Introduction
Epidermal growth factor (EGF), a crucial growth
factor, regulates cell proliferation and differentiation (1) by binding to its receptor, EGF receptor
(EGFR), on cell surfaces. Besides EGF, other ligands such as
transforming growth factor (TGF)-α and heparin-binding EGF-like
growth factor (HB-EGF) also bind to the EGFR (2–7). The
EGF activates a network of signal transduction pathways, including
activation of phosphoinositide 3-kinase (PI3K)/AKT (also known as
protein kinase B), rat sarcoma (RAS)/extracellular signal-regulated
kinase 1/2 (ERK1/2), and Janus kinase (JAK)/signal transducer and
activator of transcription (STAT) (8). These pathways regulate transcription
factors that control expression of proteins related to apoptosis
and proliferation, thereby inhibiting cell apoptosis and promoting
cell proliferation (8). EGF forms a
complex with the EGFR and then is translocated to nuclei to
activate gene expression (9,10).
Most cancer cells possess dysfunctional EGF signaling pathways that
promote cancer development, and these abnormal pathways are
exacerbated by mutations in the EGFR (11).
Cholangiocarcinomas are aggressive biliary neoplasms
arising within the intrahepatic or extrahepatic biliary tract. They
are the second most common type of primary liver cancer (12). An increasing incidence of
cholangiocarcinoma was documented. The incidence of
cholangiocarcinoma is rising, contributing to high mortality rates
due to a lack of effective therapeutic options. In total, 1/3 of
intrahepatic cholangiocarcinomas exhibit mutant KRAS and
aberrant p53 expression (13). These mutations confer resistance to
numerous chemotherapeutic agents in cholangiocarcinomas, yet there
is currently no standard therapy for this scenario (14,15).
In addition to these two gene mutations, the EGFR is highly
upregulated and mutated in patients with cholangiocarcinoma. In
total, ~85-90% of EGFR mutations occur in the tyrosine
kinase domain, while 10–15% occur in other domains (16). A total of 15% of patients with
cholangiocarcinoma display EGFR gene mutations in the
tyrosine kinase domain, leading to persistent activation of
downstream signaling, thereby enhancing cancer cell proliferation
(17). Additionally, 50% of
patients with cholangiocarcinoma exhibit EGFR overexpression, which
is correlated with cancer progression due to changes in the process
of the epithelial-mesenchymal transition (EMT) (11). In vitro studies revealed that
the EGF induces scattering of cholangiocarcinoma cells by
disrupting adherens junctions. EGF-stimulated cholangiocarcinoma
cells display internalization and decreased expression of
E-cadherin, as well as nuclear translocation of β-catenin (11).
The KRAS gene encodes a guanosine
triphosphate-binding protein, and this KRAS protein plays a crucial
role in downstream survival-promoting regulatory signaling pathways
connected to the EGFR (18). A
KRAS mutation leads to its constitutive activation, thereby
triggering activation of downstream signaling pathways, such as
rapidly accelerated fibrosarcoma/mitogen-activated protein kinase
(MAPK) kinase (mitogen-activated protein kinase)/ERK and
PI3K/AKT/mammalian target of rapamycin (mTOR), which promote cell
proliferation (19). In patients
treated with gefitinib, an EGFR inhibitor, AKT activation
correlates with disease progression in KRAS wild-type (WT)
lung adenocarcinomas. In KRAS mutant cells, administering
insulin-like growth factor 1 receptor tyrosine kinase inhibitors
may attenuate AKT signaling and potentially restore sensitivity to
gefitinib (20). Gefitinib can act
as a radiosensitizer, enhancing the radiological response of cancer
cells by inhibiting EGFR phosphorylation and activation of
subsequent downstream pathways, thereby increasing the
radiosensitivity of cholangiocarcinoma cells (21,22).
Hence, identifying alternative compounds capable of inhibiting the
cancer progression of KRAS-mutated cholangiocarcinoma cells
is crucial.
Heteronemin (Haimian jing) is a natural
marine product extracted from Hippospongia sp., a
sesterterpenoid-type secondary metabolite found in marine sponges
(23). This metabolite has
anticancer properties in several types of cancers through different
signal transduction pathways (24–28).
It was found to confer protection against carcinogenesis in
cholangiocarcinomas, prostate cancer and acute myeloid leukemia
(24–29). Heteronemin treatment induces
apoptosis via the caspase pathway and promotes the formation of
reactive oxygen species (ROS) to trigger their removal by
mitochondrial superoxide dismutase 2 (SOD2) rather than cytosolic
SOD1 (26). ROS-induced cell death
is associated with the MAPK signaling pathway. Heteronemin induces
apoptosis in hepatocellular carcinoma cells by downregulating
ERK1/2 expression and activating the p38/c-Jun N-terminal kinase
(JNK) signaling pathways (29).
Additionally, it inhibits the proliferation of colorectal cancer
cells by blocking EGF-induced PD-L1 expression through the
TGF-β1/ERK1/2 pathway (25). Given
these diverse anticancer mechanisms, it is possible that
heteronemin could have inhibitory effects on EGFR regulation in
cholangiocarcinoma cells.
In the present study, it was investigated how EGFR
regulates the proliferation of cholangiocarcinoma cells through
specific signal transduction pathways. Then, it was explored
whether and how heteronemin inhibits EGF-induced proliferation in
cholangiocarcinoma cells.
Materials and methods
Cell cultures
Two human cholangiocarcinoma cell lines, KRAS
WT SSP-25 and KRAS mutant HuCCT1, were obtained from Riken
Bioresource Research Center and were authenticated by a
next-generation sequencing (NGS) analysis. Based on the NGS
analysis, the results indicated that the intrahepatic
cholangiocarcinoma, SSP-25 cell is ETK-1:TP53; Simple; p.Arg175His
(c.524G>A) correlated to results shown on Cellosaurus website
(https://www.cellosaurus.org/). Cells
were cultured in RPMI-1640 medium (cat. no. 31800022; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(cat. no. SH30396.03; Hyclone; Cytiva) in a humidified incubator
with 5% CO2 at 37°C.
Cell treatment
After cell seeding, the cells were placed in a 0.25%
hormone-depleted, serum-supplemented medium for 2 days to induce
serum starvation. Following this, the cells were supplemented with
fresh medium containing 5% hormone-depleted serum, along with EGF
(cat. no. E9644; Sigma-Aldrich; Merck KGaA), heteronemin (purity
>98%, cat. no. 258814; Sigma-Aldrich; Merck KGaA), or LY294002
(cat. no. S1105; Selleck Chemicals) at varying concentrations and
treatment times according to the experimental design. Cells that
did not receive LY294002 or heteronemin were treated with dimethyl
sulfoxide (cat. no. D2650; Sigma-Aldrich; Merck KGaA), while cells
that did not receive EGF were treated with phosphate-buffered
saline.
Reverse-transcription quantitative PCR
(RT-qPCR)
SSP-25 and HuCCT1 cells were seeded at a density of
2×105 cells/well in six-well plates. After seeding, the
medium was replaced with 0.25% hormone-depleted serum-supplemented
medium for 48 h. The hormone-depleted serum was prepared as
previously described (30), the
hormones were removed by AG® 1–8X resin (Bio-Rad
Laboratories, Inc.). Cells were supplemented with fresh medium
containing 5% hormone-depleted serum with different concentrations
of heteronemin (0.6, 1.25, 2.5, 5 and 10 µM) for 24 h. Total RNA
was extracted and genomic DNA was removed with an Illustra RNAspin
Mini RNA Isolation Kit (Cytiva). Deoxyribonuclease I-treated total
RNA (1 µg) was reverse-transcribed into cDNA using a RevertAid H
Minus First Strand Complementary (c)DNA Synthesis Kit (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
cDNAs were used as the template for the qPCR analysis. The
thermocycling conditions for qPCR were 95°C for 3 min followed by
40 cycles of 95°C for 5 sec and 60°C for 10 sec. qPCRs were
conducted using a QuantiNovaTM SYBR® Green PCR Kit
(Qiagen) on a CFX Connect™ Real-Time PCR Detection
System (Bio-Rad Laboratories, Inc.). Primer sequences were as
follows: Homo sapiens cyclin D1 (CCND1) forward,
5′-CAAGGCCTGAACCTGAGGAG-3′ and reverse, 5′-GATCACTCTGGAGAGGAAGCG-3′
(accession no. NM_053056); H. sapiens proliferating cell
nuclear antigen (PCNA) forward, 5′-TCTGAGGGCTTCGACACCTA-3′
and reverse, 5′-TCATTGCCGGCGCATTTTAG-3′ (accession no. BC062439.1);
H. sapiens cytosol MYC proto-oncogene, bHLH transcription
factor (c-Myc) forward, 5′-TTCGGGTAGTGGAAAACCAG-3′ and
reverse, 5′-CAGCAGCTCGAATTTCTTCC-3′ (accession no. NM_002467.4);
H. sapiens EGFR forward, 5′-AATTTACAGGAAATCCTGCATGGC-3′ and
reverse, 5′-GATGCTCTCCACGTTGCACA-3′ (accession no. NM_005228); and
H. sapiens 18S ribosomal RNA (18S rRNA), forward,
5′-GTAACCCGTTGAACCCCATT-3′ and reverse, 5′-CCATCCAATCGGTAGTAGCG-3′
(accession no. NR_003286.2). Calculations of relative gene
expression (normalized to the 18S rRNA reference gene) were
performed according to the 2−ΔΔCq method (31). The fidelity of the PCR was
determined with a melting temperature analysis.
