Introduction
Melanoma is one of the most rapidly growing types of
cancer worldwide in terms of incidence and holds 5.3% of all
cancers reported in the United States in 2018(1). The majority of U.S. Food and Drug
Administration (FDA)-approved melanoma drugs target the BRAF
(V600E) mutation, which occurs in >50% of melanoma cases;
however, melanoma is associated with a poor prognosis and tumors
attain resistance to BRAF mutation inhibition (2). Even though surgical resection,
immunotherapy, radiation therapy, or chemotherapy can improve
survival rates, current trends demand effective and personalized
targeted therapeutics for melanoma.
We recently established protein kinase C-ι (PKC-ι)
as an oncogene and a prospective therapeutic target for metastatic
melanoma in vitro (3-5).
PKC belongs to the protein kinase enzyme family, which
post-translationally modifies other proteins and is involved in a
number of signal transduction cascades. In total, 15 PKC isozymes
have been identified in humans; these are divided as classical,
novel and atypical PKCs (aPKCs). aPKCs contain two structurally and
functionally distinct isozymes: PKC-ι and PKC-ζ (6-8).
PKC-ι is an oncogene that has been associated in several signaling
pathways in ovarian, glioma and prostate carcinomas (9-11).
Moreover, PKC-ι overexpression has been shown to be associated with
a poor prognosis (11).
We have previously demonstrated that PKC-ι is
overexpressed in melanoma cell lines compared to undetectable
levels in normal melanocytes using the PCS-200-013 and MEL-F-NEO
cell lines (4). Our previous
results confirmed that PKC-ι plays a vital role in inducing
epithilial-mesencymal transition (EMT), in melanoma by regulating
Vimentin dynamics (3). We have
also reported that PKC-ι is involved in the tumorigenesis,
progression and survival of melanoma. PKC-ι inhibition using
specific inhibitors or the knockdown of its expression using siRNA
significantly induces apoptosis, and reduces the migration and
invasion of melanoma (3). In our
previous study, we demonstrated that PKC-ι was heavily involved in
the phosphorylation of IκB kinase (IKKα/β) at S176/180 to activate
it. Activated phospho-IKKα/β (S176/180) then phosphorylates nuclear
factor of κ light polypeptide gene enhancer in B-cells inhibitor
(IκB), which causes IκB to dissociate from the nuclear factor
(NF)-κB complex and undergo ubiquitination. This process releases
NF-κB in the active form to translocate to the nucleus. Upon PKC-ι
inhibition using two PKC-ι specific inhibitors,
[4-(5-amino-4-carbamoylimidazol-1-yl)-2,3-dihydroxycyclopentyl]
methyl dihydrogen phosphate (ICA-1T) and
5-amino-1-((1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-4-carboxamide
(ICA-1S), we found that NF-κB nuclei translocation was blocked,
causing NF-κB activity to downregulate (3). Our previous findings on both
melanoma and prostate cancer cell lines demonstrated that PKC-ι
inhibition not only affected the pathways regulated by PKC-ι, but
also downregulated its protein expression (3,4,12).
This suggests that PKC-ι plays a role in a self-propagating cycle,
as observed in some other cycles related to cancer growth, such as
transforming growth factor (TGF)-β and CD147(13). We therefore designed the current
study to investigate the role of PKC-ι in its expression regulatory
mechanisms.
Transcription factors play a key role in gene
expression, controlling the rate of transcription of genetic
information from DNA to messenger RNA by binding to a specific DNA
sequence. Various cytokines often trigger such signaling. In this
study, we sought to determine which transcription factors are the
main regulators of PKC-ι expression in melanoma cells, giving
emphasis to cytokine stimulation and expression.
In the present study, we report the effects of the
systematic silencing of PKC-ι, NF-κB, c-Jun and Forkhead box
protein O1 (FOXO1) on PKC-ι levels in relation to NF-κB,
PI3K/AKT/FOXO1, JNK/c-Jun and signal transducer and activator of
transcription (STAT) signaling, along with cytokine production, to
establish the mechanism of PRKCI regulation. The levels of both
phosphorylated and total PKC-ι increased upon FOXO1 knockdown by
siRNA and decreased upon the knockdown of c-Jun by siRNA. The
results confirmed that c-Jun acts as a transcriptional activator
and that FOXO1 acts as a transcriptional supressor of PRKCI
expression. Going forward, we establish the roles that these
transcription factors play in an inflammation cycle that governs
PKC-ι expression and is dependent on PKC-ι for the cycle to
continue. Furthermore, we establish that major signaling pathways
such as PI3K/AKT/FOXO1 and JNK/c-Jun play a vital role in
regulation of PKC-ι expression. In addition, the cytokines IL-6 and
IL-8 promote PKC-ι expression, thereby enhancing NF-κB activity,
producing more cytokines as a part of a cycle such cancers use to
develop and spread. IL-17E and ICAM-1 induce FOXO1 to subdue PKC-ι
expression. Overall, these results suggest that PKC-ι plays a
central role in melanoma progression with a complex and tightly
regulated expression profile. The specific inhibition of PKC-ι can
distrupt its own regulatory cycle, leading to the disruption of its
oncogenic role in melanoma.
Materials and methods
Reagents and antibodies
ICA-1 nucleotide (ICA-1T) and nucleoside (ICA-1S)
were synthesized by Therachem (Jaipur, India). They were dissolved
in sterile distilled water (vehicle) prior to use. Antibodies were
purchased as follows: PKC-ι (610175) from BD Biosciences (San Jose,
CA, USA) and NF-κB p65 (sc-372-G) from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA); phospho-PKC-ι (T555; 44-968G) from
Thermo Fisher Scientific (Waltham, MA, USA); early growth response
protein 1 (EGR1; 4153S), c-Jun (9165S), phospho-c-Jun (S73; 3270S),
FOXO1 (2880S), phospho-FOXO1 (T24; 9464S) and phospho-AKT (S473;
4059S) from Cell Signaling Technology (Danvers, MA, USA); and
β-actin peroxidase (A3854) from Sigma-Aldrich (St. Louis, MO, USA).
Enhanced chemiluminescence solution (34080) was purchased from
Pierce, Inc. (Rockford, IL, USA). Dulbecco's phosphate-buffered
saline without Mg2+ and Ca2+ (D8537) and Trypsin-EDTA
(ethylenediaminetetraacetic acid) solution (T4049) were purchased
from Sigma-Aldrich. Human small interfering RNA (siRNA) for PKC-ι
(SR303741), for EGR1 (SR301358), c-Jun (SR302499), FOXO1 (SR301618)
paired box gene 3 (PAX3; SR303360), interferon regulatory factor 9
(IRF9; SR307030) and NF-κB p65 (SR321602) were purchased from
Origene Technologies, Inc. (Rockville, MD, USA). The NF-κB specific
inhibitor, 4-methyl-N1-(3-phenylpropyl)-1,2-benzenediamine (JSH-23)
(J4455) was purchased from Sigma-Aldrich.
Cells and cell culture
The SK-MEL-2 (ATCC® HTB68™) and MeWo (ATCC® HTB65™)
cell lines were purchased from the American Type Tissue Culture
Collection (ATCC; Rockville, MD, USA) in November, 2015. All cells
were frozen in liquid nitrogen immediately with early passages. The
cells of passages 2 to 5 were resuscitated from liquid nitrogen and
cultured for <3 months before re-initiating culture from the
same passage for each tested experiment. The ATCC authenticated the
cell lines using morphology, karyotyping and PCR-based approaches.
The cells were cultured at 37˚C and 5% CO2. Eagle's minimum
essential medium (90% v/v) (ATCC 30-2003) with fetal bovine serum
(10% v/v) and penicillin (5 µg/ml) were used for SK-MEL-2 and MeWo
cell culturing according to the ATTC guidelines. All cell lines
were seeded and grown as monolayers in T25 or T75 flasks.
Identification of possible
transcription factors (TFs) which bind to the PRKCI gene
The PRKCI gene sequence was obtained from
ensemble.org (ENSG00000163558) which locates in chromosome 3 from
bp170222365-170305981 (3q26.2) (14,15). The forward strand sequence was
used and compared with EPD/Eukaryotic Promoter Database
(https://epd.vital-it.ch/index.php) for its promoter sequence. A
specific sequence was then selected (chromosome 3;
170220768-170225128); the sequence contains the promoter, promoter
flank, enhancer and a motif feature. This sequence predicted
possible TFs that can bind within a dissimilarity margin ≤10% using
PROMO, which is a virtual laboratory for studying transcription
factor binding sites in DNA sequences (http://alggen.lsi.upc.es/). TF targets were then
compared with the Genomatix Matinspector results to generate the
final TF list for the following experiments.
Knockdown of TFs, PKC-i and NF-κB gene
expression by siRNA
Each siRNA contained a pool of three combined RNA
sequences for the targeted gene and respective control siRNA
contained a scrambled sequence, which did not lead to specific
degradation of any known cellular messenger RNA and whose sequence
is a proprietary of Origene Technologies, Inc. The experiments
performed with siRNA are as follows: Approximately 1x105 cells
(SK-MEL-2 and MeWo) were cultured in T25 flasks and at 24 h
post-plating, fresh medium was supplied and the cells were treated
with a 20 nM concentration of one of the transcription factor
siRNAs or scrambled siRNA as a control using ‘siTran’ siRNA
transfection reagent (TT300002) from Origene Technologies, Inc.
according to the manufacturer's recommended ratios. After 48 h of
the post-treatment period, cells were subsequently lifted and cell
lysates were collected with cell lysis buffer (C7027;
Invitrogen/Thermo Fisher Scientific). Western blot analysis was
performed as previously described in the study by Win and
Acevedo-Duncan and samples were then fractionated by SDS-PAGE and
immunoblotted (16).
Western blot analysis
The Bradford protein assay was used to measure the
protein concentrations of extracted cell lysates in each siRNA
experiment. Total protein (80 µg) was loaded into each well,
separated by 10% SDS-PAGE and electro-blotted onto supported
nitrocellulose membranes. Each blot was blocked for 1 h with 5%
bovine serum albumin (BP1600-100; Thermo Fischer Scientific) in
TTBS solution (0.1% v/v Tween in 1X TBS) at room temperature
(approximately 25˚C). Protein bands were probed with each targeted
primary antibody at 4˚C overnight followed by
horseradish-peroxidase-conjugate anti-mouse or anti-rabbit
secondary antibody for 2 h at room temperature. Immuno-reactive
bands were visualized with enhanced chemiluminescence solution
(34080) according to the manufacturer's instructions (Pierce,
Inc.). Goat anti-mouse IgG (170-6516) and goat anti-rabbit IgG
(170-6515) secondary antibodies were used from Bio-Rad Laboratories
(Hercules, CA, USA). Manufacturer's recommended concentrations were
used for all tested primary and secondary antibodies. β-actin was
used as the internal control in each western blot.
