1. Introduction
Among women with endometriosis, approximately 60%
have endometriosis-related pelvic pain, up to 50% have infertility
(1) and approximately 1% of
ovarian endometriomas give rise to ovarian cancer (2). The finding that a large number of
somatic driver mutations occur frequently in the physiologically
normal epithelial compartment of benign endometriotic lesions
without concurrent cancer was surprising (3,4).
The repeated hemorrhage in endometriotic lesions contains
erythrocytes and their degradation products, such as hemoglobin,
heme and free iron. A previous study demonstrated a genetic overlap
between the genes differentially expressed in endometriosis and
those regulated by the master oxidative stress regulator, iron
(5). Alterations in the redox
balance or the oxidant/antioxidant pathway can affect the
susceptibility to ovarian cancer (5). Oxidative stress, chronic damage and
inflammation in endometriosis can lead to epigenetic alterations
and genetic mutations of the driver genes, including tumor protein
P53 (TP53), phosphatase and tensin homolog (PTEN),
AT-rich interaction domain 1A (ARID1A),
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha (PIK3CA), KRAS and protein phosphatase 2 scaffold
subunit Aalpha (PPP2R1A) (6) by altering (epi)genetic signaling in
the endometriotic microenvironment (7). Furthermore, Guo previously
demonstrated that frequently, mutated driver genes in endometriosis
may play important roles in fibrogenesis, but do not necessarily
contribute to malignant transformation (3). This hypothesis may provide a
potential link between somatic driver mutations and the progression
of fibrosis. However, to date, at least to the best of our
knowledge, there are no studies available reporting whether these
genes are required for the development of endometriosis-related
fibrosis. At least 3 different types of examples of possibilities
on these somatic driver mutations in endometriosis were selected to
be shown in this review: i) A characteristic mutational landscape
may be an intrinsic molecular processes of normal endometrium; ii)
somatic mutations may be required to acquire a self-defined fitness
function of the endometriotic lesions under a fluctuating
environment during the initiation and progression of endometriosis;
and iii) they may not necessarily predict malignancy or
pre-malignancy, but the result of selection pressure for
fibrogenesis (3,4,8).
In this review article, we discuss the mechanisms through which
somatic mutations or the abnormal expression of
endometriosis-susceptibility driver genes can cause fibrosis in
other organ systems.
2. Data collection methods
A computerized literature search was conducted to
identify relevant studies reported in the English language. We
conducted a literature search of the PubMed and Embase databases up
to December 2018, combining the keywords ‘endometriosis’,
‘fibrosis’, ‘fibrogenesis’, ‘carcinogenesis’, ‘liver’, ‘kidney’,
‘lung’, ‘mutations’, ‘genetics’ and ‘epigenetics’. A variety of
combinations of these terms was used, depending on which database
was searched. Furthermore, the references of each article were
searched to identify potentially relevant studies. The publications
of original studies and review articles were included, while those
documenting opinions, points of view or anecdotes were
discarded.
3. The driver genes in endometriosis as a
major hallmark of fibrosis in other organ systems
This review presents the results of a literature
search for publications dealing with the association between the
endometriosis susceptibility driver gene mutations and pathological
gene alterations related to fibrosis in the liver, kidney and
lung.
Hepatic fibrosis
Hepatic fibrosis is an excessive wound-healing
repair following chronic injury to the liver, caused by virus and
parasite infections, alcohol abuse, non-alcoholic hepatosteatosis
(NASH), metabolic disorders, or autoimmune imbalances (9). Epidemiologically, progressive
hepatic fibrosis is a precursor for the development of cirrhosis
and hepatocellular carcinoma (9).
Activated hepatic stellate cells (HSCs) are the primary source of
myofibroblasts (the transdifferentiation of quiescent cells into
proliferative, fibrogenic myofibroblasts), the major players of
fibrogenesis, that produce extracellular matrix (ECM) proteins
(10). The possible molecular
mechanisms of hepatic fibrosis are associated with extrinsic and
intrinsic factors, including angiogenesis, tissue-degrading
enzymes, oxidative stress, inflammatory signaling, autophagy, or
cellular senescence. Angiogenesis-related genes are key players in
the pathogenesis of hepatic fibrosis and include the profibrotic
cytokine, transforming growth factor β (TGF-β),
platelet-derived growth factor (PDGF), connective tissue
growth factor (CTGF), hepatocyte growth factor (HGF),
tumor necrosis factor-α (TNF-α), basic fibroblast growth
factor (bFGF) and vascular endothelial growth factor
(VEGF) (11). Changes in
the fine balance between tissue-degrading matrix metalloproteinases
(MMPs) and their counterparts, tissue inhibitors of
metalloproteinases (TIMPs), which drives extracellular matrix
turnover, may be critical to maintaining wound healing and ECM
homeostasis (12). Tissue
remodeling and fibrosis might be controlled by the MMPs/TIMPs
imbalance (12). Reactive oxygen
species (ROS) can activate HSCs and the progression of fibrosis,
resulting in an increased collagen production and ECM deposition
(13). ROS are generated through
oxidative stress-related lipid peroxidation (13) and inflammatory responses via
Toll-like receptors (TLRs) (14).
TLR4 and TLR9 signaling pathways play a role in the liver
inflammation-fibrosis-carcinoma axis (14).