Western blotting
To examine signal transduction pathways involved in
EGF-induced PD-L1 expression and effects of heteronemin on
EGF-induced PD-L1 expression in SSP-25 and HuCCT1 cells, a western
blot analysis was performed to detect protein expression levels of
PD-L1 and activation of PI3K, STAT1 and STAT3 in total cell lysates
of cells that had been treated with the vehicle for 24 h. Protein
concentration was determined by bicinchoninic acid method and
protein samples were resolved by 10% sodium dodecyl-sulfate
polyacrylamide gel electrophoresis. A 20-µg quantity of protein was
loaded into each well with sample buffer, and samples were resolved
by electrophoresis at 100 V for 2 h. The resolved proteins were
transferred from the polyacrylamide gel to Millipore Immobilon-PSQ
Transfer polyvinylidene difluoride membranes (MilliporeSigma) with
the Mini Trans-Blot® Cell (Bio-Rad Laboratories, Inc.).
Membranes were blocked with a solution of 3% bovine serum albumin
(cat. no. A7030; MilliporeSigma) in Tris-buffered saline (TBS) with
0.1% Tween-20 (TBST) at room temperature for 1 h and incubated with
primary antibodies to PD-L1 (1:1,000; cat. no. GTX104763; GeneTex
International Corporation), PI3K (1:1,000; cat. no. 610046; BD
Biosciences), phosphorylated (p)-PI3K p85 (Tyr458)/p55 (Tyr199)
(1:3,000; cat. no. 4228; Cell Signaling Technology, Inc.), STAT1
(1:1,000; cat. no. 66545-1-1g; Proteintech Group, Inc.), p-STAT1
(Tyr701) (1:1,000; cat. no. 9167, Cell Signaling Technology, Inc.),
AKT (1:1,000; cat. no. 60203; Proteintech Group, Inc.), p-AKT
(Ser473) (1:1,000; cat. no. 9271, Cell Signaling Technology, Inc.),
STAT3 (1:1,000; cat. no. 610190; BD Biosciences), p-STAT3 (Tyr705)
(1:1,000; cat. no. 9145, Cell Signaling Technology, Inc.) and GAPDH
(1:20,000; cat. no. 60004-1, GeneTex International Corporation) at
4°C overnight. The antibody-probed membrane was washed with TBST
containing 5% fat-free milk (5% TBST/milk) three times for 10 min
and then probed with goat anti-mouse immunoglobulin G (IgG) (cat.
no. GTX213111-05) or goat anti-rabbit IgG (cat. no. GTX213110-04;
both from GeneTex International Corporation) horseradish peroxidase
(HRP)-conjugated secondary antibodies, which were prepared in 5%
TBST/milk at a 1:20,000 dilution at room temperature for 1 h. After
the membrane was washed three times for 10 min with TBS,
chemiluminescent detection was performed using the Immobilon
Western Chemiluminescent HRP Substrate (MilliporeSigma). Bands were
imaged with the BioSpectrum Imaging System (UVP) and quantified
using densitometry by ImageJ 1.47 software (National Institutes of
Health) according to the software instructions.
Cell viability assay
SSP-25 and HuCCT1 cells were seeded in 96-well
plates at a density of 3,000 cells/well. After 24 h for cell
attachment, cells were starved with 0.25% hormone-depleted
serum-supplemented medium for 48 h. Then, serum-starved cells were
treated with various concentrations of EGF (10, 20, 40 and 100
ng/ml) and heteronemin (0.3, 0.6, 1.25, 2.5, 5, 10 µM) or 10 µM
LY294002 in 5% hormone-depleted serum-supplemented medium for 72 h.
Medium and reagents were refreshed daily. Cell viability was
assayed with a Cell Counting Kit-8 (cat. no. 96992; Sigma-Aldrich;
Merck KGaA), according to the manufacturer's protocol. Briefly,
cells were incubated with 100 µl/well CCK-8 working solution at
37°C for 1 h, and the absorbance was measured at 450 nm.
Statistical analysis
In the present study, the statistical significance
of all data was analyzed using a one-way ANOVA followed by Tukey's
post hoc test using the SigmaPlot 14.5 software (Systat Software,
Inc.). All data are presented as the mean ± standard deviation
(SD). P<0.05 was considered to indicate a statistically
significant difference.
Results
EGF induces signal transduction and
cancer cell proliferation in cholangiocarcinoma cells
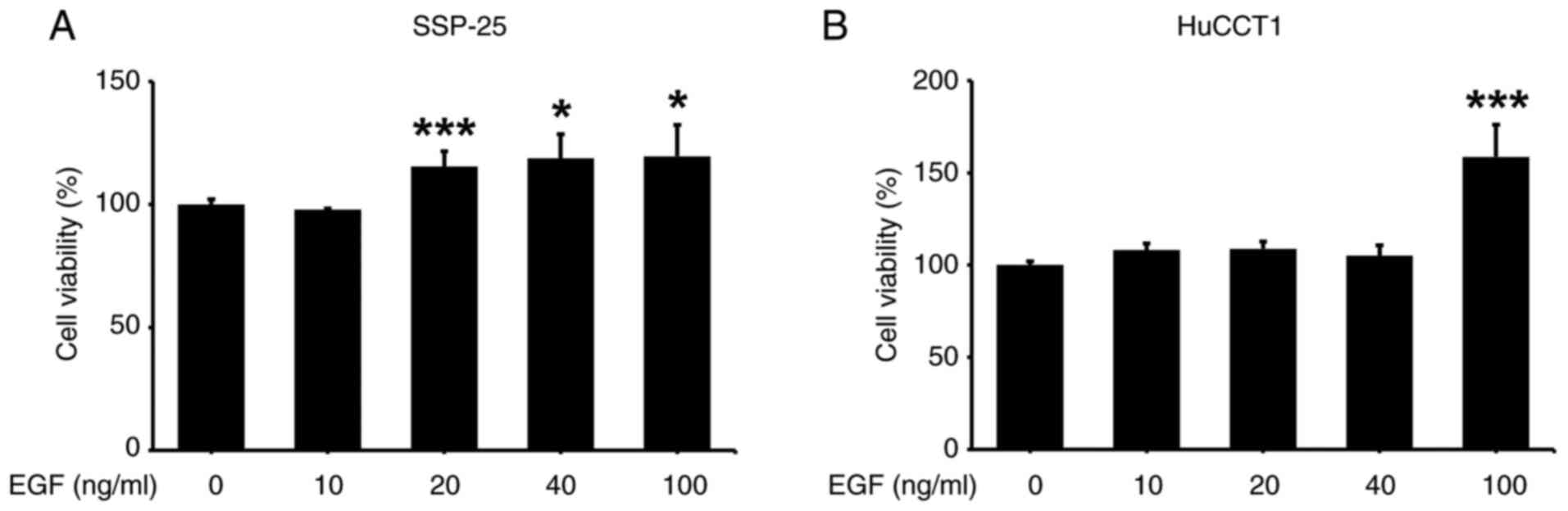
EGF stimulates cancer growth via binding to its
receptor, EGFR. Although it was reported that EGF induced growth of
the KMBC and Witt cholangiocarcinoma cell lines (32), it is unclear how cholangiocarcinoma
cells with different KRAS statuses respond to EGF stimulation. In
the present study, the proliferative effect of activated EGFR was
first examined on two cholangiocarcinoma cell lines, KRAS WT
SSP-25 and KRAS mutant HuCCT1, by stimulating them with
various EGF concentrations.
SSP-25 and HuCCT1 cells were stimulated with
different EGF concentrations for 24, 48 and 72 h to examine the
effect of EGF on their proliferation. EGF induced cell
proliferation in SSP-25 cells starting at 20 ng/ml during the 24 to
72 h treatment period (Figs. S1A
and 1A), whereas HuCCT1 cell
proliferation was significantly stimulated only at 100 ng/ml during
the same period (Figs. S1B and
1B). Although stimulation with EGF
for 24 h significantly induced cell proliferation, the noticeable
fold changes compared with the control occurred only at 72 h. Given
that EGF-stimulated cell proliferation initiates between 24 and 72
h, and knowing that growth factor stimulation leads to rapid
changes in mRNA and protein expression-often within 24 h, which is
sufficient to capture immediate signaling events-24 h were selected
to assess mRNA and protein expression and 72 h to evaluate cell
viability.
Signaling pathways activated by EGF in these two
cholangiocarcinoma cell lines were next examined. In SSP-25 cells,
EGF induced the phosphorylation of PI3K at 40 ng/ml and the
phosphorylation of STAT1 in the range of 20–40 ng/ml without
altering STAT1 expression (Fig.
2A), reflecting different mechanisms that regulate the
expression of STAT1 and the phosphorylation of STAT1. Unexpectedly,
the phosphorylation of STAT1 was decreased at 100 ng/ml EGF
treatment. This reduction may be due to an increase in negative
regulators of the STAT pathway and the induction of phosphatases
such as Src homology region 2 domain-containing phosphatase-2
(33), leading to rapid
dephosphorylation of STAT1 at Y701. Besides, EGF inhibited the
activation of another STAT member, STAT3, by suppressing its
phosphorylation without affecting its protein level (Fig. 2B). Unlike its effect in SSP-25
cells, EGF stimulation of HuCCT1 cells increased the
phosphorylation of PI3K and STAT3 in concentration-dependent
manners (Fig. 2C). EGF did not
obviously alter STAT1 expression but significantly suppressed STAT1
phosphorylation (Fig. 2C).
Additionally, EGF even at the lowest concentration stimulated PD-L1
expression, a proliferation-related protein, in both cell lines
(Fig. 2B and C). Thus, EGF
stimulation activates PI3K/STAT1 signaling to induce the
proliferation of SSP-25 cells and PI3K/STAT3 signaling to induce
the proliferation of HuCCT1 cells. To optimize the effect of
heteronemin in both cell lines under EGF stimulation, 100 ng/ml EGF
was chosen for the following experiments.