Densitometry
The intensity of western blot bands was measured
using ‘Image Studio Lite 5.x’ software developed by LI-COR
Biosciences (Lincoln, NE, USA) in which the background intensity
was subtracted from the intensity of each band to obtain the
corrected intensity of the proteins.
Analysis of cytokine expression using
enzyme-linked immunosorbent assay (ELISA)
An ELISA containing a cytokine array (cat. no.
ARY005B) was obtained from R&D Systems (Minneapolis, MN, USA).
Approximately 1x105 cells (SK-MEL-2 and MeWo) were cultured in T25
flasks and at 24 h post-plating, fresh medium was supplied and the
cells were treated with a 20 nM concentration of PKC-ι siRNA or
scrambled siRNA as a control. After 48 h of the post-treatment
period, the cells were subsequently lifted, lysed, processed and
analyzed according to manufacturer's instructions using cytokine
array kit reagents. Total protein (100 µg) from each sample was
used to expose the membranes and chemoluminescence photographs as
western blots were taken to analyze the cytokine expression
profiles of the melanoma cells upon PKC-ι siRNA knockdown.
Immunopaired antibody detection assay
(IPAD)
Approximately 1x105 cells were cultured in T25
flasks and at 24 h post-plating, fresh medium was supplied and the
cells were treated with either volume of sterile water (control) or
the IC50 concentration of ICA-1T (1 µM). Additional doses were
supplied every 24 h during a 3-day incubation period at 37˚C. The
cells were then lysed and lysates were prepared with the final
total protein concentration being >2 µg/ml and then delivered to
ActivSignal, LLC (Natick, MA, USA) for further processing and
analyzing. The ActivSignal IPAD platform is a multiplex ELISA-based
proprietary technology for analyzing the activity of multiple
signaling pathways in one reaction with high sensitivity and
specificity. The activities of >20 signaling pathways were
monitored simultaneously in a single well through assessing the
expression or protein phosphorylation of 70 target human
proteins.
Reverse transcription-quantitative PCR
(RT-qPCR)
qPCR was performed on RNA isolated from SK-MEL-2 and
MeWo cell lysates collected after siRNA treatments for PKC-ι, c-Jun
and FOXO1 against scrambled siRNA as the control. Total RNA was
isolated from the cell pellets using RNA lysis buffer which comes
with the RNeasy mini kit (74104) from Qiagen (Germantown, MD, USA).
RNA was reverse transcribed into cDNA with You-Prime First Strand
Beads (27-9264-01) form GE Healthcare UK Ltd. (Buckinghamshire,
UK). qPCR was performed on cDNA using the QuantStudio3 Real-Time
PCR system (Thermo Fisher Scientific). Gene expression was observed
for PKC-ι (primers: Forward, CACACTTTCCAAGC CAAGCG and reverse,
GGCGTCCAAGTCCCCATATT), c-Jun (primers: Forward,
GTGCCGAAAAAGGAAGCTGG and reverse, CTGCGTTAGCATGAGTTGGC), FOXO1
(primers: Forward, ATGGCTTGGTGTCTTTCTTTTCT and reverse,
TGTGGCTGACAAGACTTAACTCAA), IL-17E (primers: Forward,
GCCACCACTCCTGTCTCTTC and reverse, CCAGGGGCTCTTTCTTCTCC), IL-6
(primers: Forward, GCTCCCTACACACATGCCTT and reverse, CCT
TCCCTGTGCATGGTGAT), IL-8 (primers: Forward, CAG AGACAGCAGAGCACAC
and reverse, ATCAGGAAGGCT GCCAAGAG) and ICAM-1 (primers: Forward,
GGGAACAA CCGGAAGGTGTA and reverse, CAGTTCCACCCGTTCTG GAG). β-actin
(primers: Forward, AGAGCTACGAGCTGCCT GAC and reverse,
AGCACTGTGTTGGCGTACAG) was used as an internal control. PCR
reactions used SYBR-Green PCR Mix (Applied Biosystems, Foster City,
CA, USA). cDNA was denatured at at 95˚C for 10 min, followed by 40
cycles of denaturing at 95˚C for 20 sec and an annealing stage of
65˚C for 40 sec. QuantStudio Software 2.0 was used to quantify gene
expression using 2-ΔΔCT (Thermo Fisher Scientific) as explained by
Livak and Schmittgen (17).
Statistical analysis
All data are presented as the means ± SD.
Statistical analysis was performed with one- or two-way ANOVA
followed by Tukey's HSD test as a multiple comparisons test using
the ‘VassarStats’ web tool for statistical analysis. P-values ≤0.05
or ≤0.01 were considered to indicate statistically significant
differences.
Results
The specific sequence of the PRKCI gene (chromosome
3; 170220768-170225128), which was selected to contain the
promoter, promoter flank, enhancer and a motif feature, was 4,360
bp in length. The promoter allows TFs to bind and initiate
transcription, and the enhancer is a regulatory region in the flank
that facilitates TF binding. We narrowed down possible hits by
allowing only TFs, which can bind within a dissimilarity margin
≤10%, thereby achieving high specificity. We obtained approximately
70 TF hits to the given target after comparing the outcomes of
PROMO analysis and Genomatix Matinspector. We selected c-Jun,
ISGF3, PAX3, EGR1 and FOXO1 as the top 5 TFs with the highest
binding probability to the selected sequance of the PRKCI gene.
FOXO1 and c-Jun stand out as the main
transcriptional regulators of PKC-ι expression in melanoma
cells
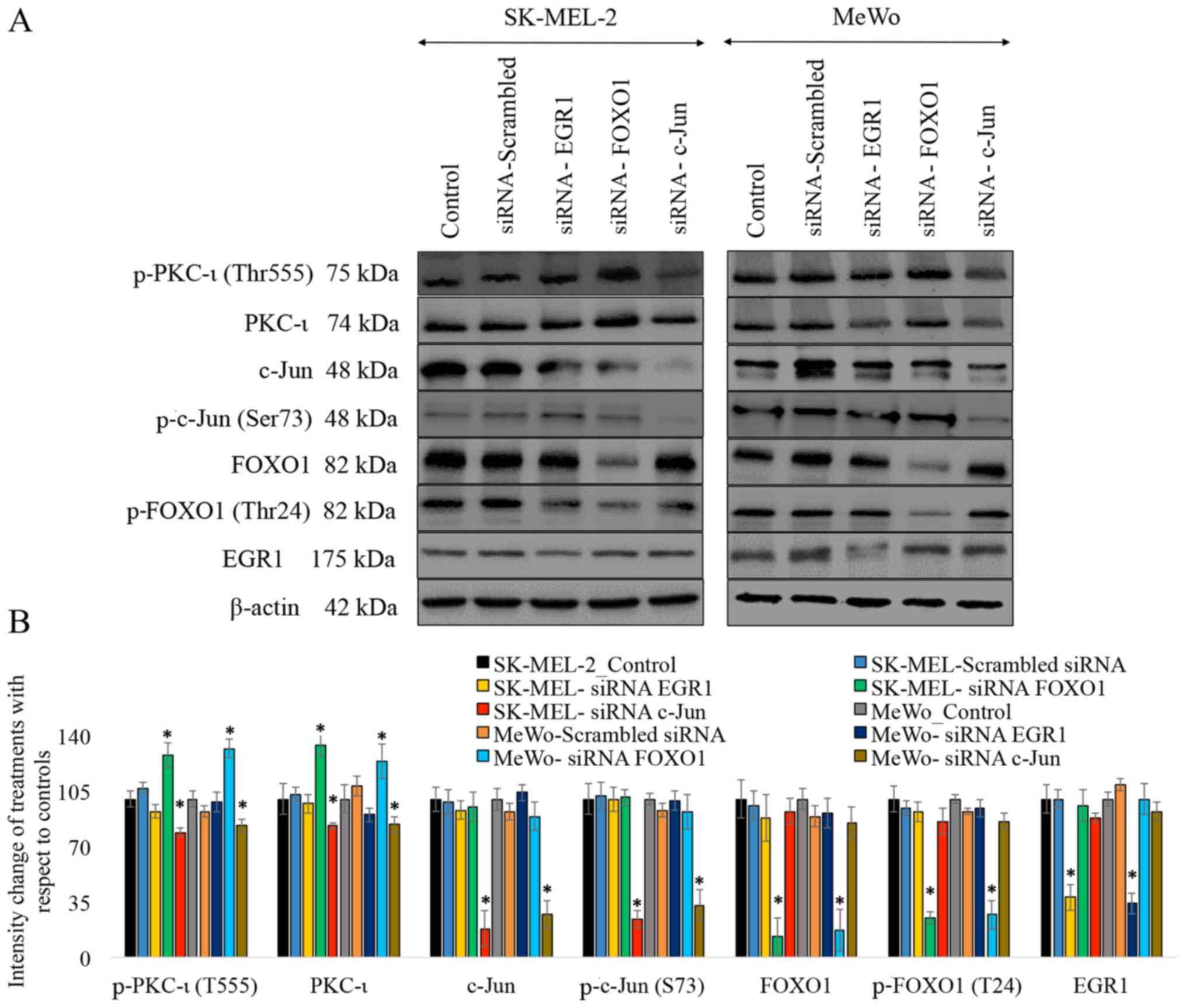
As shown in Fig.
1, the results of western blot analysis revealed that
transfection with each siRNA for EGR1, FOXO1 and c-Jun
significantly reduced expression levels of these proteins. The
siRNA for EGR1 decreased the expression of EGR1 by 72% (P≤0.05) and
76% (P≤0.05) in the SK-MEL-2 and MeWo cells, respectively. The
siRNA of FOXO1 decreased the expression of FOXO1 by 87% (P≤0.05)
and 83% (P≤0.05), while the levels of phospho-FOXO1 (T24) decreased
by 75% (P≤0.05) and 73% (P≤0.05) in the SK-MEL-2 and MeWo cells,
respectively. The siRNA of c-Jun decreased the expression of c-Jun
by 82% (P≤0.05) and 73% (P≤0.05), while the levels of phospho-c-Jun
(S73) decreased by 76% (P≤0.05) and 67% (P≤0.05) in the SK-MEL-2
and MeWo cells, respectively. These results suggested that
transfection with each of the siRNAs knocked down the expression of
its respective target. Only the knockdown of FOXO1 and c-Jun was
found to have an effect on the levels of total and phosphorylated
PKC-ι (T555) in both cell lines. The knockdown of FOXO1 by siRNA
increased the expression of total PKC-ι by 34% (P≤0.05) and 24%
(P≤0.05), while it increased the level of phospho-PKC-ι (T555) by
28% (P≤0.05) and 32% (P≤0.05) in the SK-MEL-2 and MeWo cells,
respectively. The knockdown of c-Jun by siRNA decreased the
expression of total PKC-ι by 17% (P≤0.05) and 16% (P≤0.05), and it
decreased the level of phospho-PKC-ι (T555) by 21% (P≤0.05) and 17%
(P≤0.05) in the SK-MEL-2 and MeWo cells, respectively. The
knockdown of the expression of the TFs, ISGF3, EGR1 and PAX3, by
siRNA was not found to have a significant effect on the expression
of total and phosphorylated PKC-ι (T555) in both cell lines.