The roles of driver genes in hepatic fibrosis. The
driver genes in hepatic fibrosis are as follows: i) ARID1A:
The mutation of ARID1A, an SWI/SNF chromatin-remodeling
gene, induces chromatin remodeling dysfunction and contributes to
carcinogenesis (15). The
differentially expressed genes (DEGs) and the most commonly mutated
genes observed in human hepatocellular carcinoma are TP53,
ARID1A, PTEN, cyclin-dependent kinase inhibitor 2A
(CDKN2A, p16INK4A), cyclin D1 (CCND1),
catenin beta 1 (CTNNB1), telomerase reverse transcriptase
(TERT) and axin 1 (AXIN1) (16). ARID1A deficiency has been
shown to be associated with the proliferation, invasion and
metastasis of human hepatocellular carcinoma after initiation
(15). Arid1a expression
is downregulated in regenerating tissues, and the loss of
Arid1a has been shown to increase proliferation and reduce
fibrosis following liver injury in an animal model (17). ARID1A has a negative impact
on regeneration after hepatic injury (17). Thus, ARID1A may be a
susceptible gene for hepatic damage and fibrosis. ii) PTEN:
PTEN functions as a tumor suppressor by opposing the
PI3K/AKT signaling pathway, indicating that the inactivation of
PTEN by loss-of-function mutations leads to an aggressive
cancer phenotype (18). The
PTEN gene is frequently hypermethylated in hepatocellular
carcinoma (16). The loss of
PTEN plays a role in protection against hyperglycemia and
insulin resistance, which results in the development of steatosis,
steatohepatitis and NASH (12),
as well as in the progression of hepatic fibrosis (19). iii) PIK3CA: The PI3K/Akt
signaling pathway is a downstream target of TGF-β. The
TGF-β/PI3K/Akt pathway is activated in pulmonary fibrosis through
the induction of cell proliferation and ECM synthesis (20). Therefore, the inhibition of this
signaling pathway ameliorates hepatic fibrosis (20). iv) KRAS: Activating
mutations of K-ras or the sustained activation of K-ras
expression stimulates HSCs activation, hepatic cholestasis and
fibrosis, possibly through the upregulation of TGF-β2 and
CTGF gene expression (21). v) TP53: In response to
genotoxic insults, TP53 serves multiple biological
functions, including cellular responses to DNA damage, cell cycle
arrest, DNA repair, apoptosis, senescence and fibrosis (22). TP53 promotes HSCs
activation and hepatic fibrosis via the upregulation of fibrogenic
genes, such as TGF-β, Wnt/β-catenin, CTGF, or Sonic hedgehog
(SHH) (22). vi)
BRAF: The activity of the RAF/MEK/ERK signaling pathway is
critical for cell cycle arrest, ECM synthesis and hepatic fibrosis
(23).
Considering that the majority of driver genes
observed in benign endometriosis are required for EMT, FMT, ECM
production and hepatic fibrosis, somatic mutations in
‘cancer-associated genes’ are insufficient for malignant
transformation.
Renal fibrosis
We then aimed to address whether these driver genes
are central elements of EMT, FMT, ECM production, senescence and
fibrogenesis in the kidney. Renal tubulointerstitial fibrosis is a
common pathophysiological event of chronic kidney disease (24). Chronic kidney disease causes
increased inflammation, oxidative stress and proximal tubule cell
death in the form of apoptosis or senescence, with accelerated
fibrosis and reduced renal function (24). Kidney pericytes are considered to
be a possible precursor of myofibroblasts, contributing to ECM
production, increased tissue stiffness and fibrosis (24). A number of genes that have been
reported to be commonly altered in renal fibrosis are TGF-β,
Notch, hypoxia-inducible factor 1 subunit α (HIF-1α),
protein kinase C (PKC)/ERK, PTEN, PI3K/Akt, KRAS,
TP53, nuclear factor κB subunit 1 (NF-κB), BRAF,
CDKN2A, angiotensin II/ROS and microRNAs (miRNAs or miRs)
(25).
The roles of driver genes in renal fibrosis. The
driver genes in renal fibrosis are as follows: i) ARID1A: No
data are available for this gene in this context, at least to the
best of our knowledge. ii) PTEN: TGF-β signaling upregulates
the Notch signaling pathway, which in turn promotes TGF-β
signaling, suggesting that Notch signaling plays a critical role in
TGF-β1-induced fibrosis (26).
PTEN in renal proximal cells is suppressed when TGF-β signaling is
activated (26). Both the TGF-β
and Notch pathways are involved in myofibroblast differentiation
and in the increased production of α-smooth muscle actin (α-SMA)
and ECM proteins, which results in the development of renal
fibrosis via the inhibition of PTEN expression (27). iii) PIK3CA: The TGF-β/PI3K
pathway plays a critical role in the EMT of renal tubular
epithelial cells during renal injury (28). PI3K also promotes angiotensin
II-induced renal glomerular and tubular injury, suggesting that
PI3K may be a core mediator of renal fibrosis (28). iv) KRAS: K-ras activation
has been shown to promote ECM production and stimulate renal
fibroblast proliferation in a rat model, suggesting the possible
role of K-ras activation in renal fibrosis (29). v) TP53: TP53 upregulation
induced by HIF-1α facilitates ECM production and renal fibrosis
during ischemic injury and hypoxia through the accumulation of G2/M
cells and the activation of profibrotic TGF-β, CTGF and plasminogen
activator inhibitor-1 (PAI-1)-mediated signaling pathways (30). Researches have highlighted the
critical role of the hypoxia-activated, TP53-responsive
profibrogenic pathway in the kidney. vi) BRAF: The
Raf-dependent profibrotic activity stimulates renal fibrosis
through Raf/MEK/ERK signaling, demonstrating that the Raf kinase
modulates renal fibrosis (31).
vii) CDKN2A, p16INK4a: Renal fibrosis is mediated
via the increased expression of senescence-associated markers, such
as CDKN2A (p16INK4a), CDKN1A
(p21Cip1/Waf1), TP53, RB transcriptional
corepressor 1 (Rb1) and β-galactosidase in renal tubular
cells (32). Similar results have
been observed with TGF-β, cytochrome c oxidase subunit I
(COX1) and heat shock protein A5 (HSPA5) in aging
renal fibrosis (33).
In summary, the driver genes commonly mutated in
endometriosis may cause renal fibrosis.
Pulmonary fibrosis
Damage to the alveolar epithelium or the
obliteration of alveolar-capillary structures has been proposed to
be involved in TGF-β-induced myofibroblast differentiation and
fibrosis (34). ROS are produced
by the mitochondria and NADPH oxidase induces EMT, FMT and fibrosis
by secreting TGF-β expression, which is the initiating factor in
pulmonary fibrosis (35).