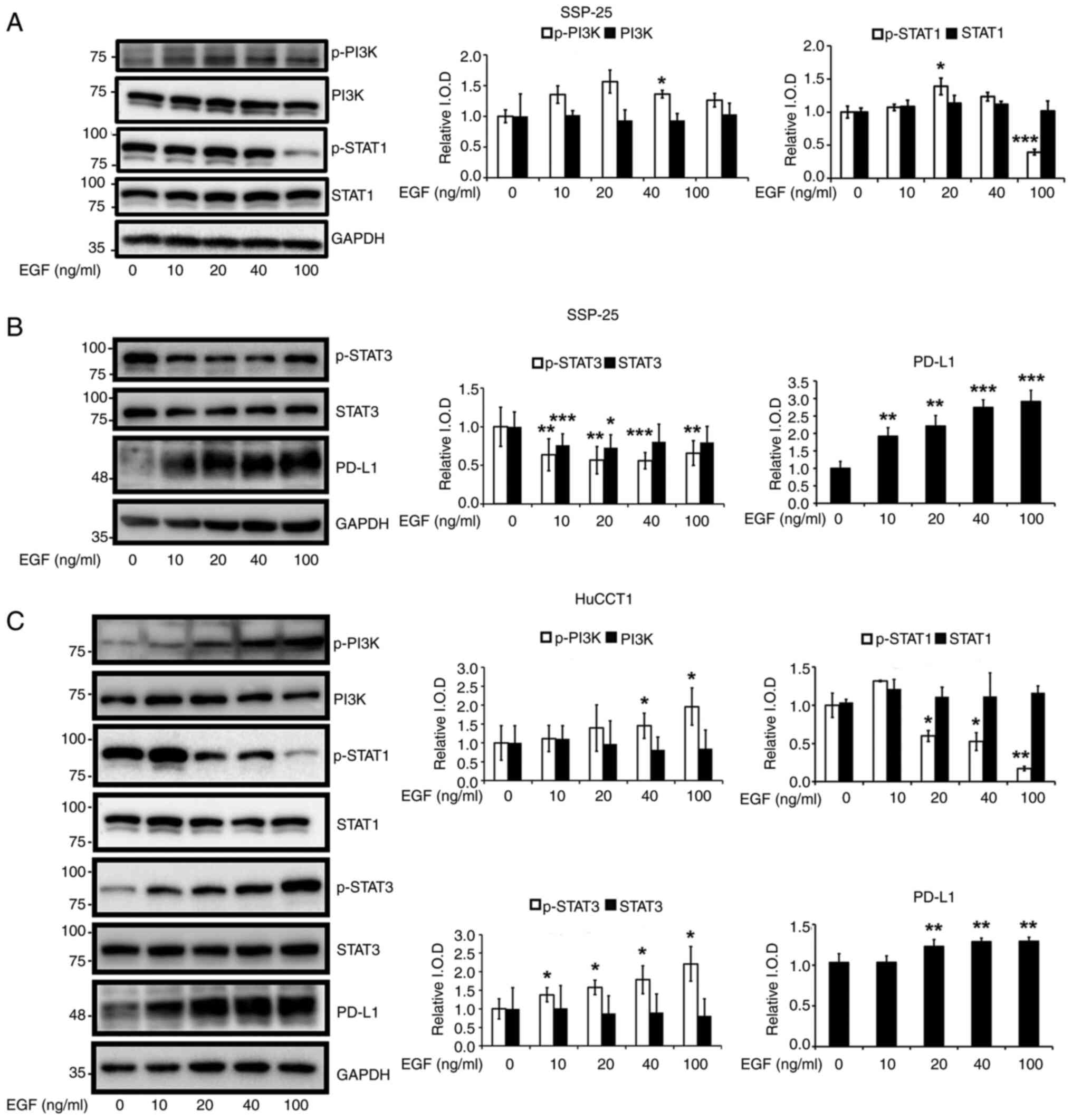 | Figure 2.EGF modulates signal transduction
pathways in cholangiocarcinoma cells. (A-C) Serum-starved (A and B)
SSP-25 and (C) HuCCT1 cells were left unstimulated or were
stimulated with various concentrations of the EGF (10, 20, 40 and
100 ng/ml) for 24 h. Cells that did not receive stimulation with
EGF were instead treated with PBS. After stimulation, the cells
were lysed, and cell lysates were subjected to western blotting to
detect p-PI3K, PI3K, p-STAT1, STAT1, p-STAT3, STAT3 and PD-L1;
GAPDH was used as a loading control for protein normalization.
Quantitative results are expressed as relative IODs by defining the
amounts of the indicated detected proteins in unstimulated cells as
1. Data are presented as the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and ***P<0.001 compared
with unstimulated cells. EGF, epidermal growth factor; p-,
phosphorylated; PD-L1, programmed cell death-ligand 1; IOD,
integrated optical density. |
Heteronemin inhibits cell
proliferation in cholangiocarcinoma cells by downregulating
expression of proliferation-related genes
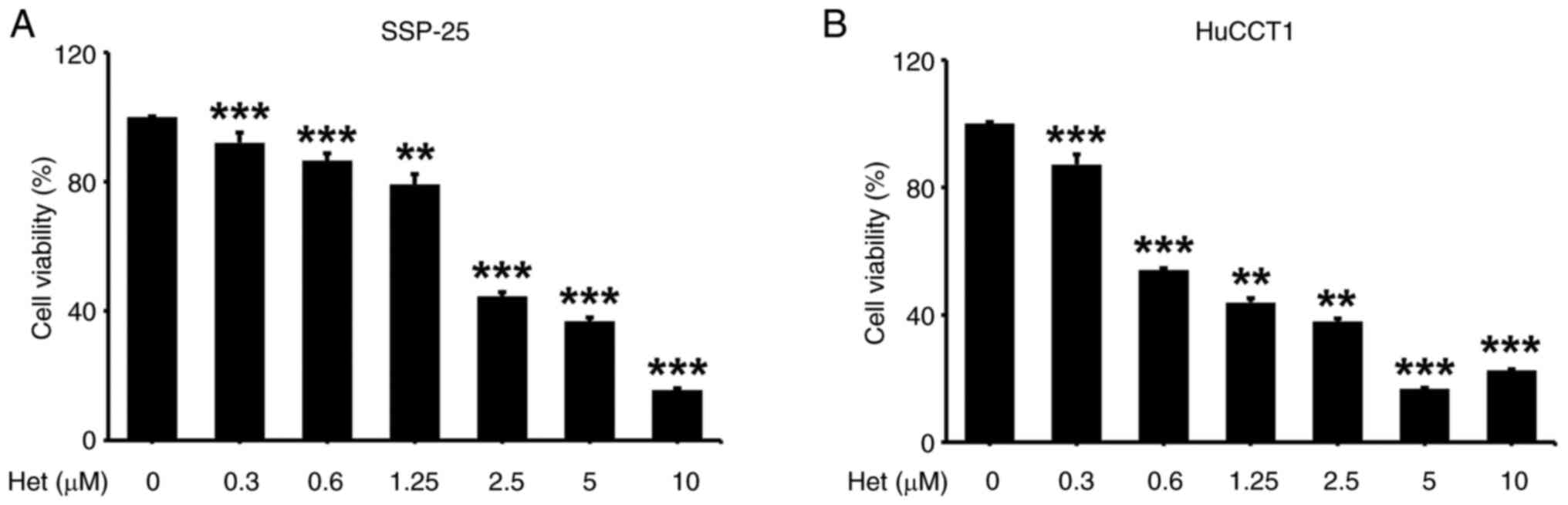
Heteronemin was shown to have antiproliferative
effects in various cancer cell lines, including cholangiocarcinoma
(23–28). However, signal transduction pathways
underlying how heteronemin causes this antiproliferative effect in
cholangiocarcinoma cells remain unknown. The proliferation
inhibitory effects of heteronemin were examined on two different
types of cholangiocarcinoma cells, SSP-25 and HuCCT1, by treating
them with various concentrations of heteronemin for 72 h.
Heteronemin caused significant, concentration-dependent cytotoxic
effects in both cell lines, starting at 0.3 µM (Fig. 3).
According to the functions of the
proliferation-related genes PCNA, CCND1 and c-Myc,
these three common proliferation genes were selected to assess the
proliferation inhibitory effect of heteronemin on
cholangiocarcinoma cells (34).
PCNA is crucial for DNA replication, CCND1 regulates
the cell cycle and is linked to cancer progression, and
c-Myc acts as a master regulator of metabolism and
proliferation, driving malignant transformation through various
oncogenic pathways. To explore the suppression of proliferation
effects by heteronemin in SSP-25 and HuCCT1 cells, particularly
related to the differential expression of PCNA, CCND1 and
c-Myc, SSP-25 and HuCCT1 cells were treated with the
indicated concentrations of heteronemin for 24 h. In SSP-25 cells,
heteronemin downregulated PCNA and CCND1 gene
expression in concentration-dependent manners (Fig. 4A). The expression of these two genes
in HuCCT1 cells was suppressed by ~90% under heteronemin treatment
in the range of 0.6–5 µM (Fig. 4B).
However, in SSP-25 and HuCCT1 cells, different concentrations of
heteronemin showed contrasting effects on c-Myc expression.
Lower concentrations (0.6 and 1.25 µM) of heteronemin exhibited an
increased effect. By contrast, higher concentrations (5 and 10 µM)
inhibited expression of this gene (Fig.