Therefore we decided to omit these 3 TFs, and only FOXO1 and c-Jun
were selected for use in the subsequent experiments. As shown in
Fig. 1, only the EGR1 negative
results were included.
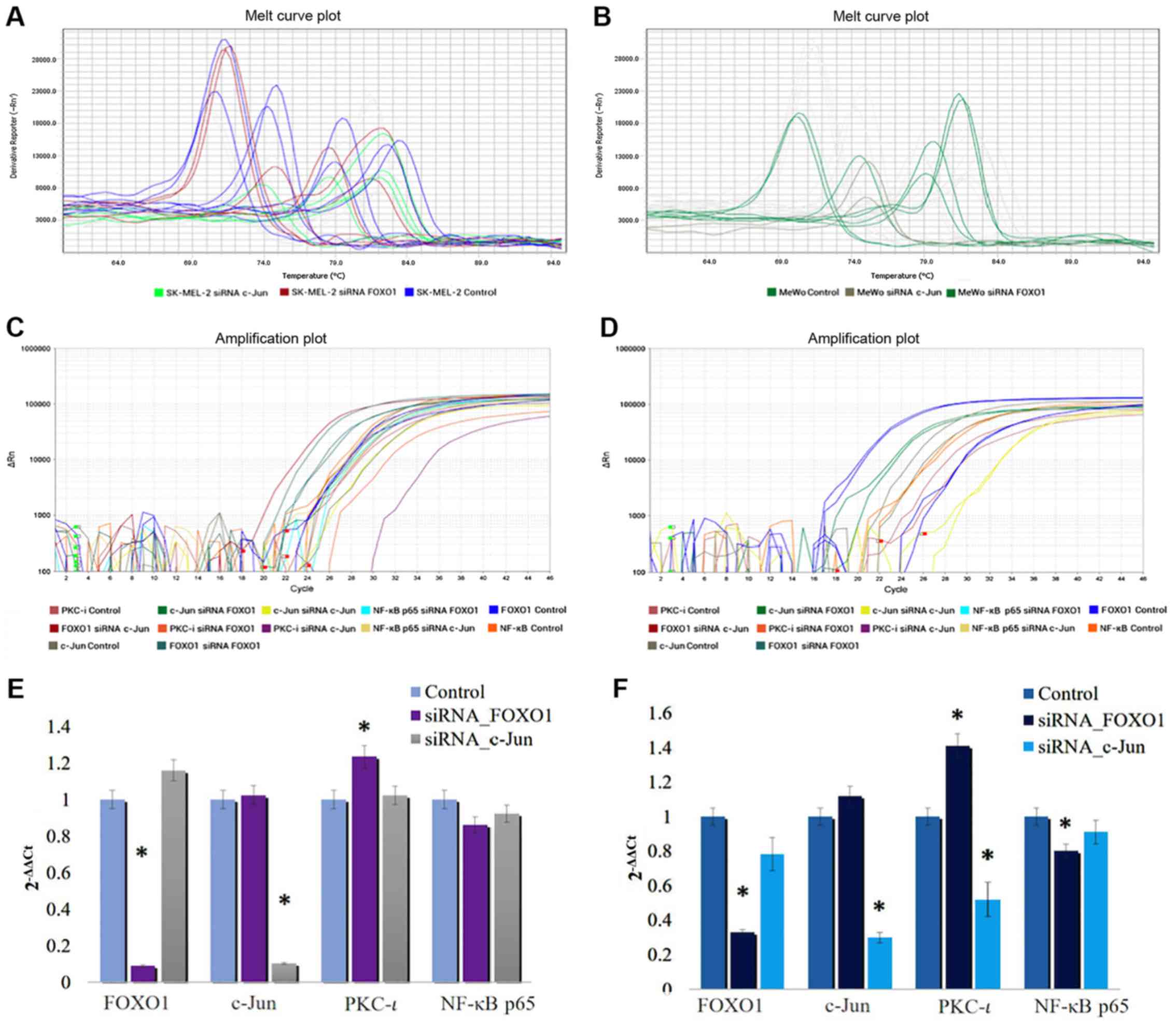
As shown in Fig.
2, the mRNA level of FOXO1 significantly decreased by 90.8 and
67.3% in the SK-MEL-2 and MeWo cells, respectively, following
transfection with FOXO1 siRNA. The mRNA level of c-Jun
significantly decreased by 89.7 and 30.15% in the SK-MEL-2 and MeWo
cells, respectively, following transfection with c-Jun siRNA. These
data confirmed the efficiencies and specificities of the applied
siRNAs. The RT-qPCR data revealed that the PKC-ι mRNA levels
decreased (by 47.2%) following transfection with siRNA for c-Jun in
the MeWo cells, even though the PKC-ι levels in the SK-MEL-2 cells
were not significantly altered. On the other hand, the PKC-ι mRNA
levels were significantly increased by 23.5 and 42% in the SK-MEL-2
and MeWo cells transfected with FOXO1 siRNA, respectively.
Therefore, the results of RT-qPCR results tally with the protein
expression, which is shown in Fig.
1. These results indicate that the downregulation of FOXO1
expression enhances PKC-ι expression, while the silencing of-Jun
expression reduces PKC-ι expression. This indicates that FOXO1
downregulates the expression of PKC-ι and c-Jun upregulates PKC-ι
expression in melanoma cells in vitro.
FOXO1 holds the key to PKC-ι
expression in melanoma cells
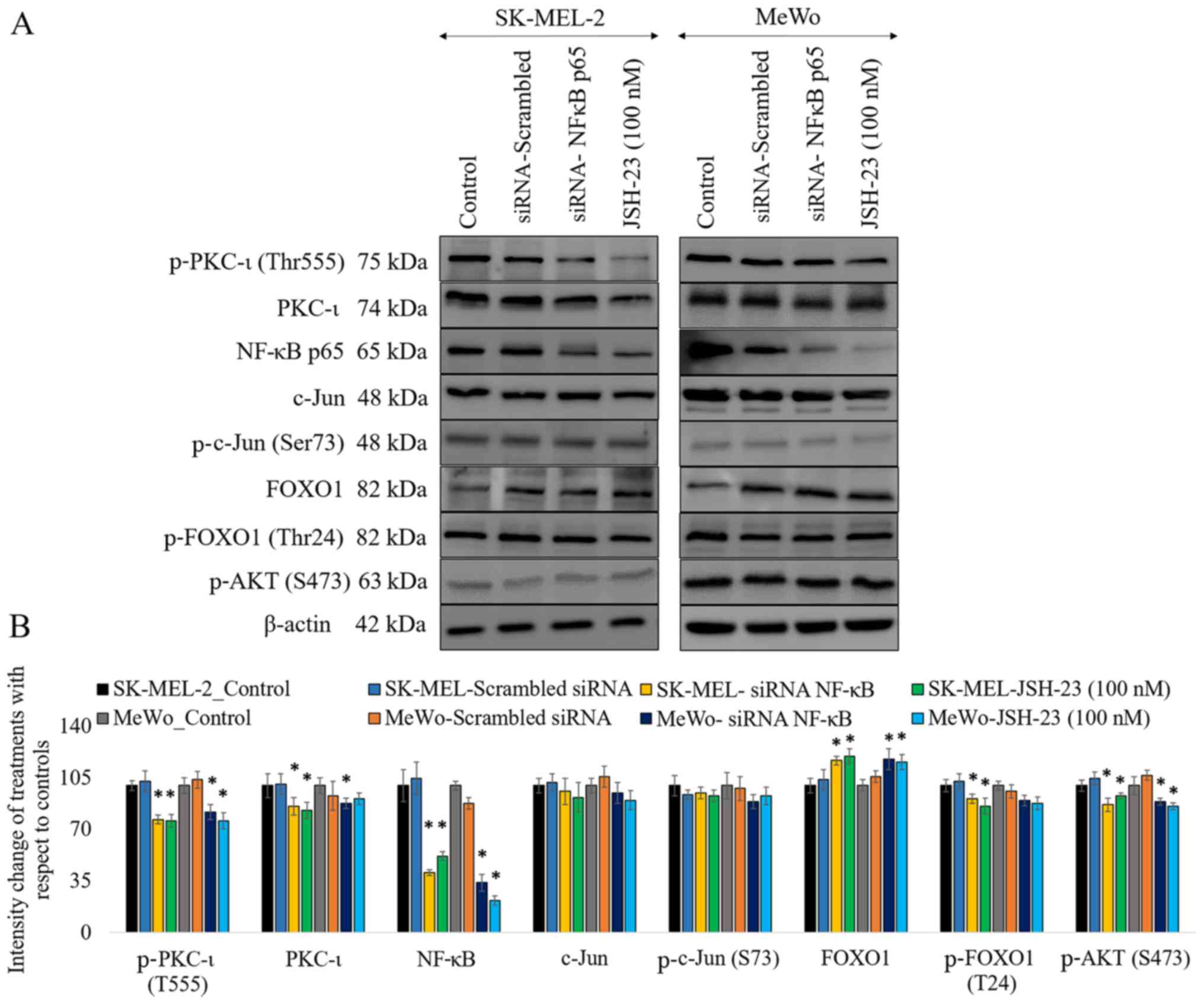
As shown in Fig.
3, the results of western blot analysis revealed that the
knockdown of the expression of NF-κB by siRNA significantly
decreased the levels of phosphorylated PKC-ι by 23% (P≤0.05) and
18% (P≤0.05) in the SK-MEL-2 and MeWo cells, respectively. The
levels of total PKC-ι significantly decreased by 14% (P≤0.05) and
12% (P≤0.05) in the SK-MEL-2 and MeWo cells, respectively. Of note,
FOXO1 expression increased by 17% (P≤0.05) and 18% (P≤0.05) in the
SK-MEL-2 and MeWo cells, whereas the levels of phosphorylated FOXO1
decreased in the cells transfected with NF-κB siRNA. As we have
reported previously, the inhibition of PKC-ι significantly
downregulated the PI3K/AKT pathway, thereby suppressing the
activation of AKT (3).
Phosphorylated AKT (S473) phosphorylates FOXO1 to cause nuclear
exclusion and thereby the degradation of FOXO1. This explains the
elevated levels of active FOXO1 that are due to the downregulation
of phospho-AKT upon the downregulation of NF-κB caused by PKC-ι
knockdown by siRNA. In this study, the silencing of NF-κB decreased
the levels of phosphorylated AKT (S473) by 13% (P≤0.05) and 11%
(P≤0.05) in the SK-MEL-2 and MeWo cells, respectively. Notably, the
levels of phosphorylated c-Jun (S73) and total c-Jun were not
significantly altered upon NF-κB knockdown. Similar results were
obtained with NF-κB inhibition using a well-known NF-κB inhibitor,
JSH-23 (100 nM).