The roles of driver genes in pulmonary fibrosis. The
driver genes in pulmonary fibrosis are as follows: i)
ARID1A: No data are available for this gene in this context,
at least to the best of our knowledge. ii) PTEN: The
overexpression of PTEN may inhibit TGF-β1-mediated myofibroblast
differentiation by attenuating signaling via the TGF-β/Smad3- and
PI3K/Akt-induced production of α-SMA, MMP-2 and MMP-9 in the lung
(36). Pulmonary fibrosis has
been shown to be associated with decreased PTEN expression,
increased senescence and the activation of NF-κB (37). Therefore, the loss of PTEN may
induce pulmonary fibrosis via alveolar epithelial cell senescence
through the DNA damage-induced NF-κB activation (37). iii) PIK3CA: The
TGF-β/PI3K/AKT/mTOR pathway in pathological myofibroblasts
contributes to the induction of pulmonary fibrosis (38). iv) KRAS: The aberrant
expression of K-ras protein and mutation in the codon 12 of
K-ras gene in type II alveolar pneumocytes are associated
with the deterioration of pulmonary fibrosis, loss of functions and
the induction of lung carcinoma in patients with idiopathic
pulmonary fibrosis (IPF) (39).
v) TP53: The senescence marker, TP53, drives the fibrotic
signaling cascade (40). The
senescence of alveolar type 2 cells is implicated in the
pathogeneses of pulmonary fibrosis through activating TP53-p21-Rb
pathway (41). TP53
(62.9%) and BRAF (17.1%) have been found to be significantly
mutated in IPF (41). vi)
CDKN2A, p16INK4a: Cellular senescence markers,
including p16INK4a, induce interstitial
remodeling, ECM deposit and pulmonary fibrosis (42). vii) BRAF: BRAF has
been shown to be overexpressed and mutated in IPF samples (43). The high incidence of BRAF
mutations has been observed in pulmonary fibrosis, most notably the
BRAFV600E mutation profile (43). viii) NOTCH1:
TGF-β/NOTCH1 signaling induces myofibroblast differentiation
in pulmonary fibrosis (44).
These data provide evidence of a role for the
endometriosis susceptibility driver genes in pulmonary
fibrosis.
In conclusion, the multifactorial etiology includes
repetitive tissue injury and repair, oxidative stress, the
overproduction of reactive oxygen species and inflammation-related
signaling processes, which promotes myofibroblast differentiation,
cellular senescence, excessive ECM synthesis along with organ
fibrosis. It is confirmed that the mutated driver pathways or gene
sets identified in endometriosis may contribute to tissue fibrosis
in the liver, kidney and lung.
4. Endometriosis as a fibrotic
condition
Under a number of pathological conditions,
endometriotic lesions promote EMT and the transdifferentiation of
fibroblasts into myofibroblasts (FMT) in response to the TGF-β/Smad
signaling pathway, resulting in the formation of exaggerated
accumulation of α-SMA and collagen, leading to increased smooth
muscle metaplasia (SMM) and fibrosis (45). Scarring or excess fibrosis may
lead to alterations of tissue function, which results in chronic
pain and infertility as endometriosis develops. Scarring regains
and maintains tissue integrity, although fibrosis induces adverse
changes in tissue architecture and the loss of physiological
functions. Fibrosis represents consistent features of 3 different
types (peritoneal, ovarian, or deep) of endometriosis (46). Older ovarian endometriomas have a
higher collagen content and a greater extent of fibrosis than fresh
ones (47). The degree of
aggregated smooth muscle component and fibrosis, so-called SMM, is
not an uncommon phenomenon in endometriosis (48). Among peritoneal, ovarian and
rectovaginal (deep infiltrating) endometriotic lesions, SMM is the
most common finding in deep infiltrating endometriosis (48).
Several elegant reviews have been published focusing
on the link between endometriosis and fibrosis thus far. The
fibrosis-related factors include TGF-β, neuropeptides
substance P (SP), neurokinin receptor 1 (NK1R),
calcitonin receptor like receptor (CRLR), calcitonin gene
related peptide (CGRP), receptor activity modifying protein
1 (RAMP-1), nuclear receptor subfamily 4 group A member 1
(NR4A1), nuclear factor, erythroid 2 like 2 (NFE2L2, also
known as Nrf2), or glutamate cysteine ligase (49,50). Among these fibrosis susceptibility
genes, TGF-β is considered to be a key mediator in a number
of fibrotic disorders, including endometriosis, and in fibrosis in
hepatic, renal, pulmonary and cardiovascular systems (1). Activated TGF-β is released by
platelets, macrophages, or myofibroblasts and positively regulates
mitochondrial NADPH oxidase 4 (NOX4), which augments ROS generation
(34). ROS signaling also leads
to the activation of TGF-β (51).
Therefore, ROS and NOX4 form a positive feedback loop to amplify
TGF-β signaling, which causes the deterioration of endometriosis
and promotes fibrogenesis. Furthermore, platelets activated by
excessive hemorrhaging drive SMM and fibrogenesis in endometriosis
through EMT and FMT (52). SP, a
neuropeptide expressed in peripheral sensory neurons, can stimulate
the development EMT, FMT, SMM and ultimately, fibrosis, in
endometriotic lesions, indicating that the main clinical features
are chronic pelvic pain (53).
It has been proposed that when normal repair
mechanisms breakdown, the injured tissues initiate proliferation,
myofibrogenesis or various diseases, including cancers, at least to
a certain extent. Cellular senescence may be one of the major
mechanisms involved in the beneficial and deleterious effects of
tissue injury-induced fibrogenesis. Tissue injury has been proposed
to promote a number of DNA damage response mechanisms and to repair
signaling cascades that control cell cycle arrest to allow DNA
damage repair and induce cellular senescence and cell fate
(54). Cellular senescence
promotes the renewal of damaged tissues. Damage accumulation
stimulates the activity of CDK inhibitors, CDKN2A
(p16Ink4a), CDKN1A
(p21Cip1/Waf1), CDKN2B
(p15Ink4b), or TP53, which antagonizes
CDKs to block cell cycle progression and induce senescence
(54). Thus, the CDKN families
are essential for the maintenance of the senescent-cell-cycle
arrest. Not only CDK inhibitors, but also the transcription factor,
hepatocyte nuclear factor (HNF)-1β, are significantly
upregulated in endometriosis and its malignant transformation,
clear cell carcinoma of the ovary (55). HNF-1β is considered to promote
G2/M cell cycle arrest in response to DNA damage through the
HNF-1β/ubiquitin specific peptidase 28 (USP28)/Claspin/Chk1
signaling pathway (56). Cellular
senescence is recognized as the state of irreversible cell cycle
arrest in response to a variety of various intrinsic and extrinsic
cellular stresses (57) and
sometimes evolves as a beneficial response to protection against
tissue fibrosis (58). On the
other hand, cellular senescence also entails irreversible
replicative arrest and acquires senescence-associated secretory
phenotype that causes endometriotic cells to become susceptible to
their own harmful microenvironment and gradually accumulates DNA
damage, and even the promotion of tumorigenesis (59). Hypotheses involving cellular
senescence will be developed, from two points of view: Senescence
appears to be beneficial or deleterious.