4). In addition, the effect of heteronemin was also examined on
EGFR expression in both cell lines. Only 5 and 10 µM
heteronemin inhibited EGFR expression in SSP-25 cells
(Fig. 4A); however, it
significantly suppressed EGFR expression in HuCCT1 cells
even at the lowest concentration (0.6 µM) (Fig. 4B). The different concentrations of
heteronemin show distinct effects on the gene expression of
CCND1, c-Myc and EGFR, which might be due to
different binding affinities. Low concentrations of heteronemin
effectively block PCNA expression through a specific target
molecule. However, the target molecules regulating CCND1,
c-Myc and EGFR have low affinity for heteronemin, which
may explain the opposite effects of low and high concentrations on
their mRNA expression. Thus, these results indicated that
heteronemin reduces expression of the PCNA, CCND1, c-Myc and
EGFR proliferation-related genes in both cholangiocarcinoma
cell lines. Additionally, KRAS mutant HuCCT1 cells exhibited
greater sensitivity to heteronemin treatment.
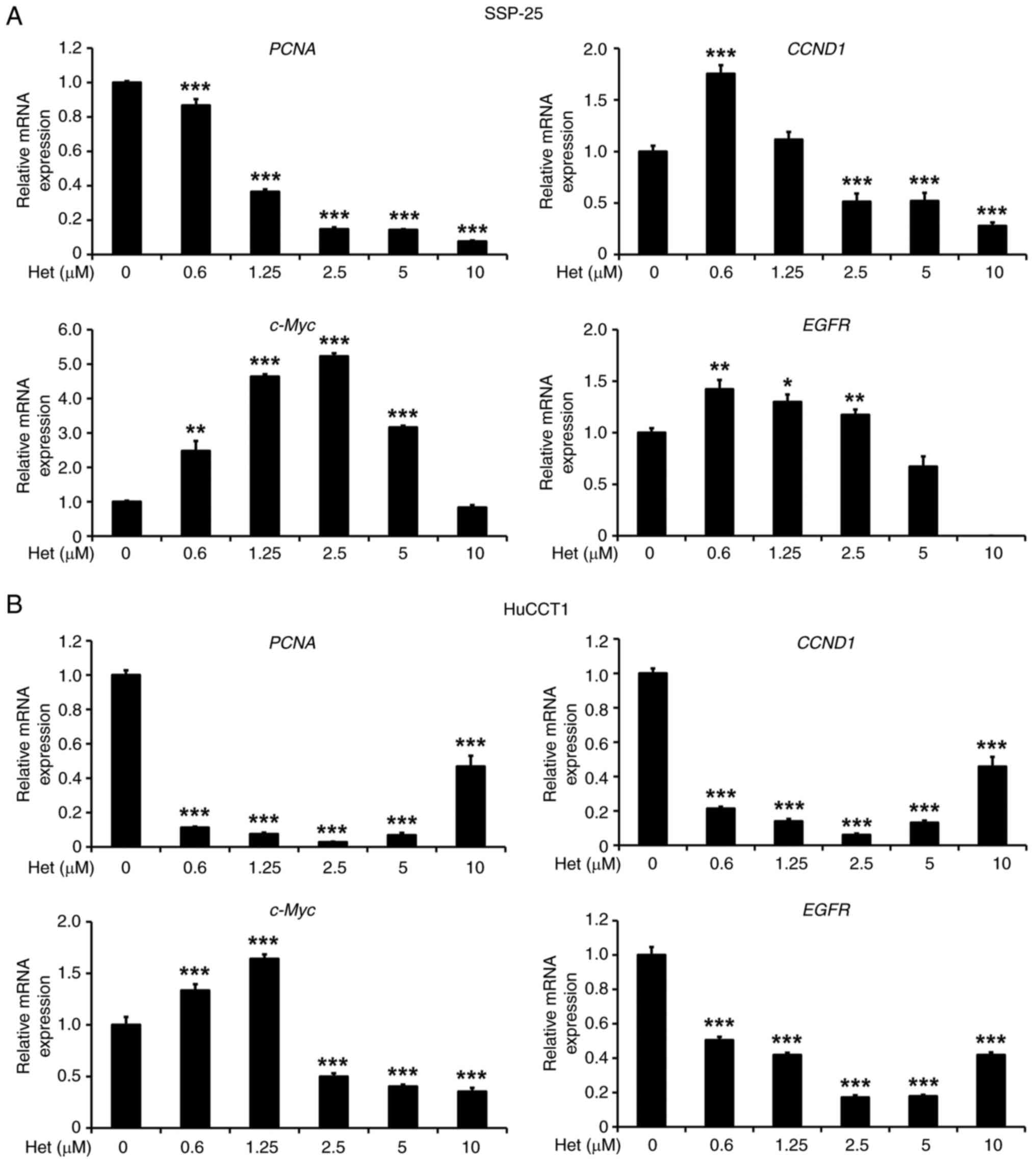 | Figure 4.Heteronemin inhibits expression of
proliferation-related genes in cholangiocarcinoma cells. (A and B)
Serum-starved (A) SSP-25 and (B) HuCCT1 cells were left untreated
or were treated with various concentrations of heteronemin (0.3,
0.6, 1.25, 2.5, 5 and 10 µM) for 24 h. Cells that did not receive
treatment with heteronemin were instead treated with DMSO. After
treatment, the cells were lysed, and mRNAs extracted from cell
lysates were subjected to a reverse-transcription reaction. mRNA
expression of PCNA, CCND1, c-Myc and EGFR, were
quantified by reverse transcription-quantitative PCR; 18S
rRNA was used as a loading control. mRNA expression of these
genes were normalized to that of 18S rRNA. Quantitative
values are expressed as relative mRNA levels by defining levels of
gene expression in the untreated group of each cell line as 1. Data
are presented as the mean ± SD of quadruplicate wells in three
independent experiments. *P<0.05, **P<0.01 and ***P<0.001
compared with untreated cells. PCNA, proliferating cell nuclear
antigen; CCND1, cyclin D1; EGFR, epidermal growth factor receptor;
18S rRNA, 18S ribosomal RNA. |
Heteronemin alters EGF-induced signal
transduction in cholangiocarcinoma
Activation of the EGFR signal transduction pathway
induces cellular motility (35) and
promotes cancer cell proliferation (1,11).
Recently, it was revealed by the authors that heteronemin inhibited
the EGFR signal transduction pathway to downregulate the
proliferation of colorectal cancer cells (25). To further investigate whether
heteronemin suppresses signal transduction pathways activated by
EGF in cholangiocarcinoma cells, SSP-25 and HuCCT1 cells were
stimulated with 100 ng/ml EGF in the presence and absence of
indicated concentrations of heteronemin for 24 h. In SSP-25 cells,
PI3K and its phosphorylated forms increased in heteronemin-treated
cells in concentration-dependent manners up to 1.25 µM (Fig. 5). After treatment of this cell line
with 1.25 and 2.5 µM heteronemin, STAT1 phosphorylation
significantly increased compared with untreated cells. In contrast
to PI3K phosphorylation, the phosphorylation pattern of STAT3
decreased in heteronemin-treated cells in a concentration-dependent
manner. Heteronemin inhibited the EGF-induced phosphorylation of
PI3K in SSP-25 cells (Fig. 5). The
phosphorylation of both STAT1 and STAT3 increased in
concentration-dependent manners under heteronemin treatment in
EGF-stimulated cells, with the exception of the effect of 2.5 µM
heteronemin on STAT3.
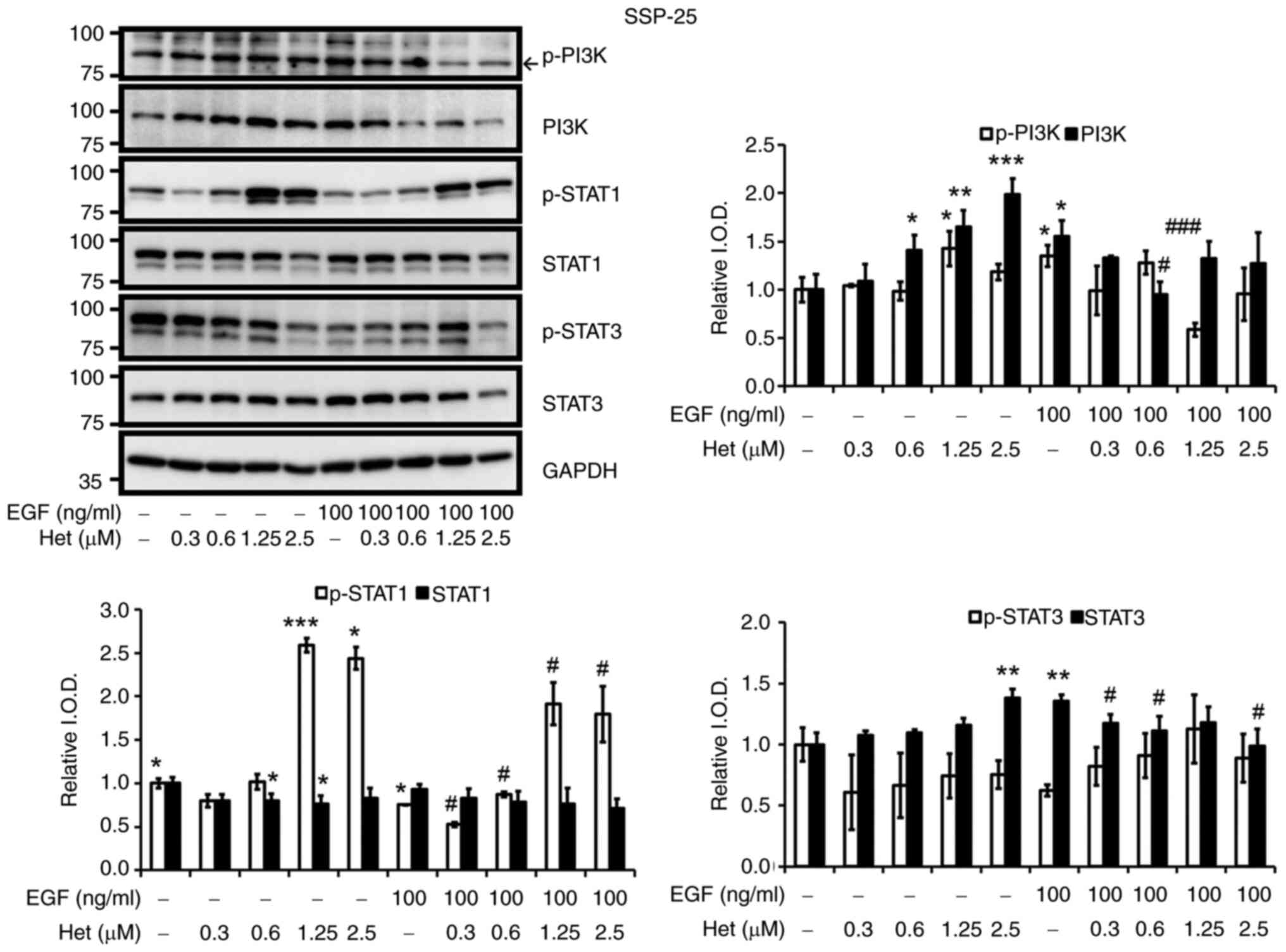 | Figure 5.Heteronemin affects EGF-induced PI3K,
STAT1 and STAT3 activation in SSP-25 cholangiocarcinoma cells.