Results of ELISA confirm an interplay
between the PI3K/AKT, JNK, NF-κB and STAT signaling pathways upon
PKC-ι inhibition to coordinate the regulation of its
expression
We used ICA-1T (with the tested IC50 concentration
of 1 µM) to specifically inhibit PKC-ι, allowing us to obtain a
broad picture of how multiple pathways may influence melanoma cells
in vitro as a result of PKC-ι inhibition related to PKC-ι
regulation. IPAD assay is an array-based ELISA allowing the
simultaneous detection of multiple proteins. As shown in Fig. 4, the caspase-3, CD44, CHOP,
E-cadherin, IκBα, Myc, NOTCH, p-4E-BP1 (T37/46), p-AKT (S473),
p-β-catenin (S33/37), p-HER3 (Y1289), p-IRS-1 (S1101), p-JNK
(T183), p-MEK1 (S217/221), p-mTOR (S2448), p-NF-κB p65 (S536),
p-SMAD1 (S463), p-SMAD2 (S465/467), p-STAT3 (Y705), p-STAT5 (Y694),
p-YAP1 (S127), p-ZAP70 (Y493), p21 and PARP levels were reported
upon ICA-1T treatments against the controls for both cell lines.
The caspase-3, E-cadherin, p-NF-κB p65 (S536), p-ZAP70 (Y493) and
p21 levels significantly increased upon PKC-ι inhibition, while the
CD44, p-4E-BP1 (T37/46), p-AKT (S473), p-IRS-1 (S1101), p-STAT3
(Y705), p-STAT5 (Y694), p-YAP1 (S127) and PARP levels significantly
decreased.
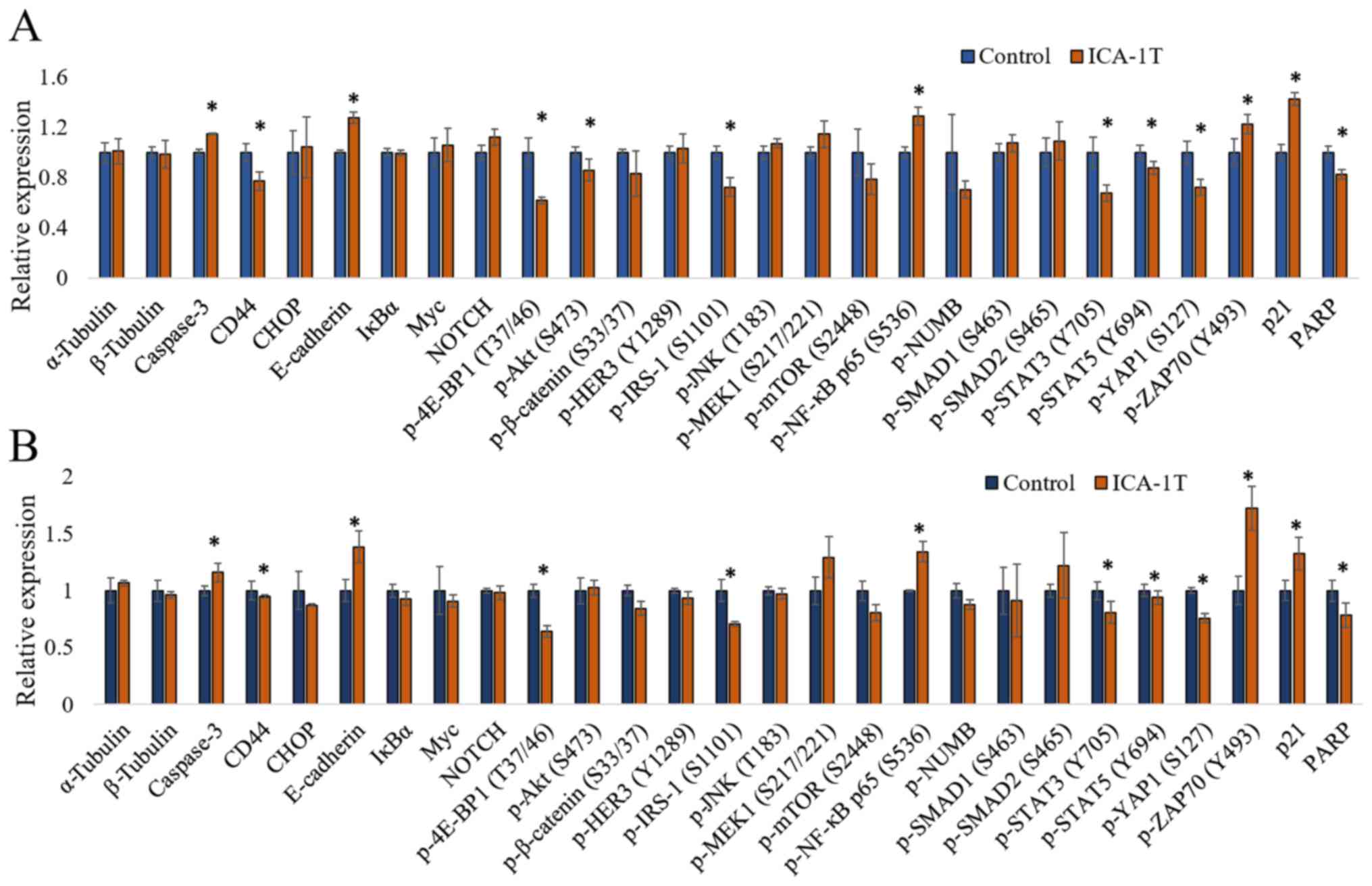 | Figure 4.Immuno-paired antibody detection
assay (IPAD) for melanoma cells (SK-MEL-2 and MeWo). (A and B) The
expression of IPAD assay targets for SK-MEL-2 and MeWo cell lines,
respectively. Approximately 1x105 cells were cultured in T75 flasks
and 24 h post-plating, fresh medium was supplied and the cells were
treated with either volume of sterile water (control) or IC50
concentration of ICA-1T (1 µM). Additional doses were supplied
every 24 h during a 3-day incubation period. The cells were then
lysed and prepared lysates with the final total protein
concentration to be >2 µg/ml and then sent them to ActivSignal,
LLC facility to conduct the IPAD assay. IPAD platform is a
proprietary multiplexed ELISA technology for analyzing the activity
of multiple signaling pathways in one reaction. Activities of
multiple signaling pathways were monitored simultaneously in a
single well through assessing the expression or protein
phosphorylation of 25 target human proteins, such as caspase-3,
CD44, CHOP, E-cadherin, IκBα, Myc, NOTCH, p-4E-BP1, p-AKT (S473),
p-β-catenin, p-HER3, p-IRS-1, p-JNK, p-MEK1, p-mTOR, p-NF-κB,
p-NUMB, p-SMAD1, p-SMAD2, p-STAT3, p-STAT5, p-YAP1, p-ZAP70, p21
and PARP. α-tubulin and β-tubulin were used as internal controls in
each trial. Experiments (n=3) were performed in each cell lines and
the means ± SD are plotted. Statistical significance is indicated
by an asterisk (*P≤0.05). |
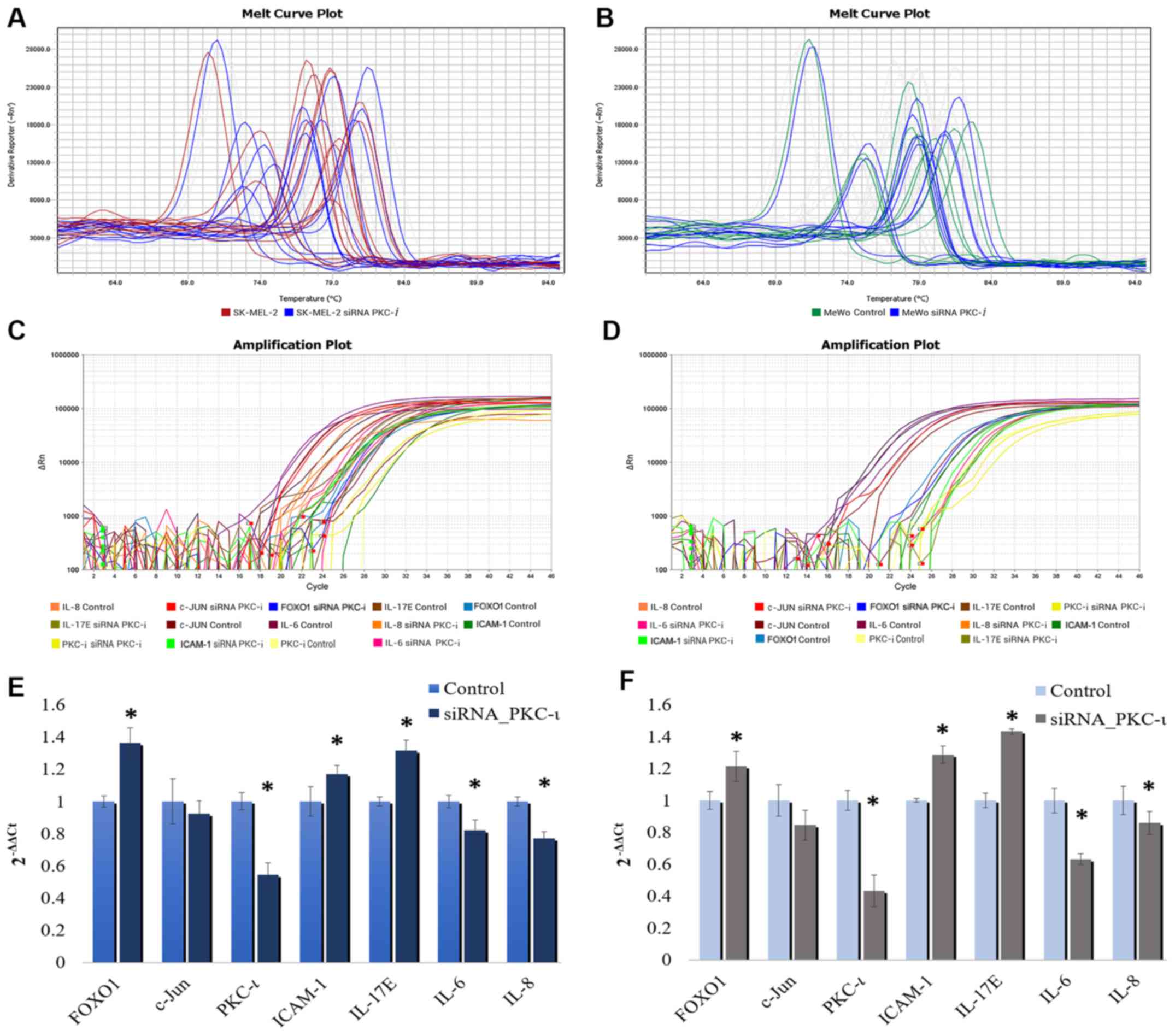
IL-6, IL-8, IL-17E and ICAM-1 may
participate in PKC-ι regulation in an autocrine manner through
transcriptional activation/deactivation
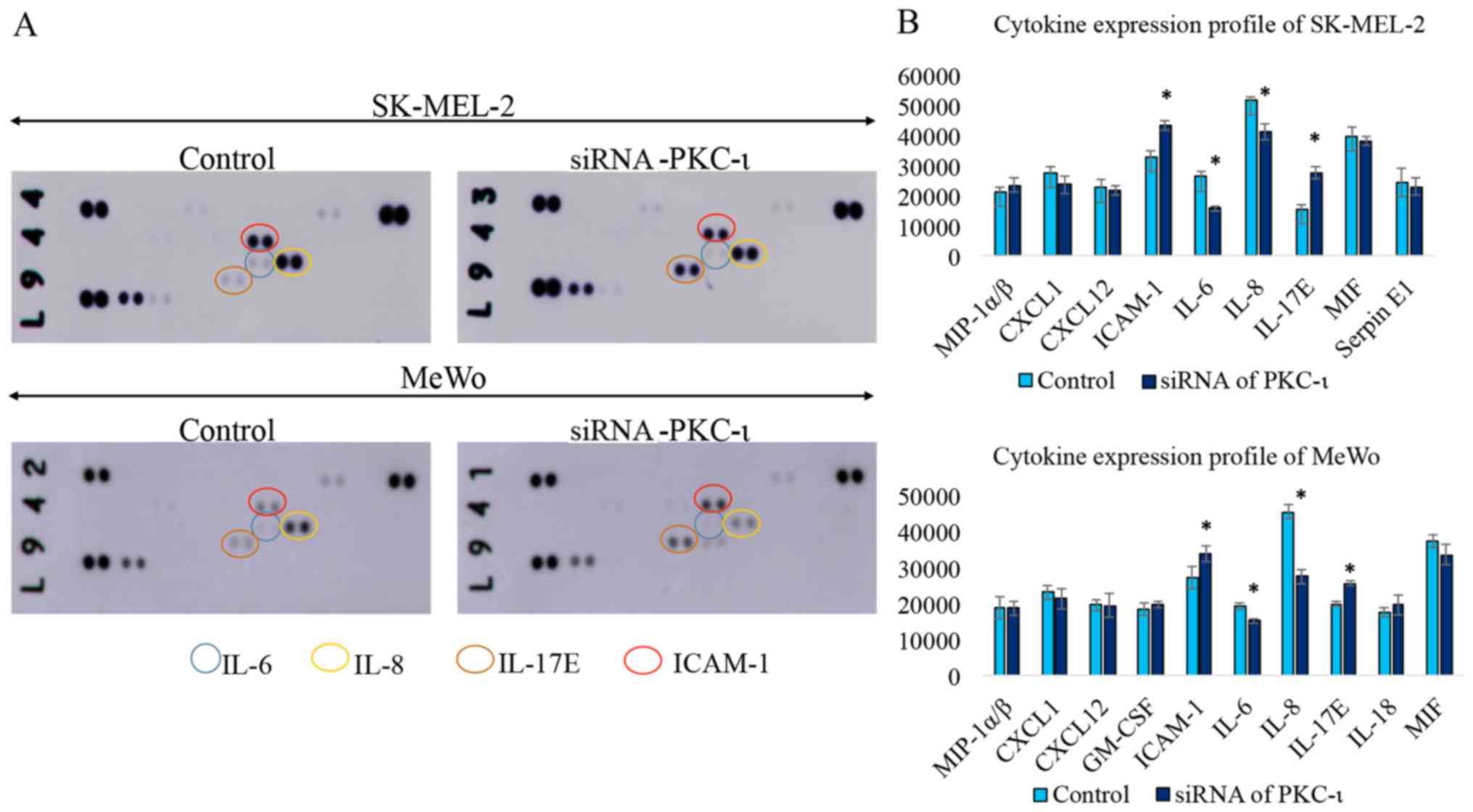
As shown in Fig.
5, the western blot cytokine expression profile for the two
melanoma cell lysates exhibited a significant decrease in the
levels of IL-6 and IL-8, while the levels of IL-17E and ICAM-1
increased in the cells transfected with PKC-ι siRNA (siRNA for
PKC-ι, 20 nM) compared to the control (scrambled siRNA, 20 nM). We
also found CXCL-1, CXCL-12, GM-SCF, MIF and Serpin in detectable
levels, although these were not significantly altered due to PKC-ι
knockdown. The results of the RT-qPCR analysis of the same lysates
are shown in Fig. 6. Since only
IL-6, IL-8, IL-17E and ICAM-1 exhibited a significant change in
expression upon PKC-ι silencing by siRNA, we only tested these for
the mRNA levels in the RT-qPCR experiments. These RT-qPCR data
revealed how the mRNA levels of PKC-ι significantly decreased by
35.6 and 56.7% in the SK-MEL-2 and MeWo cells, respectively