An interesting study demonstrated that numerous
somatic mutations were identified, not only in benign
endometriosis, but also in the normal endometrium (8). Suda et al discussed the
impact of intrinsic molecular processes on mutation acquisition in
the normal endometrium (8).
Repeated hemorrhaging during menstruation and in endometriotic
lesions leads to severe hemolysis that results in high levels of
hemoglobin, heme and free iron. Autoxidation and fenton reaction of
hemoglobin from the ferrous Fe2+ (oxyhemoglobin) state
to the ferric Fe3+ (methemoglobin) state lead to the
production of excess ROS, such as O2- and ·OH, which
mediate oxidative stress and promote DNA damage and mutations
(60). DNA mutations provide
benefits that are essential for the survival of endometriotic cells
and normal endometrial cells exposed regularly to menstrual blood.
These cells may be adapted or selected to survive under the
conditions of oxidative stress.
Recent data have indicated a fundamental role of
oxidative stress, secondary to the influx of hemoglobin, heme and
free iron, in expressing CpG demethylases, ten-eleven translocation
(TET) and jumonji (JMJ) genes (61). Both TET and JMJ
genes recognize a wide range of endogenous DNA methyltransferases
(DNMTs) (61). The expression
levels of DNMTs may be involved in the subsequent epigenetic
modulation of endometriosis susceptibility genes (61). Indeed, 8-OHdG immunohistochemical
staining, a marker of oxidative DNA damage, has been found in
normal ovarian cortex surrounding ovarian endometriomas (62). Elevated levels of ROS may lead to
the increased damage of DNA and induce cell cycle arrest
accompanied by the acquisition of replication stress,
senescence-associated characteristics and carcinogenesis (63). This review supports the hypothesis
that there are at least 2 distinct phases of epigenetic and genetic
modifications in endometriosis: The initial wave of
hemoglobin-induced oxidative stress and (epi)genetic DNA damage and
mutations would be followed by the second big wave of cell
senescence, fibrogenesis and finally, carcinogenesis.
5. Endometriosis as a premalignant
condition
The epidemiological, histological, genetic and
molecular alterations in endometriosis may suggest that
endometriosis is a premalignant condition (64,65). First, there is evidence of a broad
overlap between gene mutation clusters for benign endometriosis and
its malignant transformation. The candidate mutated driver gene
sets, including ARID1A, PIK3CA, PTEN, TP53, KRAS and
PPP2R1A, have been identified in endometriosis (6,7).
More recently, Lac et al reported that hotspot mutations in
KRAS, erb-b2 receptor tyrosine kinase 2 (ERBB2),
PIK3CA and CTNNB1, heterogeneous PTEN loss and
ARID1A loss were also identified in endometriosis (66). Genetic studies have demonstrated
that endometriotic lesions have somatic mutations in genes directly
related to endometriosis-associated ovarian cancer (6,7,64-66).
It has been established that gene mutations are found in benign
endometriotic lesions adjacent to invasive carcinomas having
mutations (67). However, the
same mutations have also been described in benign endometriosis
that do not coincide with synchronous carcinoma specimens or that
never progress to malignancy (66). Studies have shown a significant
overlap in driver mutations between endometriosis with or without
coexistent malignancy (6,7,66,67).
Second, a recent study by Suda et al reported
that numerous somatic mutations were identified in both
endometriotic and normal epithelium samples (8). The eutopic endometrium may share
similarities with the ectopic endometrium at the environmental
level: Erythrocytes release some inflammatory mediators, including
oxidative stress and ROS, into the uterine cavity, peritoneal
cavity, or into cysts. A previous study demonstrated a global
overlap in the genes differentially expressed in endometriosis and
those modulated by the oxidative stress regulator, iron (5). Alterations in the redox balance can
explain a part of the somatic oncogene mutations (5). For example, oxidative stress, in the
form of lipid peroxidation and 4-hydroxy-2-nonenal, has been linked
to site-specific mutations of the TP53 gene (68). A positive association has also
been shown between heme iron intake and the risk of colorectal
carcinoma with activating G>A mutations in KRAS (69). The iron-induced genetic mutations
that may arise from the formation of 8-OHdG involve GC → TA
transversions (70).
Epidemiological, biological and molecular data suggest that
endometriosis is a risk factor for the later development of several
types of cancer, including endometrial cancer and ovarian cancer
(64,65). The key genetic alterations occur
as a normal endometrium and progress to endometriosis, and
ultimately tumor cells, which may explain the reasons and
mechanisms though which the normal endometrium can change into
endometriosis and subsequently into several types of cancer.
However, the exact molecular mechanisms underlying the conversion
of the normal endometrium to endometriosis and then cancers remain
uncertain.
Finally, it is of interest that cancer,
endometriosis and the normal endometrium often share common
pathogenetic determinants, such as DNA damage, oxidative stress,
chronic inflammation and other chronic conditions, such as the
longer duration of menstruation and hemorrhaging. This article
outlined the possibility of endometriosis fibrogenesis, possibly
through driver genes controlling malignant transformation. This
review supports novel conceptual ideas that frequently mutated
driver genes in endometriosis may initially play dominant roles in
the initiation and progression of fibrogenesis, although these
genetic abnormalities alone are insufficient for malignant
transformation.
6. Conclusions and future
considerations
Given that somatic driver mutations occur frequently
in the physiologically normal endometrium and benign endometriosis,
herein, we reviewed the functions of these driver genes (ARID1A,
PTEN, PI3K, KRAS, TP53, CDKN2A and BRAF) in EMT, FMT,
ECM production and increased SMM during fibrosis in liver, kidney
and lung (6,7,64-66).