Serum-starved SSP-25 cells were left untreated or were treated with
various concentrations of heteronemin (0.3, 0.6, 1.25 and 2.5 µM)
together with or without 100 ng/ml EGF for 24 h. Cells that did not
receive treatment with heteronemin and/or EGF were instead treated
with DMSO and/or PBS, respectively. After treatment, the cells were
lysed, and cell lysates were subjected to western blotting to
detect p-PI3K, PI3K, p-STAT1, STAT1, p-STAT3 and STAT3; GAPDH was
used as a loading control for protein normalization. Quantitative
results are expressed as relative IOD by defining amounts of the
indicated detected proteins in unstimulated cells in the absence of
heteronemin treatment to be 1. Data are presented as the mean ± SD
of three independent experiments. *P<0.05, **P<0.01 and
***P<0.001 compared with untreated cells, in the absence of
heteronemin treatment. #P<0.05 and
###P<0.001 compared with EGF-stimulated cells, in the
absence of heteronemin treatment. EGF, epidermal growth factor;
PI3K, phosphoinositide 3-kinase; STAT, STAT, signal transducer and
activator of transcription; p-, phosphorylated; IOD, integrated
optical density. |
A parallel experiment was conducted in HuCCT-1
cells, and it was found that heteronemin suppressed EGF-induced
signal transduction pathways. Heteronemin at up to 1.25 µM induced
the phosphorylation of PI3K, STAT1 and STAT3 in HuCCT-1 cells
(Fig. 6). On the other hand, 100
ng/ml EGF increased concentrations of PI3K, STAT1 and STAT3, and
their phosphorylated forms, compared with unstimulated cells.
Heteronemin inhibited the EGF-induced phosphorylation of PI3K,
STAT1 and STAT3 in concentration-dependent manners (Fig. 6). In addition, PI3K, STAT1 and STAT3
levels decreased under treatment with 2.5 µM heteronemin in HuCCT1
cells. To confirm that PI3K plays an important role in regulating
signaling pathways induced by the EGF in SSP-25 and HuCCT1 cells,
10 µM LY294002, an inhibitor of PI3K, was applied to EGF-stimulated
cells. As shown in Fig. S2,
inhibition of PI3K activity decreased levels of PD-L1 and p-STAT1
and p-STAT3 in both EGF-stimulated cell lines. This pattern was
similar to the effect of heteronemin on EGF-stimulated SSP-25 and
HuCCT1 cells. These results indicated that heteronemin inhibits the
proliferation of SSP-25 and HuCCT1 cells by suppressing the
PI3K-mediated signaling pathways induced by EGF.
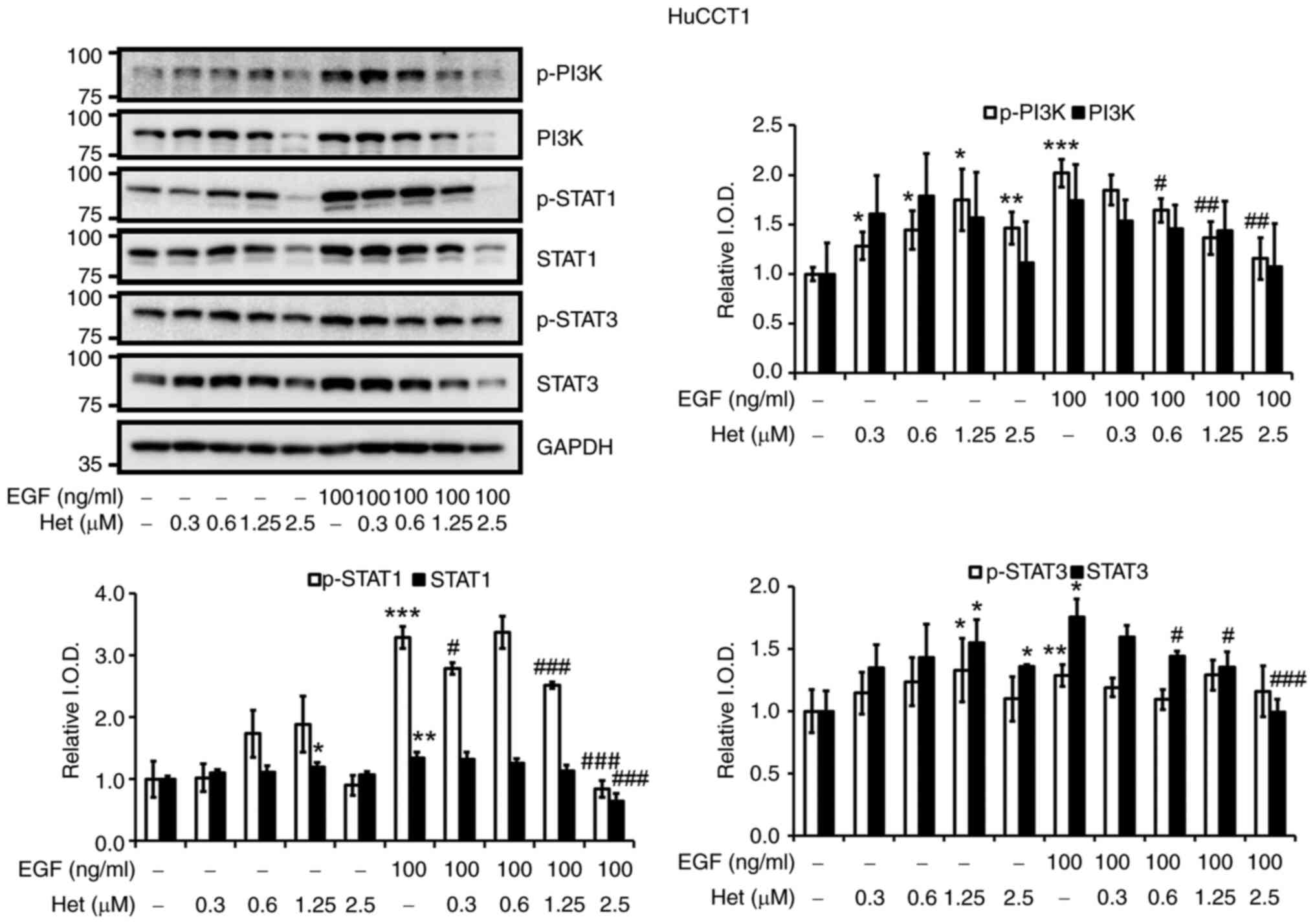 | Figure 6.Heteronemin affects EGF-induced
activation of PI3K, STAT1 and STAT3 in HuCCT1 cholangiocarcinoma
cells. Serum-starved HuCCT1 cells were left untreated or were
treated with various concentrations of heteronemin (0.3, 0.6, 1.25,
and 2.5 µM) together with or without 100 ng/ml EGF for 24 h. Cells
that did not receive treatment with heteronemin and/or EGF were
instead treated with DMSO and/or PBS, respectively. After
treatment, the cells were lysed, and cell lysates were subjected to
western blotting to detect p-PI3K, PI3K, p-STAT1, STAT1, p-STAT3
and STAT3; GAPDH was used as a loading control for protein
normalization. Quantitative results are expressed as relative IOD
by defining amounts of the indicated detected proteins in
unstimulated cells, in the absence of heteronemin treatment to be
1. Data are presented as the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and ***P<0.001 compared
with untreated cells, in the absence of heteronemin treatment.
#P<0.05, ##P<0.01 and
###P<0.001 compared with EGF-stimulated cells, in the
absence of heteronemin treatment. EGF, epidermal growth factor;
PI3K, phosphoinositide 3-kinase; STAT, STAT, signal transducer and
activator of transcription; p-, phosphorylated; IOD, integrated
optical density. |
Heteronemin inhibits EGF-induced PD-L1
expression and cell proliferation in cholangiocarcinoma cells
As demonstrated in Figs.