following transfection with PKC-ι siRNA. We observed a significant
decrease in the mRNA levels of both IL-6 and IL-8, with a
significant increase in the levels of IL-17E and ICAM-1 upon the
knockdown of expression of PKC-ι in both cell lines. In addition,
we found that the FOXO1 mRNA levels increased significantly by 36.1
and 21.5% in the SK-MEL-2 and MeWo cells, respectively; the c-Jun
mRNA levels decreased by 8 and 15.5% in the SK-MEL-2 and MeWo
cells, respectively. These data confirm the association between
PKC-ι, FOXO1 and c-Jun presented in Figs. 1 and 2.
Fig. 7 shows a
schematic summary of the regulation of the expression of PKC-ι in
melanoma based on the current and previous data (3,4).
This model depicting the interactions between NF-κB,
PI3K/AKT/FOXO1, JNK/c-Jun and STAT3/5 signaling pathways during the
PKC-ι regulation. It is shown that PKC-ι plays a very important
role in the regulation of its expression through the
transcriptional activation/deactivation of c-Jun and FOXO1. PKC-ι
is overexpressed as a result of c-Jun transcriptional activity with
the help of pro-survival, oncogenic PI3K/AKT, NF-κB, STAT3/5
signaling cascades in melanoma cells. Specific inhibitors of PKC-ι
initiates a disruption to rapid PKC-ι expression cycle where the
reduced activity of PKC-ι downregulates the NF-κB pathway and its
transcriptional activity thereby decreases the expression of IL-6
and IL-8. The activity of AKT decreases as a result of lacking the
stimulation from cytokines, such as IL-6, IL-8 and TNF-α, which
leads to the upregulation of FOXO1, which turns out to be the most
important TF regulating PKC-ι expression after the disruption
initiated as a result of PKC-ι inhibition. FOXO1 negatively
regulates the expression of PKC-ι and also diminishes the JNK
activity to retard its activation of c-Jun. We found c-Jun as the
transcription component which upregulates PKC-ι expression. This
process continues and leads to the further downregulation of NF-κB,
c-Jun and the upregulation of FOXO1, which leads to the
continuation of the diminution of PKC-ι expression. As a result,
the total PKC-ι level decreases in melanoma cells. These findings
strongly support our previous data where a reduction in total PKC-ι
levels was observed upon the specific inhibition using PKC-ι
inhibitors, such as ICA-1T and ICA-1S (3,4).
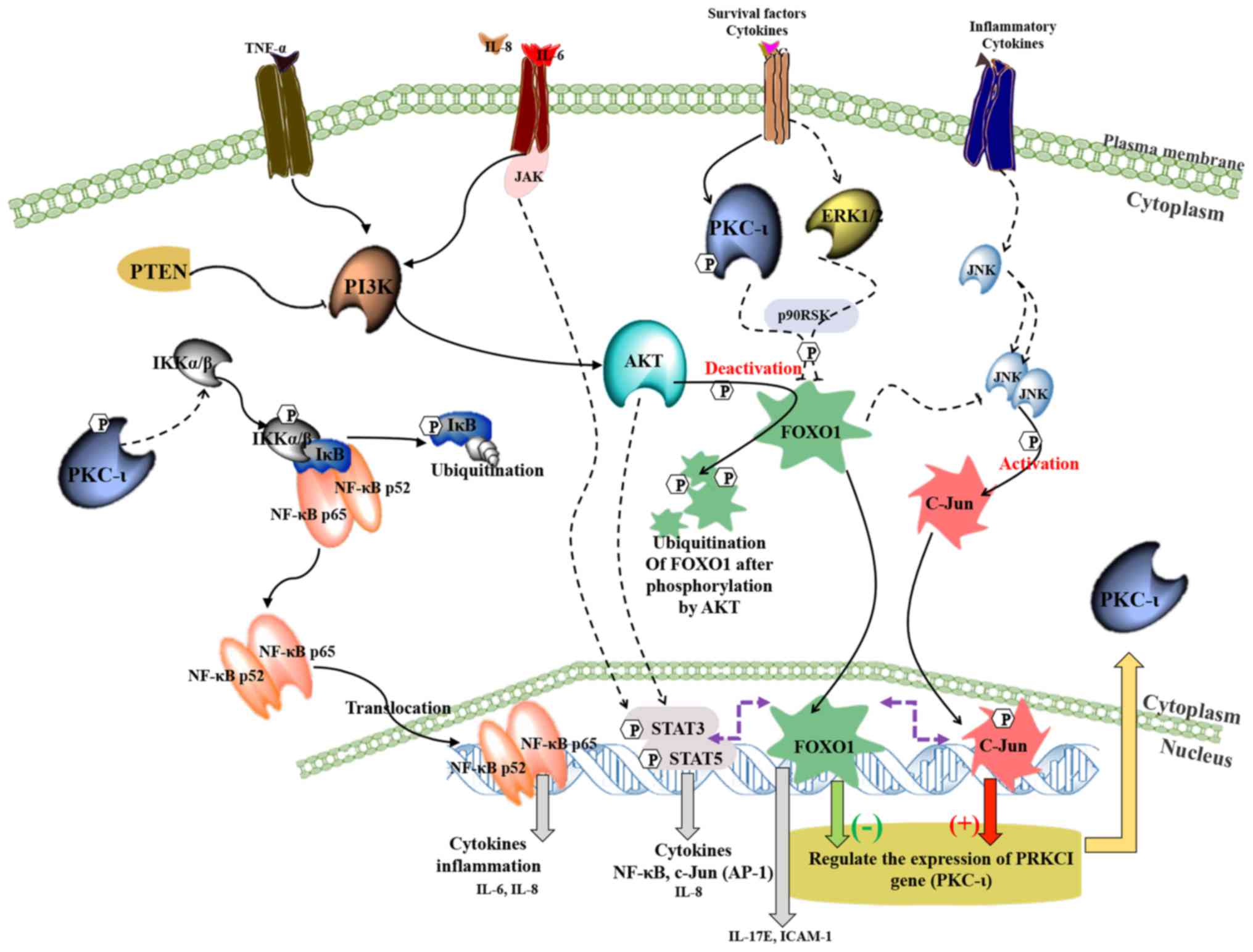 | Figure 7.A schematic summary of the regulation
of the expression of PKC-ι in melanoma. This model depicting how
the crosstalk takes place between NF-κB, PI3K/AKT/FOXO1, JNK/c-Jun
and STAT3/5 signaling pathways during the PKC-ι regulation. It is
shown that PKC-ι plays a very important role in the regulation of
its expression in a complex signaling network through the
transcriptional activation/deactivation of c-Jun and FOXO1. In
melanoma cancer cells, PKC-ι is overexpressed as a result of c-Jun
transcriptional activity with the help of pro-survival, oncogenic
PI3K/AKT, NF-κB, STAT3/5 signaling cascades. The inhibition of
PKC-ι initiates a disruption to rapid PKC-ι expression cycle where
the reduced activity of PKC-ι downregulates the NF-κB pathway and
its transcriptional activity, which in turn depletes the expression
of IL-6 and IL-8. The activity of AKT decreases as a result of
lacking the stimulation from cytokines, such as IL-6, IL-8 and
TNF-α, which leads to the upregulation of FOXO1, which turns out to
be the most important TF regulating PKC-ι expression after the
disruption initiated as a result of PKC-ι inhibition. FOXO1
negatively regulates the expression of PKC-ι and also diminishes
the JNK activity to retard its activation of c-Jun. We found c-Jun
as the transcription component which upregulates PKC-ι expression.