To date, much has been shown about a complex pathway model for
fibrogenesis based on common driver genes. The driver molecules
related to the progression of fibrosis are TGF-β, Notch, HIF-1α,
PTEN, PIK3CA, PI3K/Akt, KRAS, TP53, NF-κB, BRAF, CDKN2A and
ARID1A that can activate myofibrogenesis, resulting in
increased ECM production, collagen deposition and fibrosis
(11-13,16,22,25,32,49,50,54).
A significant overlap was observed between 2 sets, epigenetic
alterations and genetic mutations of the driver genes in
endometriosis and the driver molecules related to the progression
of fibrosis in the liver, kidney and lung. This review provides
support for the hypothesis that these driver genes can accelerate
the process of fibrogenesis in endometriosis (Fig. 1).
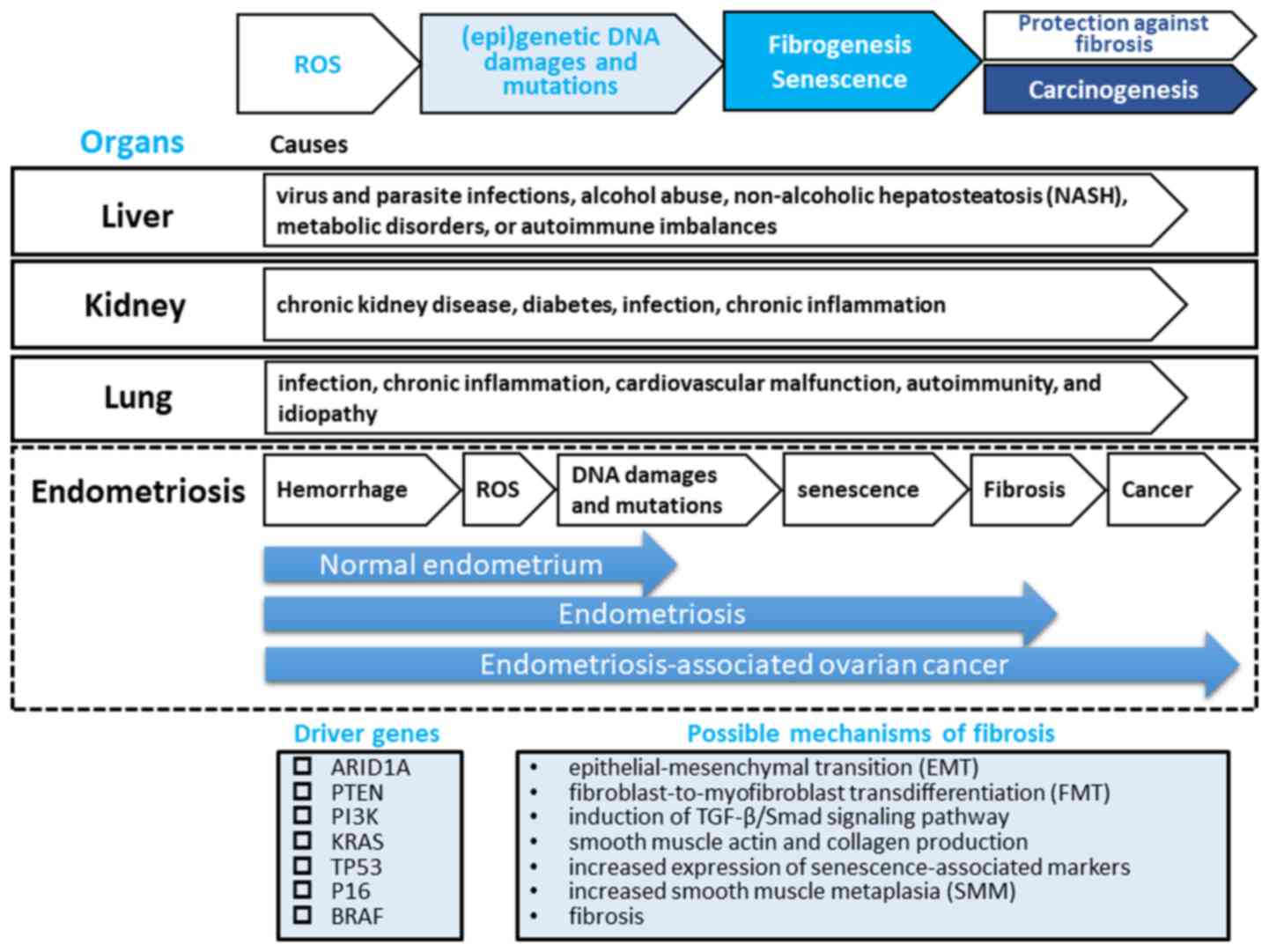 | Figure 1.A hypothesis for the pathogenesis of
endometriosis fibrogenesis. Genetic studies have demonstrated that
endometriotic lesions have somatic mutations in driver genes,
including AT-rich interaction domain 1A (ARID1A),
phosphatase and tensin homolog (PTEN), phosphoinositide
3-kinase (PI3K), KRAS, tumor protein P53
(TP53), P16, or B-Raf proto-oncogene,
serine/threonine kinase (BRAF), directly related to
neoplasms. Tissue injury and repair induce inflammation and
oxidative stress, which induces (epi)genetic DNA damage and
mutations, promotes epithelial-mesenchymal transition (EMT) and
fibroblast-to-myofibroblast transdifferentiation (FMT) through the
induction of the transforming growth factor (TGF)-β/Smad signaling
pathway, resulting in α-smooth muscle actin (α-SMA) and collagen
production and increased cellular contractility, leading to
increased smooth muscle metaplasia (SMM) and fibrosis (45).
Elevated levels of reactive oxygen species (ROS) may lead to
increased damage to DNA and induce cell cycle arrest accompanied by
the acquisition of replication stress, senescence-associated
characteristics and carcinogenesis, at least to a certain extent
(63). This process may be a major hallmark of fibrogenesis of
liver, kidney and lung. There may be at least 2 distinct phases of
epigenetic and genetic modifications in endometriosis: the initial
wave of hemoglobin-induced oxidative stress and (epi)genetic DNA
damage and mutations would be followed by the second big wave of
cell senescence, fibrogenesis and finally carcinogenesis.