1 and 2, PI3K may play a
crucial role in promoting EGF-triggered proliferation in
cholangiocarcinoma cells. Moreover, EGF led to a
concentration-dependent elevation in PD-L1 expression, as depicted
in Fig. 1. Notably, heteronemin was
observed to suppress EGF-induced PI3K activation (Figs. 5 and 6). To determine whether heteronemin
inhibits the proliferation of cholangiocarcinoma cells via the PI3K
signaling transduction pathway activated by the EGF, PD-L1
expression in SSP-25 and HuCCT1 cells was examined by stimulating
them with 100 ng/ml of EGF in the presence of different
concentrations of heteronemin for 24 h. Heteronemin treatment
increased PD-L1 expression in SSP-25 cells and decreased PD-L1
expression in HuCCT1 cells in concentration-dependent manners
(Fig. 7). EGF induced PD-L1
expression in both cholangiocarcinoma cell lines, and this effect
was concentration-dependently inhibited by heteronemin (Fig. 7).
 | Figure 7.Heteronemin blocks EGF-induced PD-L1
expression in cholangiocarcinoma cells. (A and B) Serum-starved (A)
SSP-25 and (B) HuCCT1 cells were left untreated or were treated
with various concentrations of heteronemin (0.3, 0.6, 1.25 and 2.5
µM) together with or without 100 ng/ml EGF for 24 h. Cells that did
not receive treatment with heteronemin and/or EGF were instead
treated with DMSO and/or PBS, respectively. After treatment, the
cells were lysed, and cell lysates were subjected to western
blotting to detect PD-L1; GAPDH was used as a loading control for
protein normalization. Quantitative results are expressed as
relative IOD by defining the amounts of PD-L1 in untreated cells,
in the absence of heteronemin treatment to be 1. Data are presented
as the mean ± SD of three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 compared with untreated cells, in the
absence of heteronemin treatment. #P<0.05,
##P<0.01 and ###P<0.001 compared with
EGF-stimulated cells, in the absence of heteronemin treatment. EGF,
epidermal growth factor; PD-L1, programmed cell death-ligand 1;
IOD, integrated optical density. |
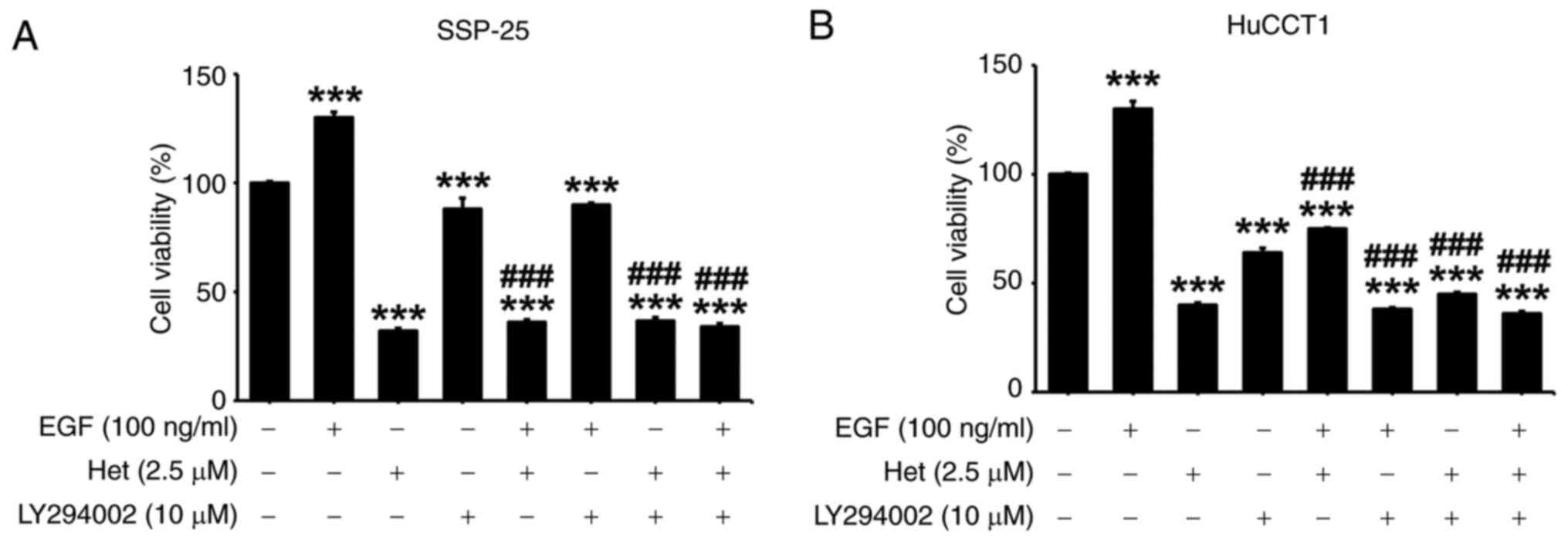
The role of PI3K in the heteronemin-induced
inhibitory effect was next investigated on EGF-induced
proliferation in cholangiocarcinoma cells. The two
cholangiocarcinoma cell lines were treated with EGF and
heteronemin, either alone or in the presence of LY294002 for 72 h.
In both cell lines, LY294002 not only suppressed cell proliferation
on its own but also inhibited EGF-induced cancer cell proliferation
(Fig. 8). Heteronemin similarly
inhibited cancer cell proliferation in both control and
EGF-stimulated cells. Although the combined treatment of
heteronemin and LY294002 did not show an additive effect on cell
viability, their inhibitory effect was the same as that of
treatment with heteronemin, LY294002 or combined treatment in
SSP-25 cells but not in HuCCT1 cells. It is possible that the
KRAS mutation in HuCCT1 cells constitutively activates other
signaling pathways, such as ERK1/2, to offset the inhibitory effect
of heteronemin. This situation is similar to previous studies,
where a combination treatment (heteronemin plus another inhibitor)
does not show an additive effect on cell viability but does
suppress target protein activation (25,36).
Taken together with the inhibitory effect of heteronemin on PI3K
phosphorylation (Figs. 5,6), these results revealed that the
inhibitory effect of heteronemin on EGF-induced cell proliferation
depends on suppression of PI3K activity.
Discussion
The results of the present showed that EGF
stimulates the proliferation of cholangiocarcinoma KRAS WT
SSP-25 cells starting at 20 ng/ml and KRAS mutant HuCCT1
cells starting at 100 ng/ml. PI3K is identified as a critical
kinase inducing cell proliferation activated by the EGF in these
two cell lines. Heteronemin suppressed EGF-stimulated cell
proliferation and PD-L1 expression through inhibiting the PI3K
signaling pathway. Additionally, ERK1/2 activation is observed
under EGF stimulation in cholangiocarcinoma (37). This EGF-stimulated ERK1/2 activation
is diminished by heteronemin treatment (27). The current study indicated that EGF
activated ERK1/2 and PI3K to induce cholangiocarcinoma cell
proliferation. However, the current research landscape presented a
challenge. It remains to be differentiated whether EGF-activated
ERK1/2 and PI3K act independently or via crosstalk with the
FAK-dependent mechanism. Using inhibitors or knockdown strategies
might inadvertently affect other signal transduction pathways and
cell viability. This complexity is further exemplified in our
research on the combined treatment of EGF and heteronemin, where it
was difficult to determine which signal transduction pathways were
more vital for proliferation or anti-cancer growth. These
limitations underscored the urgent need for further investigation
into the specific mechanisms at play.
Heteronemin inhibited the proliferation of SSP-25
and HuCCT1 cells by targeting different signaling pathways between
KRAS WT and KRAS mutant cells. It inhibited
EGF-induced phosphorylation of PI3K and STAT3 in SSP-25 cells, but
not STAT1 (Fig. 5). By contrast, in
HuCCT1 cells, it downregulated the phosphorylation of both PI3K and
STAT1 (Fig. 6). These observations
are consistent with the expression patterns of PD-L1 (Fig. 7) and cell viability results
(Fig. 8). As shown in the Fig. S2, inhibition of PI3K activity
suppressed EGF-stimulated PD-L1 expression in both these two cells.
In addition, a previous study by the authors indicated that
heteronemin treatment suppresses the expression of EGFR and
downstream genes in KRAS mutant HCT-116 colon cancer cells
(25). It also suppresses PD-L1
expression in both KRAS WT HT-29 cells and KRAS
mutant HCT-116 cells (25).
Therefore, it is likely that heteronemin inhibits the viability of
these two cell lines through different EGF-induced signaling
pathways. In EGF-stimulated SSP-25 cells, it inhibits the
PI3K/STAT3/PD-L1 pathway, while it reduces EGF-induced viability of
HuCCT1 cells by targeting the PI3K/STAT1/PD-L1 pathway.
Cisplatin and gemcitabine are frequently used as
standard chemotherapeutic agents for cholangiocarcinoma. Gefitinib
acts as a radiosensitizer in cholangiocarcinoma therapy by
inhibiting radiation-induced EGFR phosphorylation and subsequent
pathway activation, thereby enhancing radiosensitivity (17,38).
On the other hand, combined treatment with lovastatin, a
3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, and
gefitinib significantly inhibited cell proliferation in the SSP-25
and HuH-28 cholangiocarcinoma cell lines (39). The synergistic effect of gefitinib
and gemcitabine combination therapy was observed to suppress
cholangiocarcinoma cell line proliferation via inhibiting ERK1/2
activation (40), which is pivotal
for cholangiocarcinoma cell proliferation, migration and invasion
(41). In vivo studies
further demonstrated the synergistic efficacy of this combination
in cholangiocarcinoma HuCCT1 cell xenografts (40). Thus, combining EGFR inhibitors and
additional chemicals could be a new therapeutic approach for
cholangiocarcinoma (38).
Heteronemin is known to inhibit cholangiocarcinoma proliferation
and motility by blocking the TGF-β pathway (24), and it also suppressed ERK1/2
activation in renal carcinoma cells (24,36).
It is worth noting that EGF/EGFR and TGF-β pathways stimulate
ERK1/2 activation to enhance cell proliferation and migration
(41,42). Cancer cell motility, linked to the
EMT, has been implicated in cancer invasion and metastasis
(43). EGFR and STAT3 are involved
in the EMT, thus playing crucial roles in tumor metastasis
(25,44). The current data, together with
previous findings aforementioned, support the idea that combining
EGFR inhibitors and heteronemin could be a new therapeutic approach
for inhibiting the proliferation and/or metastasis of
cholangiocarcinoma cells.