The downregulation of IL-6 and IL-8 expression leads to the
impaired STAT3/5 signaling, which causes c-Jun transcriptional
reduction. This whole process continues and leads to the further
downregulation of NF-κB, c-Jun and the upregulation of FOXO1, which
leads to the continuation of the diminution of PKC-ι expression. As
a result, the total PKC-ι level decreases in melanoma cells. These
findings strongly support our previous data where a reduction in
total PKC-ι levels was observed upon the specific inhibition using
PKC-ι inhibitors, such as ICA-1T and ICA-1S (3,4). |
Discussion
In our previous study, we demonstrated that ICA-1T
and ICA-1S inhibited PKC-ι by selectively binding to a druggable
allosteric site within the C-lobe of PKC-ι kinase domain, thereby
blocking the activity of PKC-ι. These inhibitors reduced cell
proliferation, migration and invasion, while inducing apoptosis in
melanoma in vitro via the downregulation of the NF-κB pathway.
Therefore, we identified PKC-ι as a major component responsible for
inducing cell growth, differentiation, survival and EMT promotion
in melanoma, as a result of PKC-ι specific inhibitor applications
(3,4). Apart from these findings, we noted
that the inhibition of PKC-ι leads to a decrease in its own
expression. This indicates that PKC-ι plays a role in its
expression in melanoma. The goal of the current study was therefore
to identify the PKC-ι regulatory mechanisms in melanoma in
vitro.
The PRKCI gene is located on chromosome 3 (3q26.2),
a region identified as an amplicon with the tendency to undergo
replication events (18). To
identify TFs which regulate the PRKCI gene, we selected a specific
sequence which includes the PRKCI promoter with a motif feature,
promoter flank and an enhancer. This area provides the optimal
platform for TFs to bind to regulate the transcription, so the
selection of this sequence is highly justified. Possible TF
bindings were predicted using two different systems: PROMO and
Genomatix Matinspector. Through these, we identified 5 TFs,
including FOXO1 and c-Jun. We systematically silenced these TFs to
analyze the downstream effect on PKC-ι expression.
c-Jun is a transcription factor that combines with
AP-1 and c-Fos to form an early response transcription factor
complex (19). c-Jun is activated
by phosphorylation at S63 and S73 by c-Jun N-terminal kinases
(JNKs), and c-Jun expression is regulated by various extracellular
stimuli, such as cytokines (20).
Extracellular signal-regulated kinase (ERK) increases c-Jun
transcription (21). c-Jun is the
first discovered oncogenic TF that is associated with metastatic
breast cancer, non-small cell lung cancer and several other types
of cancer (22-24).
Phosphorylation at S63 and S73 activates c-Jun, thereby increasing
transcription of c-Jun targeted genes. c-Jun promotes the oncogenic
transformation of 'ras' and 'fos' in several types of cancer
(25,26). In addition, FOXO1 regulates
gluconeogenesis, insulin signaling and adipogenesis.
Phosphorylation plays a key role in the function of FOXO1 (27,28). AKT phosphorylates FOXO1 at T24,
which causes FOXO1 to drive nuclear exclusion, leading to
ubiquitination (29,30). Therefore, the phosphorylation of
FOXO1 is an indication of its downregulation. FOXO1 is a
well-known, bona fide tumor suppressor (31-33).
It plays a regulatory role in both the intrinsic and extrinsic
pathways of apoptosis in many types of cancer, exhibiting an
association between FOXO dysregulation and cancer progression
(34,35). Furthermore, the overexpression of
FOXO1 decreases cancer cell proliferation and inhibits migration
and tumorigenesis. In vitro and in vivo experiments have proven
this tumor suppressing activity (36). Importantly, FOXO1 can also be
downregulated by ERK1/2 and PKC-ι, in addition to AKT (33). In the current study, we
demonstrate that, due to PKC-ι inhibition, the availability of
active PKC-ι decreases so that it becomes ineffective at
deactivating FOXO1 through phosphorylation. Importantly, this is
one of the direct involvements of PKC-ι in its own expression
regulation and PKC-ι inhibition that leads to continuous
upregulation of FOXO1.
On the other hand our previous data showed that
PKC-ι inhibition significantly downregulated the PI3K/AKT pathway,
thereby suppressing the activation of AKT (3,4).
In this study, we provide additional data for the downregulation of
NF-κB, which reduces the activity of AKT. This affects FOXO1, as
shown by the significantly higher levels of total FOXO1, while a
reduction in its phosphorylated levels was observed, suggesting
that NF-κB downregulation upregulates FOXO1 activity. The elevated
levels of FOXO1 negatively influenced PKC-ι expression and
phosphorylation as shown in Fig.
3 as result of NF-κB depletion. This further confirms our
previous observations with PKC-ι inhibition with ICA-1T and ICA-1S,
where total PKC-ι, phosphorylated PKC-ι, NF-κB activation and
activated AKT (S473) were significantly reduced (3). These results could be due to the
tight regulation of PKC-ι expression by FOXO1, which retards PRKCI
from transcription. Such results confirmed that FOXO1 is a major
regulator which suppresses the expression of PKC-ι regulation. Of
note, the c-Jun and phosphorylated c-Jun (S63) levels were not
significantly altered as a result of NF-κB siRNA knockdown. This
suggests that NF-κB downregulation does not affect PKC-ι expression
through c-Jun. Instead, c-Jun protects cancer cells from apoptosis
by cooperating with NF-κB to prevent apoptosis upon TNF-α
stimulation (19). We have
previously shown how TNF-α upregulates NF-κB, phospho-AKT and PKC-ι
expression in these two melanoma cell lines (3). However, the data from the current
study suggest that the TNF-α downstream target is mainly FOXO1,
where it ‘switches off’ through the phosphorylation of elevated
AKT. The inhibition of PKC-ι diminishes this AKT activation,
thereby upregulating FOXO1 activity.
On the other hand, our systematic silencing of
c-Jun, FOXO1 and EGR1 revealed that (Figs. 1 and 2) c-Jun also seemed to regulate PKC-ι
expression, apart from FOXO1. To explain the crosstalk between
these cell signaling pathways in relation to PKC-ι regulation, we
conducted two other in vitro experiments, ELISA using IPAD assay
and a cytokine array. These findings demonstrated links between
PKC-ι expression with the cytokines, IL-6, IL-8, IL-17E and ICAM-1,
along with some other key cellular signaling points.
As shown in Fig.
4, the IPAD ELISA data revealed a significant increase in the
levels of caspase-3, E-cadherin, p-NF-κB p65 (S536), p-ZAP70 (Y493)
and p21 levels upon PKC-ι inhibition, while the levels of CD44,
p-4E-BP1 (T37/46), p-AKT (S473), p-IRS-1 (S1101), p-STAT3 (Y705),
p-STAT5 (Y694), p-YAP1 (S127) and PARP levels significantly
decreased. We have already shown in our previous study, using
various apoptotic markers, that PKC-ι inhibition induced the
apoptosis of melanoma cells (3,4).
This is again evident from increases in the levels of caspase-3 and
the cleavage of PARP in the IPAD assay. Additionally, PKC-ι
inhibition delayed EMT in melanoma and we observed elevated levels
of E-cadherin with downregulation of Vimentin and CD44 expression
(3,4). Therefore, this IPAD assay provided
additional data for the results we have shown in our previous
studies. Phosphorylation at S536 on the NF-κB p65 transactivation
domain is an indication of dimerization of NF-κB subunits. Since
PKC-ι inhibition downregulates NF-κB translocation to the nucleus,
phospho-NF-κB levels increase in order to diminish the effect of
PKC-ι inhibition. However, elevated FOXO1 does not allow NF-κB to
take over the control since it is missing the crucial assistance
needed from PKC-ι due to its inhibition from ICA-1T and ICA-1S
inhibitors. Aberrant STAT3/5 activity has been shown to be
connected to multiple types of cancer (37-42).
The cytokines, IL-6 and IL-5, are known to upregulate STAT
signaling, which induces cell survival in many types of cancer
(37,38,43). Upregulated STAT3 increases the
transcription of c-Jun (37,44). Our IPAD results indicated that
STAT3 and STAT5 activities were downregulated due to PKC-ι
inhibition, suggesting that c-Jun expression can be retarded. Few
studies, including the one by Hornsveld et al, have provided
connections between the JNK pathway and FOXO1, elaborating its
tumor suppressing features for weakening JNK activity (33,45,46). However, JNK activates c-Jun. The
data from our western blot and RT-qPCR analysis demonstrated that
c-Jun depletion diminished PKC-ι expression, which suggested that
c-Jun acts as an activator of PKC-ι expression. It is therefore
evident that both FOXO1 and c-Jun are involved in regulating PKC-ι
expression. The results suggest that FOXO1 plays a major role over
c-Jun only upon PKC-ι inhibition, possibly through multiple
mechanisms, such as the reduction of JNK signaling, retarding PKC-ι
expression and cell cycle arrest. Upregulated FOXO1 is well known
to induce cell cycle arrest by promoting the transcription of cell
cycle kinase inhibitors or cyclin-dependent kinase inhibitor (CKI).
p21 and p27 are two of the most well-known downstream CKIs induced
by FOXOs (33,45). Notably, FOXO1 is also believed to
induce anoikis, which is apoptosis that occurs when cells detach
from the extracellular matrix. Our IPAD-ELISA results revealed
significantly elevated levels of p21 by approximately 25% in both
cell lines, suggesting that the inhibition of PKC-ι induces cell
cycle arrest through FOXO1. This is another indirect downstream
effect of PKC-ι involvement in its expression where inhibition of
PKC-ι enhances FOXO1 anti-tumor activity.