Therefore, endometriosis may represent a hemorrhage-induced
pre-fibrotic and low-grade neoplastic lesion that may be a
precursor lesion of a subset of malignant transformation of
endometriosis. Our hypothesis is not inconsistent with available
evidence that a type I tumor exhibits an adaptive stepwise
progression from normal endometrium, through endometriosis and
fibrosis, to endometriosis-associated ovarian cancer. |
In both endometriotic and normal epithelium
samples, numerous somatic mutations have been identified within
genes frequently mutated in endometriosis-associated ovarian
cancers (8). Perhaps the greatest
uncertainty is not why endometriosis spontaneously develops into
fibrosis, but rather why the normal endometrium with similar driver
mutations does not. Endometriosis, but not the normal endometrium,
are prone to repetitive hemorrhage-induced tissue injury and
repair, which persistently induces oxidative stress, the
overproduction of ROS and inflammation-related signaling. We made
the following hypothesis that long-term damage to the endometriotic
lesions leads to excess oxidative stress, inflammation, senescence,
myofibroblast differentiation and ultimately fibrosis, whereas
excessive menstruation may cause a series of repetitive injuries to
the eutopic endometrial tissue resulting in an altered healing
process, which changes the architecture of the uterine myometrium,
leading to the development of adenomyosis.
In conclusion, the aberrant expression or somatic
genetic mutations in TP53, PTEN, ARID1A, PIK3CA and
KRAS identified in endometriosis may not be limited to early
stages of cancer evolution, but cause fibrosis in various organs.
Future studies are warranted to summarize the mechanisms and
functions of the somatic driver landscape of endometriosis
affecting fibrogenesis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Author's contribution
HK performed the literature search, and collected
the data using the PubMed and Embase databases. HK conceived this
review article. HK designed this review article and interpreted the
included research studies. The final version of the manuscript has
been read and approved by HK.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that there are no competing
interests.
References
|
1
|
Giudice LC: Clinical practice.
Endometriosis. N Engl J Med. 362:2389–2398. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Anglesio MS and Yong PJ:
Endometriosis-associated Ovarian Cancers. Clin Obstet Gynecol.
60:711–727. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Guo SW: Cancer driver mutations in
endometriosis: Variations on the major theme of fibrogenesis.
Reprod Med Biol. 17:369–397. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Anglesio MS, Papadopoulos N, Ayhan A,
Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T,
et al: Cancer-associated mutations in endometriosis without cancer.
N Engl J Med. 376:1835–1848. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kobayashi H, Yamada Y, Kanayama S,
Furukawa N, Noguchi T, Haruta S, Yoshida S, Sakata M, Sado T and Oi
H: The role of iron in the pathogenesis of endometriosis. Gynecol
Endocrinol. 25:39–52. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kurman RJ and Shih IeM: Molecular
pathogenesis and extraovarian origin of epithelial ovarian cancer -
shifting the paradigm. Hum Pathol. 42:918–931. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xie H, Chen P, Huang HW, Liu LP and Zhao
F: Reactive oxygen species downregulate ARID1A expression via its
promoter methylation during the pathogenesis of endometriosis. Eur
Rev Med Pharmacol Sci. 21:4509–4515. 2017.PubMed/NCBI
|
|
8
|
Suda K, Nakaoka H, Yoshihara K, Ishiguro
T, Tamura R, Mori Y, Yamawaki K, Adachi S, Takahashi T, Kase H, et
al: Clonal expansion and diversification of cancer-associated
mutations in endometriosis and normal endometrium. Cell Rep.
24:1777–1789. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Iwaisako K, Brenner DA and Kisseleva T:
What's new in liver fibrosis? The origin of myofibroblasts in liver
fibrosis. J Gastroenterol Hepatol. 27 (Suppl 2):65–68.
2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Seki E and Brenner DA: Recent advancement
of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat
Sci. 22:512–518. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wight TN and Potter-Perigo S: The
extracellular matrix: An active or passive player in fibrosis? Am J
Physiol Gastrointest Liver Physiol. 301:G950–G955. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Guo J and Friedman SL: Hepatic
fibrogenesis. Semin Liver Dis. 27:413–426. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ghatak S, Biswas A, Dhali GK, Chowdhury A,
Boyer JL and Santra A: Oxidative stress and hepatic stellate cell
activation are key events in arsenic induced liver fibrosis in
mice. Toxicol Appl Pharmacol. 251:59–69. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Song IJ, Yang YM, Inokuchi-Shimizu S, Roh
YS, Yang L and Seki E: The contribution of toll-like receptor
signaling to the development of liver fibrosis and cancer in
hepatocyte-specific TAK1-deleted mice. Int J Cancer. 142:81–91.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Abe H, Hayashi A, Kunita A, Sakamoto Y,
Hasegawa K, Shibahara J, Kokudo N and Fukayama M: Altered
expression of AT-rich interactive domain 1A in hepatocellular
carcinoma. Int J Clin Exp Pathol. 8:2763–2770. 2015.PubMed/NCBI
|
|
16
|
Khemlina G, Ikeda S and Kurzrock R: The
biology of hepatocellular carcinoma: Implications for genomic and
immune therapies. Mol Cancer. 16(149)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sun X, Chuang JC, Kanchwala M, Wu L, Celen
C, Li L, Liang H, Zhang S, Maples T, Nguyen LH, et al: Suppression
of the SWI/SNF component Arid1a promotes mammalian regeneration.
