells, tumor and stroma cells exposed to
irradiation and MAPK inhibitor treatment. We found pERK expression
of malignant cells HNSCCUM-02T and A549 almost completely
suppressed by U0126 either alone or in combination with IR. In this
context, it was interesting to note that NIH3T3 fibroblasts showed
marginally diminished pERK levels only after treatment with the
maximum dose of 30 Gy plus inhibitor. Detrimental effects of
combination therapy on nearby healthy tissue might not be as
substantial as on the tumor mass itself. We recently demonstrated a
significantly decreased survival in colony assays for HNSCCUM-02T
after application of IR and U0126, whereas A549 showed increased
colony counts presumably due to an upstream K-Ras mutation and
activation of PI3K/Akt (1).
In the present study, we assayed the colony forming
ability of NIH3T3, which displayed analogous responses to
HNSCCUM-02T with a relevant reduction of colonies after combined
treatment. However, despite the fact that the colony forming
ability was suppressed by IR and U0126, the effect was comparable
to HNSCCUM-02T, as anticipated from western blotting data. MTT
assays revealed similar patterns in HNSCCUM-02T, A549, NIH3T3 and
normal fibroblasts. Proliferation was impeded by a dosage of 30 Gy
with or without U0126 in these lines. Combined application of IR
and MAPK inhibition sufficiently suppressed pERK levels in SCC
cells and did not exert any dose-limiting effects on fibroblasts,
which is an essential condition for the preservation of healthy
surrounding tissue under therapy. However, CAFs did not exhibit any
impairment of their proliferation activity, either by treatment
with maximum doses of IR or U0126 alone, or after application of
both.
In this regard, the responses of tumor-associated
fibroblasts to radiation and chemotherapy must be considered. The
tumor stroma and particularly CAFs are capable of modulating tumor
growth and determine the response to therapy (10). However, only limited data on the
potential impact of CAFs on therapy outcome are available. Overall,
very few studies have been undertaken using freshly isolated
fibroblasts from human cancer specimens (11,12).
It is known that breast CAFs stimulated by physiological
concentrations of estradiol rapidly induce ERK phosphorylation
mediated by estrogen receptor GPR30 (13). If increased expression levels of
activated ERK caused by physiological stimuli are existent in oral
CAFs as well, this might explain why proliferation of CAFs could
not be influenced by MAPK suppression and/or radiation in our
system. Beyond that, irradiated CAFs have been reported to release
soluble mediators that enhance the invasive potential in pancreatic
cancer cells (14). Thus, instead
of targeting them, IR seems to lend tumor promoting potential to
CAFs. CAFs appear to be intangible to combined therapy approaches
successfully affecting tumor cells. This might be due to the fact
that these cells are conditioned by their tumor environment in a
complex mutual interplay, possibly even gaining more resistance
towards therapy than tumor cells. Moreover, CAFs are known to
harbor a high frequency of genetic alterations that may have
already existed independently of the original tumor (15).
We may conclude from our findings that CAFs pose a
great challenge for novel strategies targeting cancer. Recent
studies even commend using CAFs as therapeutic targets for
different carcinoma types (16).
Additional studies are needed to better assess the role played by
CAFs in initiating and promoting tumorigenic alterations in cancer
cells as well as in modifying the response of tumors to therapy.
Enhanced understanding of the Gordian network of cancer cell-stroma
interactions and the signaling cascades involved in CAF-mediated
protection is the prerequisite to modulate treatment resistance of
tumor and microenvironment by designing appropriate multitargeting
strategies.
Acknowledgements
The authors thank Ms. Simone Mendler and Ms. Bettina
Mros for their excellent technical support. This study was funded
by a grant provided by the Foundation Head and Neck Tumor Research,
Wiesbaden, Germany.
References
|
1
|
Affolter A, Drigotas M, Fruth K, et al:
Increased radioresistance via G12S K-Ras by compensatory
upregulation of MAPK and PI3K pathways in epithelial cancer. Head
Neck. Feb 2–2012.(Epub ahead of print). View Article : Google Scholar
|
|
2
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2. Role in cell survival following oxidant
injury. J Biol Chem. 271:4138–4142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Affolter A, Fruth K, Brochhausen C,
Schmidtmann I, Mann WJ and Brieger J: Activation of
mitogen-activated protein kinase extracellular signal-related
kinase in head and neck squamous cell carcinomas after irradiation
as part of a rescue mechanism. Head Neck. 33:1448–1457. 2011.
View Article : Google Scholar
|
|
5
|
Bissell MJ and Radisky D: Putting tumours
in context. Nat Rev Cancer. 1:46–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsai KK, Chuang JJ, Little JB and Yuan ZM:
Cellular mechanisms for low-dose ionizing radiation-induced
perturbation of the breast tissue microenvironment. Cancer Res.
65:6734–6744. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsai KK, Stuart J, Chuang JJ, Little JB
and Yuan ZM: Low-dose radiation-induced senescent stromal
fibroblasts render nearby breast cancer cells radioresistant.
Radiat Res. 172:306–313. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Welkoborsky HJ, Jacob R, Riazimand SH,
Bernauer HS and Mann WJ: Molecular biologic characteristics of
seven new cell lines of squamous cell carcinomas of the head and
neck and comparison to fresh tumor tissue. Oncology. 65:60–71.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gupta AK, Bakanauskas VJ, Cerniglia GJ, et
al: The Ras radiation resistance pathway. Cancer Res. 61:4278–4282.
2001.PubMed/NCBI
|
|
10
|
Chometon G and Jendrossek V: Targeting the
tumour stroma to increase efficacy of chemo- and radiotherapy. Clin
Transl Oncol. 11:75–81. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hwang RF, Moore T, Arumugam T, et al:
Cancer-associated stromal fibroblasts promote pancreatic tumor
progression. Cancer Res. 68:918–926. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hawasawi NM, Ghebeh H, Hendrayani SF, et
al: Breast-carcinoma-associated fibroblasts and their counterparts
display neoplastic-specific changes. Cancer Res. 68:2717–2725.
2008. View Article : Google Scholar
|
|
13
|
Madeo A and Maggiolini M: Nuclear
alternate estrogen receptor GPR30 mediates 17β-estradiol-induced
gene expression and migration in breast cancer-associated
fibroblasts. Cancer Res. 70:6036–6046. 2010.PubMed/NCBI
|
|
14
|
Ohuchida K, Mizumoto K, Murakami M, et al:
Radiation to stromal fibroblasts increases invasiveness of
pancreatic cancer cells through tumor-stromal interactions. Cancer
Res. 64:3215–3222. 2004. View Article : Google Scholar
|
|
15
|
Littlepage LE, Egeblad M and Werb Z:
Coevolution of cancer and stromal cellular responses. Cancer Cell.
7:499–500. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar
|