As shown in Fig.
7, our results summarize that PRKCI expression is negatively
regulated by FOXO1 and positively regulated by c-Jun. When PKC-ι is
inhibited, the following series of downstream effects take place.
The downregulation of NF-κB activity decreases the levels of
phospho-AKT (S473). As a result of a low activity of AKT along with
diminished levels of PKC-ι, the phosphorylation of FOXO1 is reduced
and therefore active FOXO1 (unphosphorylated FOXO1) levels are
being elevated. Elevated FOXO1 suppresses PRKCI gene expression
similar to 'switch off' effect. The downregulation of PKC-ι also
diminishes STAT3/5 activity, as shown by the IPAD assay data. STAT3
and STAT5 are known to upregulate the transcription of c-Jun and
NF-κB (44,47). Therefore, it is evident that PKC-ι
inhibition induces the downregulation of STAT3/5, which decreases
the transcription and activation c-Jun. These data suggest that
PKC-ι levels were decreased when c-Jun expression is silenced by
siRNA (Fig. 1), and as shown by
RT-qPCR in Fig. 6, the knockdown
of PKC-ι decreased c-Jun mRNA expression, but not
significantly.
As shown in Fig.
5, further in vitro experiments demonstrated changes in
cytokine expression (IL-6, IL-8, IL-17E and ICAM-1) in melanoma
cells upon PKC-ι knockdown. As shown by the results of both western
blot and RT-qPCR analyses, the protein levels of IL-6 and IL-8 (as
well as their mRNA levels) decreased, while the levels of IL-17E
and ICAM-1 increased significantly in both cell lines upon PKC-ι
knockdown by siRNA. This suggests that the PKC-ι self-regulated
expression cycle is involved in autocrine signaling. The cellular
environment of a tumor, and in particular melanoma, is frequently
exposed to various inflammatory factors and immune cells. The
effect of these factors function to either promote chronic
inflammation or engage in anti-tumor activity (48). Cytokines are examples of these
inflammatory factors; they play an essential role in regulating
tumor microenvironments (49).
Cytokines utilize several signaling pathways to carry out their
functions. They act in order to promote or dysregulate tumor
progression and metastasis. Cytokines, such as CXCL-1, CXCL-12,
IL-18, CXCL-10, IL-6 and IL-8 promote cancer progression by
facilitating metastasis. CXCL1, also known as melanoma
growth-stimulatory activity/growth-regulated protein α, is secreted
by melanoma cells and is associated with roles in wound healing,
angiogenesis and inflammation. In particular, it has been linked to
tumor formation (50). High
levels of CXCL10/CXCR3 expressed in melanoma have been linked to
metastasis regulation (50).
CXCL10 plays an important role in promoting tumor growth and
metastasis (51). CXCL12, also
known as stromal-derived factor-1, utilizes the receptors CXCR4 and
CXCR7. CXCL12 and its receptors have been linked to roles in
regulating tumor metastasis. CXCL-1, CXCL-10, CXCL-12 and IL-18
levels were not significantly altered due to PKC-ι depletion.
IL-6 is a very important cytokine and contributes to
the degradation of IκB-α, leading to the upregulation of NF-κB
translocation. We have previously shown that PKC-ι stimulates NF-κB
translocation through IκB-α degradation (3). The translocation of NF-κB to the
nucleus induces cell survival through the transcription of various
survival factors as well as other cytokines (37,43,52). IL-8 is an example of such a
cytokine. IL-8 plays a role in regulating polymorphonuclear
neutrophil mobilization. In melanoma, IL-8 has been attributed to
extravasation, a key step in metastasis. Studies have shown that
the expression of IL-8 in melanoma is regulated via NF-κB. When
NF-κB is translocated to the nucleus, IL-8 expression increases,
leading to the promotion of a more favorable microenvironment for
metastasis (53,54). The results of this study indicated
that both IL-6 and IL-8 expression levels decrease upon
transfection with PKC-ι siRNA. As is summarized by the diagram in
Fig. 7, IL-6 expression is
regulated by NF-κB, and IL-8 expression is regulated by both NF-κB
and STATs. Our data justified how the PKC-ι inhibition/knockdown
downregulates the NF-κB and STAT signaling pathways. The results
suggested that IL-6 and IL-8 play an important role in upregulating
PKC-ι expression, activating c-Jun, while deactivating FOXO1.
Some cytokines promote anti-tumor activity by
utilizing an immune response. ICAM-1 plays a key role in the immune
response, including antigen recognition and lymphocyte activation
(55,56). ICAM-1 has been linked to the
inhibition of tumor progression through the inhibition of the
PI3K/AKT pathway. The inhibition of this pathway via ICAM-1 exposes
tumor cells to death via cytotoxic T-lymphocytes (56). Clinical research has also proven
that, within the first 5 years of ovarian cancer diagnosis, ICAM-1
expression inhibition is associated with an increased risk of
metastasis (55,56). Another anti-tumor cytokine is
IL-17E. IL-17E belongs to a family of cytokines known as IL-17. In
various forms of cancer, including melanoma and pancreatic cancers,
treatment with recombinant IL-17E has been shown to decrease tumor
growth (57,58). The upregulation of IL-17E is
linked to the increased expression of TH17 cells. T cells, such as
TH17 have been implicated in the inhibition of tumor-infiltrating
effector T cells. The exact mechanism of IL-17E function in the
anti-tumor effect has not been thoroughly explored (59). Notably, the results of our western
blot and RT-qPCR analyses indicated that ICAM-1 and IL-17E protein
and mRNA expression increased upon the silencing of PKC-ι by siRNA.
This confirms that anti-tumor/pro-apoptotic signaling is
upregulated upon the knockdown of oncogenic PKC-ι via an autocrine
manner through IL-17E and ICAM-1. Moreover, the results suggest
that IL-17E and ICAM-1 play an important downregulatory role in the
regulation of PKC-ι expression along with FOXO1, opposite to c-Jun,
IL-6 and IL-8.
In conclusion, our overall results demonstrate PKC-ι
itself to play an important role in its expression in a complex
signaling network through the transcriptional activation/
deactivation of c-Jun and FOXO1. The reduced activity of PKC-ι due
to its specific inhibition, downregulates the NF-κB pathway and its
transcriptional activity, which turns out to 'strike' the
expression of IL-6 and IL-8. As a result, the activity of AKT
decreases, which leads to the upregulation of FOXO1. FOXO1 is the
most important TF regulating PKC-ι expression upon receiving
stimulation from IL-17E and ICAM-1. FOXO1 negatively regulates the
expression of PKC-ι, diminishing JNK activity to retard the
activation of c-Jun. IL-6 and IL-8 expression are downregulated via
PKC-ι-mediated NF-κB transcriptional activity reduction. This leads
to STAT3/5 signaling downregulation, reducing c-Jun expression.
This whole process continues and leads to the further
downregulation of NF-κB, c-Jun and upregulation of FOXO1, which
leads to the continuation of the depletion of PKC-ι expression. As
a result of this sequence of events, the total PKC-ι level
decreases in melanoma cells, which began as a result of PKC-ι
inhibition. These results indicate that PKC-ι is being regulated in
a rather complex manner, which involves itself as a key component.
PKC-ι inhibition leads to a decrease in its own production, and
during this process, PKC-ι inhibition also triggers multiple
anti-tumor/pro-apoptotic signaling. This makes PKC-ι one of the
central key point of interest to specifically target and diminish
as a means of treating melanoma in vitro. Therefore, our overall
results confirm that PKC-ι inhibition using specific and effective
inhibitors, such as ICA-1T and ICA-1S, is significantly effective
in the treatment of melanoma. The results also strongly suggest
that PKC-ι is a prime novel biomarker that can be targeted to
design and develop personalized and targeted therapeutics for
melanoma.
Acknowledgements
The authors would like to thank Dr Meera Nanjundan
and Ms. Stephanie Rockfield for their valuable suggestions on the
PROMO and Genomatix applications and data interpretation.
Funding
The authors acknowledge the generous financial
contributions from the Frederick H. Leonhardt Foundation, David
Tanner Foundation, Bradley Zankel Foundation, Inc., Kyrias
Foundation, Brotman Foundation of California, Baker Hughes
Foundation, Irving S. Cooper Family Foundation, and the Creag
Foundation.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AHA, WSR and MAD were involved in the
conceptualization of the study. WSR and CAA were involved in the
data analysis. MAD and WSR were involved in the investigation; WSR
and SB were involved in cell culturing. WSR was involved in western
blot analysis. WSR, SB and CAA were involved in the cytokine
analysis and sample preparation for IPAD assay and RT-qPCR. CAA, SB
and WSR were involved in the RT-qPCR analysis. WSR and SB were
involved in the writing of the original draft. MAD was involved in
the writing and reviewing of the manuscript. WSR and CAA edited the
manuscript. MAD were involved in obtaining resources and was also
involved in the supervision of the study and funding acquisition.
All authors have read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cronin KA, Lake AJ, Scott S, Sherman RL,
Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, et
al: Annual Report to the Nation on the Status of Cancer, part I:
National cancer statistics. Cancer. 124:2785–2800. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ratnayake WS, Apostolatos CA, Apostolatos
AH, Schutte RJ, Huynh MA, Ostrov DA and Acevedo-Duncan M: Oncogenic
PKC-ι activates Vimentin during epithelial-mesenchymal transition
in melanoma; a study based on PKC-ι and PKC-ζ specific inhibitors.
Cell Adhes Migr. 0:1–17. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ratnayake WS, Apostolatos AH, Ostrov DA
and Acevedo-Duncan M: Two novel atypical PKC inhibitors; ACPD and
DNDA effectively mitigate cell proliferation and epithelial to
mesenchymal transition of metastatic melanoma while inducing
apoptosis. Int J Oncol. 51:1370–1382. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ratnayake WS and Acevedo-Duncan M:
Abstract 862: Atypical protein kinase c inhibitors can repress
epithelial to mesenchymal transition (type III) in malignant
melanoma. Cancer Res. 77((Suppl 13)): 862. 2017. View Article : Google Scholar
|
|
6
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Regala RP, Weems C, Jamieson L, Khoor A,
Edell ES, Lohse CM and Fields AP: Atypical protein kinase C iota is
an oncogene in human non-small cell lung cancer. Cancer Res.
65:8905–8911. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Murray NR and Fields AP: Atypical protein
kinase C iota protects human leukemia cells against drug-induced
apoptosis. J Biol Chem. 272:27521–27524. 1997.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Desai S, Pillai P, Win-Piazza H and
Acevedo-Duncan M: PKC-ι promotes glioblastoma cell survival by
phosphorylating and inhibiting BAD through a phosphatidylinositol
3-kinase pathway. Biochim Biophys Acta. 1813:1190–1197.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Win HY and Acevedo-Duncan M: Role of
protein kinase C-iota in transformed non-malignant RWPE-1 cells and
androgen-independent prostate carcinoma DU-145 cells. Cell Prolif.