Cell Stem Cell. 18:456–466. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guigon CJ, Zhao L, Willingham MC and Cheng
SY: PTEN deficiency accelerates tumour progression in a mouse model
of thyroid cancer. Oncogene. 28:509–517. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yu F, Chen B, Dong P and Zheng J: HOTAIR
epigenetically modulates PTEN expression via microRNA-29b: A novel
mechanism in regulation of liver fibrosis. Mol Ther. 25:205–217.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Son MK, Ryu YL, Jung KH, Lee H, Lee HS,
Yan HH, Park HJ, Ryu JK, Suh JK, Hong S, et al: HS-173, a novel
PI3K inhibitor, attenuates the activation of hepatic stellate cells
in liver fibrosis. Sci Rep. 3(3470)2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Makino Y, Hikita H, Kodama T, Shigekawa M,
Yamada R, Sakamori R, Eguchi H, Morii E, Yokoi H, Mukoyama M, et
al: CTGF mediates tumor-stroma interactions between hepatoma cells
and hepatic stellate cells to accelerate HCC progression. Cancer
Res. 78:4902–4914. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kodama T, Takehara T, Hikita H, Shimizu S,
Shigekawa M, Tsunematsu H, Li W, Miyagi T, Hosui A, Tatsumi T, et
al: Increases in p53 expression induce CTGF synthesis by mouse and
human hepatocytes and result in liver fibrosis in mice. J Clin
Invest. 121:3343–3356. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Guo X, Cen Y, Wang J and Jiang H:
CXCL10-induced IL-9 promotes liver fibrosis via Raf/MEK/ERK
signaling pathway. Biomed Pharmacother. 105:282–289.
2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Portilla D: Apoptosis, fibrosis and
senescence. Nephron Clin Pract. 127:65–69. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu M, Ning X, Li R, Yang Z, Yang X, Sun S
and Qian Q: Signalling pathways involved in hypoxia-induced renal
fibrosis. J Cell Mol Med. 21:1248–1259. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lan R, Geng H, Polichnowski AJ, Singha PK,
Saikumar P, McEwen DG, Griffin KA, Koesters R, Weinberg JM, Bidani
AK, et al: PTEN loss defines a TGF-β-induced tubule phenotype of
failed differentiation and JNK signaling during renal fibrosis. Am
J Physiol Renal Physiol. 302:F1210–F1223. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bielesz B, Sirin Y, Si H, Niranjan T,
Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, et al:
Epithelial Notch signaling regulates interstitial fibrosis
development in the kidneys of mice and humans. J Clin Invest.
120:4040–4054. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhou T, Luo M, Cai W, Zhou S, Feng D, Xu C
and Wang H: Runt-related transcription factor 1 (RUNX1) promotes
TGF-β-induced renal tubular epithelial-to-mesenchymal transition
(EMT) and renal fibrosis through the PI3K subunit p110δ.
EBioMedicine. 31:217–225. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Rodríguez-Peña AB, Santos E, Arévalo M and
López-Novoa JM: Activation of small GTPase Ras and renal fibrosis.
J Nephrol. 18:341–349. 2005.PubMed/NCBI
|
|
30
|
Liu L, Zhang P, Bai M, He L, Zhang L, Liu
T, Yang Z, Duan M, Liu M, Liu B, et al: p53 upregulated by HIF-1α
promotes hypoxia-induced G2/M arrest and renal fibrosis in vitro
and in vivo. J Mol Cell Biol. Jul 18. 2018.(Epub ahead of print).
PubMed/NCBI View Article : Google Scholar
|
|
31
|
Buchholz B, Klanke B, Schley G, Bollag G,
Tsai J, Kroening S, Yoshihara D, Wallace DP, Kraenzlin B, Gretz N,
et al: The Raf kinase inhibitor PLX5568 slows cyst proliferation in
rat polycystic kidney disease but promotes renal and hepatic
fibrosis. Nephrol Dial Transplant. 26:3458–3465. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Adijiang A, Shimizu H, Higuchi Y,
Nishijima F and Niwa T: Indoxyl sulfate reduces klotho expression
and promotes senescence in the kidneys of hypertensive rats. J Ren
Nutr. 21:105–109. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Melk A, Schmidt BM, Takeuchi O, Sawitzki
B, Rayner DC and Halloran PF: Expression of p16INK4a and other cell
cycle regulator and senescence associated genes in aging human
kidney. Kidney Int. 65:510–520. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jain M, Rivera S, Monclus EA, Synenki L,
Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR and
Chandel NS: Mitochondrial reactive oxygen species regulate
transforming growth factor-β signaling. J Biol Chem. 288:770–777.
2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Leask A and Abraham DJ: TGF-beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xie B, Zheng G, Li H, Yao X, Hong R, Li R,
Yue W and Chen Y: Effects of the tumor suppressor PTEN on the
pathogenesis of idiopathic pulmonary fibrosis in Chinese patients.
Mol Med Rep. 13:2715–2723. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen
J and Cai H: Loss of PTEN induces lung fibrosis via alveolar
epithelial cell senescence depending on NF-κB activation. Aging
Cell. 18(e12858)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su
K and Hung SC: Involvement of ER stress, PI3K/AKT activation, and
lung fibroblast proliferation in bleomycin-induced pulmonary
fibrosis. Sci Rep. 7(14272)2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Takahashi T, Munakata M, Ohtsuka Y,
Nisihara H, Nasuhara Y, Kamachi-Satoh A, Dosaka-Akita H, Homma Y
and Kawakami Y: Expression and alteration of ras and p53 proteins
in patients with lung carcinoma accompanied by idiopathic pulmonary
fibrosis. Cancer. 95:624–633. 2002.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Álvarez D, Cárdenes N, Sellarés J, Bueno
M, Corey C, Hanumanthu VS, Peng Y, D'Cunha H, Sembrat J, Nouraie M,
et al: IPF lung fibroblasts have a senescent phenotype. Am J
Physiol Lung Cell Mol Physiol. 313:L1164–L1173. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Higgins SP, Tang Y, Higgins CE, Mian B,
Zhang W, Czekay RP, Samarakoon R, Conti DJ and Higgins PJ:
TGF-β1/p53 signaling in renal fibrogenesis. Cell Signal. 43:1–10.
2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Kuwano K, Kunitake R, Kawasaki M, Nomoto
Y, Hagimoto N, Nakanishi Y and Hara N: P21Waf1/Cip1/Sdi1 and p53
expression in association with DNA strand breaks in idiopathic
pulmonary fibrosis. Am J Respir Crit Care Med. 154:477–483.
1996.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hwang JA, Kim D, Chun SM, Bae S, Song JS,
Kim MY, Koo HJ, Song JW, Kim WS, Lee JC, et al: Genomic profiles of
lung cancer associated with idiopathic pulmonary fibrosis. J
Pathol. 244:25–35. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Hu B, Wu Z, Bai D, Liu T, Ullenbruch MR
and Phan SH: Mesenchymal deficiency of Notch1 attenuates
bleomycin-induced pulmonary fibrosis. Am J Pathol. 185:3066–3075.