PI3K activity is oppositely affected by the
combination of heteronemin and EGF. As demonstrated in Figs. 5 and 6, heteronemin could increase the levels of
p-PI3K, p-STAT1 and p-STAT3, while a combination of heteronemin and
EGF inhibited the phosphorylation of PI3K, STAT1 and STAT3. These
results are similar to our previous study in colorectal cancer
cells (25), where heteronemin
induced the phosphorylation of PI3K, and a combination of
heteronemin and tetraiodothyroacetic acid (tetrac), a derivative of
a thyroid hormone, suppressed the phosphorylation of PI3K. The
apparent activation of PI3K, STAT1 and STAT3 by heteronemin can be
attributed to compensatory mechanisms. Previous studies have shown
that inhibition of mTOR, a molecule involved in the PI3K/AKT
signaling pathway, increases the phosphorylation of AKT at T308
(45–48). Thus, anticancer drugs or inhibitors
specific to the PI3K/AKT/mTOR signaling pathway could cause an
increased phosphorylation level of PI3K or AKT through compensatory
mechanisms. Heteronemin is a potential inhibitor specific to PI3K.
When cells are exposed to heteronemin, they may initially perceive
it as a stressor, triggering a compensatory signaling response that
includes other non-PI3K proliferation-related signaling pathways
(23). Moreover, PI3K and its
downstream molecules are involved in these signaling pathways.
Thus, this might explain why it was observed that heteronemin
induces the phosphorylation of PI3K, STAT1 and STAT3. On the other
hand, heteronemin can effectively inhibit EGF-induced
phosphorylation of PI3K, STAT1 and STAT3 without the effect of
compensatory signaling responses. This is because the stimulatory
effect of EGF, a growth factor, is sufficient to overcome the
compensatory mechanism triggered by heteronemin.
The patient with aggressive cancer exhibits an
ineffective response to therapy for KRAS mutant-driven
tumor. KRAS mutation notably contributes to therapeutic
resistance observed in patients with cholangiocarcinoma and is
associated with poor prognosis (49). Although heteronemin inhibited
EGF-induced proliferation in KRAS mutant cholangiocarcinoma
HuCCT1 cells through the PD-L1-mediated PI3K signaling pathway
(Figs. 3 and S2), another study indicated that
inhibiting cell surface PD-L1 with a specific antibody led to a
notable decrease in tumor-sphere formation, but did not hinder
sphere growth, suggesting that cell surface PD-L1 might be an
adhesive molecule for colon and breast cancer stem cells (50). It is possible that heteronemin has
multiple inhibitory effects on KRAS mutant cancer
progression. In KRAS mutant HCT-116 colon cancer cells,
heteronemin, tetrac and their combined treatment induced an
antiproliferative effect through suppression of the expression of
EGFR and downstream genes, including PD-L1 (25). The combination of heteronemin and
tetrac in this KRAS mutant colon cancer cells appeared to
reduce expression levels of signal transduction pathways involved
in EGFR/PD-L1 signaling, including those related to cell
proliferation, migration and the EMT in tumor metastasis, compared
with heteronemin alone (24,25).
Indeed, combination therapies, such as inhibitors of PD-L1 and
cytotoxic T-lymphocyte-associated protein 4, demonstrate improved
inhibitory efficacy in patients with KRAS WT and KRAS mutant
cholangiocarcinoma (51). Thus,
heteronemin combined with other potential compounds such as tetrac
could be a new approach to treating KRAS mutant cancers.
In summary, EGF initiates diverse signal
transduction pathways and elevates PD-L1 expression, thereby
promoting the proliferation of cholangiocarcinoma cells.
Alternatively, heteronemin likely regulates expression of PI3K,
STAT1 and STAT3 and their phosphorylation to inhibit EGF-induced
growth. Crucially, heteronemin suppresses the growth of
EGF-stimulated cholangiocarcinoma by inhibiting PI3K activation and
PD-L1 expression.
Supplementary Material
Supporting Data
Acknowledgements
The authors express their profound appreciation for
the excellent secretarial performance of Ms Y.-J. Shih in our
group. The corresponding author also wishes to thank Dr L.-F. Liu,
(Academia Sinica, Taiwan) for her spiritual support in pursuing a
research career.
Funding
The present study was supported in part by the grants from E-Da
Hospital, Taiwan (grant no. EDAHS113020 and EDAHI113001), the Chair
Professor Research Funds, the TMU Research Center of Cancer
Translational Medicine from The Featured Areas Research Center
Program within the framework of the Higher Education Sprout
Project, by the Ministry of Education (MOE) in Taiwan (grant no.
DP2-107-20000), the Ministry of Science and Technology, Taiwan
(grant nos. NSTC112-2811-B-038-037, NSTC112-2314-B-038-098 and
NSTC112-2320-B-038-021) and the Ministry of Science and Technology,
Taiwan (grant nos. NSTC112-2314-B-038-004, NSTC112-2314-B-038-005
and NSTC112-2314-B-038-003).
Availability of data and materials
The data generated in the present study are
included in the figures and/or tables of this article.
Authors' contributions
YCSHY, CCT, YNY, HYL, JWP and KW conceptualized the
study. CCT, FCL, HCC, MCL, ZLL, YCC and TYC designed the study and
performed experiments. FCL, SYL, JCY, DRC, YCC, TYC, HCC and MCL
analyzed data and performed figure visualization. YCSHY, CCT, SYL
and JCY wrote the original draft. DRC, HYL, JWP and KW wrote and
edited the manuscript. YNY, HYL, JWP and KW supervised the study
and acquired funding. YCSHY, CCT, YNY and HYL confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
18S rRNA
|
18S ribosomal RNA
|
|
CCND1
|
cyclin D1
|
|
cDNA
|
complementary DNA
|
|
c-Myc
|
cytosol MYC proto-oncogene, bHLH
transcription factor
|
|
EGF
|
epidermal growth factor
|
|
EGFR
|
EGF receptor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
HB-EGF
|
heparin-binding EGF-like growth
factor
|
|
HRP
|
horseradish peroxidase
|
|
IgG
|
immunoglobulin G
|
|
JAK
|
Janus kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
mTOR
|
mammalian target of rapamycin
|
|
MYC
|
MYC proto-oncogene, bHLH
transcription factor
|
|
NGS
|
next-generation sequencing
|
|
PCNA
|
proliferating cell nuclear
antigen
|
|
PD-1
|
programmed cell death protein 1
|
|
PD-L1
|
programmed cell death-ligand 1
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
RAS
|
rat sarcoma
|
|
ROS
|
reactive oxygen species
|
|
RT-qPCR
|
reverse-transcription quantitative
polymerase chain reaction
|
|
SD
|
standard deviation
|
|
SOD
|
superoxide dismutase
|
|
STAT
|
signal transducer and activator of
transcription
|
|
TBS
|
Tris-buffered saline
|
|
TBST
|
TBS with 0.1% Tween-20
|
|
tetrac
|
tetraiodothyroacetic acid
|
|
TGF
|
transforming growth factor
|
|
WT
|
wild-type
|
References
|
1
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: Implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marquardt H, Hunkapiller MW, Hood LE and
Todaro GJ: Rat transforming growth factor type 1: Structure and
relation to epidermal growth factor. Science. 223:1079–1082. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: Correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Higashiyama S, Abraham JA, Miller J,
Fiddes JC and Klagsbrun M: A heparin-binding growth factor secreted
by macrophage-like cells that is related to EGF. Science.
251:936–939. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shing Y, Christofori G, Hanahan D, Ono Y,
Sasada R, Igarashi K and Folkman J: Betacellulin: A mitogen from
pancreatic beta cell tumors. Science. 259:1604–1607. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Toyoda H, Komurasaki T, Uchida D, Takayama
Y, Isobe T, Okuyama T and Hanada K: Epiregulin. A novel epidermal
growth factor with mitogenic activity for rat primary hepatocytes.
J Biol Chem. 270:7495–7500. 1995.PubMed/NCBI
|
|
7
|
Rayego-Mateos S, Rodrigues-Díez R,
Morgado-Pascual JL, Rodrigues Díez RR, Mas S, Lavoz C, Alique M,
Pato J, Keri G, Ortiz A, et al: Connective tissue growth factor is
a new ligand of epidermal growth factor receptor. J Mol Cell Biol.
5:323–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Normanno N, De Luca A, Bianco C, Strizzi
L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F and
Salomon DS: Epidermal growth factor receptor (EGFR) signaling in
cancer. Gene. 366:2–16. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wieduwilt MJ and Moasser MM: The epidermal
growth factor receptor family: Biology driving targeted
therapeutics. Cell Mol Life Sci. 65:1566–1584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brand TM, Iida M, Li C and Wheeler DL: The
nuclear epidermal growth factor receptor signaling network and its
role in cancer. Discov Med. 12:419–432. 2011.PubMed/NCBI
|
|
11
|
Clapéron A, Mergey M, Nguyen Ho-Bouldoires
TH, Vignjevic D, Wendum D, Chrétien Y, Merabtene F, Frazao A,
Paradis V, Housset C, et al: EGF/EGFR axis contributes to the
progression of cholangiocarcinoma through the induction of an
epithelial-mesenchymal transition. J Hepatol. 61:325–332. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sarcognato S, Sacchi D, Fassan M, Fabris
L, Cadamuro M, Zanus G, Cataldo I, Capelli P, Baciorri F,
Cacciatore M and Guido M: Cholangiocarcinoma. Pathologica.