42:182–194. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Eder AM, Sui X, Rosen DG, Nolden LK, Cheng
KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, et al:
Atypical PKCiota contributes to poor prognosis through loss of
apical-basal polarity and cyclin E overexpression in ovarian
cancer. Proc Natl Acad Sci USA. 102:12519–12524. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Apostolatos AH, Ratnayake WS, Win-Piazza
H, Apostolatos CA, Smalley T, Kang L, Salup R, Hill R and
Acevedo-Duncan M: Inhibition of atypical protein kinase C-ι
effectively reduces the malignancy of prostate cancer cells by
downregulating the NF-κB signaling cascade. Int J Oncol.
53:1836–1846. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wu J, Lu M, Li Y, Shang YK, Wang SJ, Meng
Y, Wang Z, Li ZS, Chen H, Chen ZN, et al: Regulation of a
TGF-β1-CD147 self-sustaining network in the differentiation
plasticity of hepatocellular carcinoma cells. Oncogene.
35:5468–5479. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Venter JC, Adams MD, Myers EW, Li PW,
Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al:
The sequence of the human genome. Science. 291:1304–1351.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fagerberg L, Hallström BM, Oksvold P,
Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S,
Danielsson A, Edlund K, et al: Analysis of the human
tissue-specific expression by genome-wide integration of
transcriptomics and antibody-based proteomics. Mol Cell Proteomics.
13:397–406. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Win HY and Acevedo-Duncan M: Atypical
protein kinase C phosphorylates IKKalphabeta in transformed
non-malignant and malignant prostate cell survival. Cancer Lett.
270:302–311. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Butler AM, Scotti Buzhardt ML, Erdogan E,
Li S, Inman KS, Fields AP and Murray NR: A small molecule inhibitor
of atypical protein kinase C signaling inhibits pancreatic cancer
cell transformed growth and invasion. Oncotarget. 6:15297–15310.
2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wisdom R, Johnson RS and Moore C: c-Jun
regulates cell cycle progression and apoptosis by distinct
mechanisms. EMBO J. 18:188–197. 1999.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Angel P, Hattori K, Smeal T and Karin M:
The jun proto-oncogene is positively autoregulated by its product,
Jun/AP-1. Cell. 55:875–885. 1988.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lopez-Bergami P, Huang C, Goydos JS, Yip
D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson
S, et al: Rewired ERK-JNK signaling pathways in melanoma. Cancer
Cell. 11:447–460. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Vogt PK: Fortuitous convergences: The
beginnings of JUN. Nat Rev Cancer. 2:465–469. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Szabo E, Riffe ME, Steinberg SM, Birrer MJ
and Linnoila RI: Altered cJUN expression: An early event in human
lung carcinogenesis. Cancer Res. 56:305–315. 1996.PubMed/NCBI
|
|
24
|
Vleugel MM, Greijer AE, Bos R, van der
Wall E and van Diest PJ: c-Jun activation is associated with
proliferation and angiogenesis in invasive breast cancer. Hum
Pathol. 37:668–674. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Behrens A, Sibilia M and Wagner EF:
Amino-terminal phosphorylation of c-Jun regulates stress-induced
apoptosis and cellular proliferation. Nat Genet. 21:326–329.
1999.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Nateri AS, Spencer-Dene B and Behrens A:
Interaction of phosphorylated c-Jun with TCF4 regulates intestinal
cancer development. Nature. 437:281–285. 2005.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rena G, Guo S, Cichy SC, Unterman TG and
Cohen P: Phosphorylation of the transcription factor forkhead
family member FKHR by protein kinase B. J Biol Chem.
274:17179–17183. 1999.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nakae J, Kitamura T, Kitamura Y, Biggs WH
III, Arden KC and Accili D: The forkhead transcription factor Foxo1
regulates adipocyte differentiation. Dev Cell. 4:119–129.
2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Matsuzaki H, Daitoku H, Hatta M, Tanaka K
and Fukamizu A: Insulin-induced phosphorylation of FKHR (Foxo1)
targets to proteasomal degradation. Proc Natl Acad Sci USA.
100:11285–11290. 2003.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lu H and Huang H: FOXO1: A potential
target for human diseases. Curr Drug Targets. 12:1235–1244.
2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Borkhardt A, Repp R, Haas OA, Leis T,
Harbott J, Kreuder J, Hammermann J, Henn T and Lampert F: Cloning
and characterization of AFX, the gene that fuses to MLL in acute
leukemias with a t(X;11)(q13;q23). Oncogene. 14:195–202.
1997.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Anderson MJ, Viars CS, Czekay S, Cavenee
WK and Arden KC: Cloning and characterization of three human
forkhead genes that comprise an FKHR-like gene subfamily. Genomics.
47:187–199. 1998.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Farhan M, Wang H, Gaur U, Little PJ, Xu J
and Zheng W: FOXO signaling pathways as therapeutic targets in
cancer. Int J Biol Sci. 13:815–827. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fu Z and Tindall DJ: FOXOs, cancer and
regulation of apoptosis. Oncogene. 27:2312–2319. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang Y, Zhang L, Sun H, Lv Q, Qiu C, Che
X, Liu Z and Jiang J: Forkhead transcription factor 1 inhibits
endometrial cancer cell proliferation via sterol regulatory
element-binding protein 1. Oncol Lett. 13:731–737. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hodge DR, Hurt EM and Farrar WL: The role
of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer.
41:2502–2512. 2005.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yue P and Turkson J: Targeting STAT3 in
cancer: How successful are we? Expert Opin Investig Drugs.
18:45–56. 2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jing N and Tweardy DJ: Targeting Stat3 in
cancer therapy. Anticancer Drugs. 16:601–607. 2005.PubMed/NCBI
|
|
40
|
Page BDG, Khoury H, Laister RC, Fletcher
S, Vellozo M, Manzoli A, Yue P, Turkson J, Minden MD and Gunning
PT: Small molecule STAT5-SH2 domain inhibitors exhibit potent
antileukemia activity. J Med Chem. 55:1047–1055. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Pardanani A, Lasho T, Smith G, Burns CJ,
Fantino E and Tefferi A: CYT387, a selective JAK1/JAK2 inhibitor:
In vitro assessment of kinase selectivity and preclinical studies
using cell lines and primary cells from polycythemia vera patients.
Leukemia. 23:1441–1445. 2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Rani A and Murphy JJ: STAT5 in cancer and
immunity. J Interferon Cytokine Res. 36:226–237. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Korneev KV, Atretkhany KN, Drutskaya MS,
Grivennikov SI, Kuprash DV and Nedospasov SA: TLR-signaling and
proinflammatory cytokines as drivers of tumorigenesis. Cytokine.
89:127–135. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang X, Wrzeszczynska MH, Horvath CM and
Darnell JE Jr: Interacting regions in Stat3 and c-Jun that
participate in cooperative transcriptional activation. Mol Cell
Biol. 19:7138–7146. 1999.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hornsveld M, Dansen TB, Derksen PW and
Burgering BM: Re-evaluating the role of FOXOs in cancer. Semin
Cancer Biol. 50:90–100. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Sunters A, Madureira PA, Pomeranz KM,
Aubert M, Brosens JJ, Cook SJ, Burgering BM, Coombes RC and Lam EW:
Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer
cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res.
66:212–220. 2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yuan ZL, Guan YJ, Wang L, Wei W, Kane AB
and Chin YE: Central role of the threonine residue within the p+1
loop of receptor tyrosine kinase in STAT3 constitutive
phosphorylation in metastatic cancer cells. Mol Cell Biol.
24:9390–9400. 2004.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Antonicelli F, Lorin J, Kurdykowski S,
Gangloff SC, Le Naour R, Sallenave JM, Hornebeck W, Grange F and
Bernard P: CXCL10 reduces melanoma proliferation and invasiveness
in vitro and in vivo. Br J Dermatol. 164:720–728. 2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zaynagetdinov R, Sherrill TP, Gleaves LA,
McLoed AG, Saxon JA, Habermann AC, Connelly L, Dulek D, Peebles RS
Jr, Fingleton B, et al: Interleukin-5 facilitates lung metastasis
by modulating the immune microenvironment. Cancer Res.
75:1624–1634. 2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Sun X, Cheng G, Hao M, Zheng J, Zhou X,
Zhang J, Taichman RS, Pienta KJ and Wang J: CXCL12/CXCR4/CXCR7
chemokine axis and cancer progression. Cancer Metastasis Rev.
29:709–722. 2010.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wightman SC, Uppal A, Pitroda SP, Ganai S,
Burnette B, Stack M, Oshima G, Khan S, Huang X, Posner MC, et al:
Oncogenic CXCL10 signalling drives metastasis development and poor
clinical outcome. Br J Cancer. 113:327–335. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ishiguro H, Akimoto K, Nagashima Y, Kojima
Y, Sasaki T, Ishiguro-Imagawa Y, Nakaigawa N, Ohno S, Kubota Y and
Uemura H: aPKClambda/ι promotes growth of prostate cancer cells in
an autocrine manner through transcriptional activation of
interleukin-6. Proc Natl Acad Sci USA. 106:16369–16374.
2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Peng H, Chen P, Cai Y, Chen Y, Wu QH, Li
Y, Zhou R and Fang X: Endothelin-1 increases expression of
cyclooxygenase-2 and production of interlukin-8 in hunan pulmonary
epithelial cells. Peptides. 29:419–424. 2008.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Timani KA, Győrffy B, Liu Y, Mohammad KS
and He JJ: Tip110/SART3 regulates IL-8 expression and predicts the
clinical outcomes in melanoma. Mol Cancer. 17(124)2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yang M, Liu J, Piao C, Shao J and Du J:
ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2
polarization through blockade of efferocytosis. Cell Death Dis.
6(e1780)2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
de Groote ML, Kazemier HG, Huisman C, van
der Gun BT, Faas MM and Rots MG: Upregulation of endogenous ICAM-1
reduces ovarian cancer cell growth in the absence of immune cells.
Int J Cancer. 134:280–290. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Benatar T, Cao MY, Lee Y, Lightfoot J,
Feng N, Gu X, Lee V, Jin H, Wang M, Wright JA, et al: IL-17E, a
proinflammatory cytokine, has antitumor efficacy against several
tumor types in vivo. Cancer Immunol Immunother. 59:805–817.
2010.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Benatar T, Cao MY, Lee Y, Li H, Feng N, Gu
X, Lee V, Jin H, Wang M, Der S, et al: Virulizin induces production
of IL-17E to enhance antitumor activity by recruitment of
eosinophils into tumors. Cancer Immunol Immunother. 57:1757–1769.
2008.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wei C, Sirikanjanapong S, Lieberman S,
Delacure M, Martiniuk F, Levis W and Wang BY: Primary mucosal
melanoma arising from the eustachian tube with CTLA-4, IL-17A,
IL-17C, and IL-17E upregulation. Ear Nose Throat J. 92:36–40.
2013.PubMed/NCBI
|