2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhang Q, Duan J, Olson M, Fazleabas A and
Guo SW: Cellular changes consistent with epithelial-mesenchymal
transition and fibroblast-to-myofibroblast transdifferentiation in
the progression of experimental endometriosis in baboons. Reprod
Sci. 23:1409–1421. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Vigano P, Candiani M, Monno A, Giacomini
E, Vercellini P and Somigliana E: Time to redefine endometriosis
including its pro-fibrotic nature. Hum Reprod. 33:347–352.
2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Guo SW, Ding D, Shen M and Liu X: Dating
endometriotic ovarian cysts based on the content of cyst fluid and
its potential clinical implications. Reprod Sci. 22:873–883.
2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kim HS, Yoon G, Ha SY and Song SY: Nodular
smooth muscle metaplasia in multiple peritoneal endometriosis. Int
J Clin Exp Pathol. 8:3370–3373. 2015.PubMed/NCBI
|
|
49
|
Yan D, Liu X and Guo SW: Neuropeptides
substance P and calcitonin gene related peptide accelerate the
development and fibrogenesis of endometriosis. Sci Rep.
9(2698)2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Marcellin L, Santulli P, Chouzenoux S,
Cerles O, Nicco C, Dousset B, Pallardy M, Kerdine-Römer S, Just PA,
Chapron C, et al: Alteration of Nrf2 and Glutamate Cysteine Ligase
expression contribute to lesions growth and fibrogenesis in ectopic
endometriosis. Free Radic Biol Med. 110:1–10. 2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Gonzalez-Gonzalez FJ, Chandel NS, Jain M
and Budinger GRS: Reactive oxygen species as signaling molecules in
the development of lung fibrosis. Transl Res. 190:61–68.
2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang Q, Liu X and Guo SW: Progressive
development of endometriosis and its hindrance by anti-platelet
treatment in mice with induced endometriosis. Reprod Biomed Online.
34:124–136. 2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Liu X, Yan D and Guo SW: Sensory
nerve-derived neuropeptides accelerate the development and
fibrogenesis of endometriosis. Hum Reprod. 34:452–468.
2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
van Deursen JM: The role of senescent
cells in ageing. Nature. 509:439–446. 2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kato N, Sasou S and Motoyama T: Expression
of hepatocyte nuclear factor-1beta (HNF-1beta) in clear cell tumors
and endometriosis of the ovary. Mod Pathol. 19:83–89.
2006.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Ito F, Yoshimoto C, Yamada Y, Sudo T and
Kobayashi H: The HNF-1β-USP28-Claspin pathway upregulates DNA
damage-induced Chk1 activation in ovarian clear cell carcinoma.
Oncotarget. 9:17512–17522. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Kadota T, Fujita Y, Yoshioka Y, Araya J,
Kuwano K and Ochiya T: Emerging role of extracellular vesicles as a
senescence-associated secretory phenotype: Insights into the
pathophysiology of lung diseases. Mol Aspects Med. 60:92–103.
2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Regulski MJ: Cellular senescence: What,
why, and how. Wounds. 29:168–174. 2017.PubMed/NCBI
|
|
59
|
Watanabe S, Kawamoto S, Ohtani N and Hara
E: Impact of senescence-associated secretory phenotype and its
potential as a therapeutic target for senescence-associated
diseases. Cancer Sci. 108:563–569. 2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Iwabuchi T, Yoshimoto C, Shigetomi H and
Kobayashi H: Oxidative stress and antioxidant defense in
endometriosis and its malignant transformation. Oxid Med Cell
Longev. 2015(848595)2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Ito F, Yamada Y, Shigemitsu A, Akinishi M,
Kaniwa H, Miyake R, Yamanaka S and Kobayashi H: Role of oxidative
stress in epigenetic modification in endometriosis. Reprod Sci.
24:1493–1502. 2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Di Emidio G, D'Alfonso A, Leocata P,
Parisse V, Di Fonso A, Artini PG, Patacchiola F, Tatone C and Carta
G: Increased levels of oxidative and carbonyl stress markers in
normal ovarian cortex surrounding endometriotic cysts. Gynecol
Endocrinol. 30:808–812. 2014.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Baker DJ, Alimirah F, van Deursen JM,
Campisi J and Hildesheim J: Oncogenic senescence: A
multi-functional perspective. Oncotarget. 8:27661–27672.
2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Painter JN, O'Mara TA, Morris AP, Cheng
THT, Gorman M, Martin L, Hodson S, Jones A, Martin NG, Gordon S, et
al: Genetic overlap between endometriosis and endometrial cancer:
Evidence from cross-disease genetic correlation and GWAS
meta-analyses. Cancer Med. 7:1978–1987. 2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Ramalingam P: Morphologic,
immunophenotypic, and molecular features of epithelial ovarian
cancer. Oncology (Williston Park). 30:166–176. 2016.PubMed/NCBI
|
|
66
|
Lac V, Verhoef L, Aguirre-Hernandez R,
Nazeran TM, Tessier-Cloutier B, Praetorius T, Orr NL, Noga H, Lum
A, Khattra J, et al: Iatrogenic endometriosis harbors somatic
cancer-driver mutations. Hum Reprod. 34:69–78. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Siufi Neto J, Kho RM, Siufi DF, Baracat
EC, Anderson KS and Abrão MS: Cellular, histologic, and molecular
changes associated with endometriosis and ovarian cancer. J Minim
Invasive Gynecol. 21:55–63. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Petersen DR: Alcohol, iron-associated
oxidative stress, and cancer. Alcohol. 35:243–249. 2005.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Gilsing AM, Fransen F, de Kok TM, Goldbohm
AR, Schouten LJ, de Bruïne AP, van Engeland M, van den Brandt PA,
de Goeij AF and Weijenberg MP: Dietary heme iron and the risk of
colorectal cancer with specific mutations in KRAS and APC.
Carcinogenesis. 34:2757–2766. 2013.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Valko M, Izakovic M, Mazur M, Rhodes CJ
and Telser J: Role of oxygen radicals in DNA damage and cancer
incidence. Mol Cell Biochem. 266:37–56. 2004.PubMed/NCBI
|