113:158–169. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsu M, Sasaki M, Igarashi S, Sato Y and
Nakanuma Y: KRAS and GNAS mutations and p53 overexpression in
biliary intraepithelial neoplasia and intrahepatic
cholangiocarcinomas. Cancer. 119:1669–1674. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu CC, Yu CTR, Chang GC, Lai JM and Hsu
SL: Aurora-A promotes gefitinib resistance via a NF-κB signaling
pathway in p53 knockdown lung cancer cells. Biochem Biophys Res
Commun. 405:168–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Massarelli E, Varella-Garcia M, Tang X,
Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS and Wistuba II:
KRAS mutation is an important predictor of resistance to therapy
with epidermal growth factor receptor tyrosine kinase inhibitors in
non-small-cell lung cancer. Clin Cancer Res. 13:2890–2896. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amelia T, Kartasasmita RE, Ohwada T and
Tjahjono DH: Structural insight and development of EGFR tyrosine
kinase inhibitors. Molecules. 27:8192022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leone F, Cavalloni G, Pignochino Y,
Sarotto I, Ferraris R, Piacibello W, Venesio T, Capussotti L, Risio
M and Aglietta M: Somatic mutations of epidermal growth factor
receptor in bile duct and gallbladder carcinoma. Clin Cancer Res.
12:1680–1685. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Serna-Blasco R, Sánchez-Herrero E,
Sanz-Moreno S, Rodriguez-Festa A, García-Veros E, Casarrubios M,
Sierra-Rodero B, Laza-Briviesca R, Cruz-Bermúdez A, Mielgo-Rubio X,
et al: KRAS p.G12C mutation occurs in 1% of EGFR-mutated advanced
non-small-cell lung cancer patients progressing on a first-line
treatment with a tyrosine kinase inhibitor. ESMO Open.
6:1002792021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jeannot V, Busser B, Brambilla E, Wislez
M, Robin B, Cadranel J, Coll JL and Hurbin A: The PI3K/AKT pathway
promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by
a deacetylase-dependent mechanism. Int J Cancer. 134:2560–2571.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanaka T, Munshi A, Brooks C, Liu J, Hobbs
ML and Meyn RE: Gefitinib radiosensitizes non-small cell lung
cancer cells by suppressing cellular DNA repair capacity. Clin
Cancer Res. 14:1266–1273. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yabuuchi S, Katayose Y, Oda A, Mizuma M,
Shirasou S, Sasaki T, Yamamoto K, Oikawa M, Rikiyama T, Onogawa T,
et al: ZD1839 (IRESSA) stabilizes p27Kip1 and enhances
radiosensitivity in cholangiocarcinoma cell lines. Anticancer Res.
29:1169–1180. 2009.PubMed/NCBI
|
|
23
|
Wang K, Chen YF, Yang YCSH, Huang HM, Lee
SY, Shih YJ, Li ZL, Whang-Peng J, Lin HY and Davis PJ: The power of
heteronemin in cancers. J Biomed Sci. 29:412022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin HY, Tey SL, Ho Y, Chin YT, Wang K,
Whang-Peng J, Shih YJ, Chen YR, Yang YN, Chen YC, et al:
Heteronemin induces anti-proliferation in cholangiocarcinoma cells
via inhibiting TGF-β pathway. Mar Drugs. 16:4892018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Unson S, Chang TC, Yang YN, Wang SH, Huang
CH, Crawford DR, Huang HM, Li ZL, Lin HY, Whang-Peng J, et al:
Heteronemin and tetrac induce anti-proliferation by blocking
EGFR-mediated signaling in colorectal cancer cells. Mar Drugs.
20:4822022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang YCSH, Li ZL, Huang TY, Su KW, Lin CY,
Huang CH, Chen HY, Lu MC, Huang HM, Lee SY, et al: Effect of
estrogen on heteronemin-induced anti-proliferative effect in breast
cancer cells with different estrogen receptor status. Front Cell
Dev Biol. 9:6886072021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang CH, Huang TY, Chang WJ, Pan YS, Chu
HR, Li ZL, Unson S, Chin YT, Lin CY, Huang HM, et al: Combined
treatment of heteronemin and tetrac induces antiproliferation in
oral cancer cells. Mar Drugs. 18:3482020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chung CC, Huang TY, Chu HR, De Luca R,
Candelotti E, Huang CH, Yang YCSH, Incerpi S, Pedersen JZ, Lin CY,
et al: Heteronemin and tetrac derivatives suppress non-small cell
lung cancer growth via ERK1/2 inhibition. Food Chem Toxicol.
161:1128502022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang WT, Bow YD, Fu PJ, Li CY, Wu CY,
Chang YH, Teng YN, Li RN, Lu MC, Liu YC and Chiu CC: A Marine
terpenoid, heteronemin, induces both the apoptosis and ferroptosis
of hepatocellular carcinoma cells and involves the ROS and MAPK
pathways. Oxid Med Cell Longev. 2021:76890452021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samuels HH, Stanley F and Casanova J:
Depletion of L-3,5,3′-triiodothyronine and L-thyroxine in euthyroid
calf serum for use in cell culture studies of the action of thyroid
hormone. Endocrinology. 105:80–85. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yoon JH, Gwak GY, Lee HS, Bronk SF,
Werneburg NW and Gores GJ: Enhanced epidermal growth factor
receptor activation in human cholangiocarcinoma cells. J Hepatol.
41:808–814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chong ZZ and Maiese K: The Src homology 2
domain tyrosine phosphatases SHP-1 and SHP-2: Diversified control
of cell growth, inflammation, and injury. Histol Histopathol.
22:1251–1267. 2007.PubMed/NCBI
|
|
34
|
Whitfield ML, George LK, Grant GD and
Perou CM: Common markers of proliferation. Nat Rev Cancer.
6:99–106. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Garcia R, Franklin RA and McCubrey JA: EGF
induces cell motility and multi-drug resistance gene expression in
breast cancer cells. Cell Cycle. 5:2820–2826. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu SY, Sung PJ, Chang YL, Pan SL and Teng
CM: Heteronemin, a spongean sesterterpene, induces cell apoptosis
and autophagy in human renal carcinoma cells. Biomed Res Int.
2015:7382412015.PubMed/NCBI
|
|
37
|
Lan T, Li Y, Wang Y, Mu CY, Tao AB, Gong
JL, Zhou Y, Xu H, Li SB, Gu B, et al: Increased endogenous PKG I
activity attenuates EGF-induced proliferation and migration of
epithelial ovarian cancer via the MAPK/ERK pathway. Cell Death Dis.
14:392023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Silini A, Ghilardi C, Figini S, Sangalli
F, Fruscio R, Dahse R, Pedley RB, Giavazzi R and Bani M: Regulator
of G-protein signaling 5 (RGS5) protein: A novel marker of cancer
vasculature elicited and sustained by the tumor's proangiogenic
microenvironment. Cell Mol Life Sci. 69:1167–1178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang SH, Lin HY, Chang VH, Chen CC, Liu
YR, Wang J, Zhang K, Jiang X and Yen Y: Lovastatin overcomes
gefitinib resistance through TNF-α signaling in human
cholangiocarcinomas with different LKB1 statuses in vitro and in
vivo. Oncotarget. 6:23857–23873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakajima Y, Takagi H, Kakizaki S,
Horiguchi N, Sato K, Sunaga N and Mori M: Gefitinib and gemcitabine
coordinately inhibited the proliferation of cholangiocarcinoma
cells. Anticancer Res. 32:5251–5262. 2012.PubMed/NCBI
|
|
41
|
Sritananuwat P, Sueangoen N, Thummarati P,
Islam K and Suthiphongchai T: Blocking ERK1/2 signaling impairs
TGF-β1 tumor promoting function but enhances its tumor suppressing
role in intrahepatic cholangiocarcinoma cells. Cancer Cell Int.
17:852017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Indramanee S, Sawanyawisuth K,
Silsirivanit A, Dana P, Phoomak C, Kariya R, Klinhom-On N, Sorin S,
Wongkham C, Okada S and Wongkham S: Terminal fucose mediates
progression of human cholangiocarcinoma through EGF/EGFR activation
and the Akt/Erk signaling pathway. Sci Rep. 9:172662019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Choudhary KS, Rohatgi N, Halldorsson S,
Briem E, Gudjonsson T, Gudmundsson S and Rolfsson O: EGFR
signal-network reconstruction demonstrates metabolic crosstalk in
EMT. PLoS Comput Biol. 12:e10049242016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue
P, Fu H and Khuri FR: Activation of Akt and eIF4E survival pathways
by rapamycin-mediated mammalian target of rapamycin inhibition.
Cancer Res. 65:7052–7058. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shi Y, Yan H, Frost P, Gera J and
Lichtenstein A: Mammalian target of rapamycin inhibitors activate
the AKT kinase in multiple myeloma cells by up-regulating the
insulin-like growth factor receptor/insulin receptor
substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther.
4:1533–1540. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cloughesy TF, Yoshimoto K, Nghiemphu P,
Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, et al:
Antitumor activity of rapamycin in a phase I trial for patients
with recurrent PTEN-deficient glioblastoma. PLoS Med. 5:e82008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bergholz JS and Zhao JJ: How compensatory
mechanisms and adaptive rewiring have shaped our understanding of
therapeutic resistance in cancer. Cancer Res. 81:6074–6077. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guo L, Zhou F, Liu H, Kou X, Zhang H, Chen
X and Qiu J: Genomic mutation characteristics and prognosis of
biliary tract cancer. Am J Transl Res. 14:4990–5002.
2022.PubMed/NCBI
|
|
50
|
Chen M, Sharma A, Lin Y, Wu Y, He Q, Gu Y,
Xu ZP, Monteiro M and Gu W: Insluin and epithelial growth factor
(EGF) promote programmed death ligand 1(PD-L1) production and
transport in colon cancer stem cells. BMC Cancer. 19:1532019.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Harding JJ, Khalil DN, Fabris L and
Abou-Alfa GK: Rational development of combination therapies for
biliary tract cancers. J Hepatol. 78:217–228. 2023. View Article : Google Scholar : PubMed/NCBI
|