1. Introduction
Protein arginine methyltransferases (PRMTs) are
methylases that are widely present in numerous organisms. Their
main biological function is to mediate arginine methylation
modification, a post-translational modification that commonly
occurs in the nucleus and cytoplasm. In recent years, the
modification of arginine methylation and its related mechanisms
have received increasing attention. Although arginine methylation
products were detected in nuclear extracts in 1996, the genes
encoding PRMT-related proteins were only identified in recent years
(1).
Cardiovascular disease (CVD) is currently among the
most serious threats to human life and health worldwide (2). Its morbidity and mortality exceed
those of other tumor diseases and are ranked first. Common CVDs
include hypertension, atherosclerosis (AS), heart failure,
hyperlipidemia and coronary heart disease, which are associated
with high morbidity, disability, mortality and serious
complications (3). As PRMTs and CVD
are closely related, clarifying their relationship and the
mechanisms of PRMTs is important for reducing the incidence of CVD
and improving human health.
In addition, arginine methylation is increasingly
associated with cancer progression. As they perform arginine
methylation, PRMTs regulate post-translational modification of
proteins that are vital for increasing proteome diversity and
maintaining cellular homeostasis (4). Protein arginine methylation is an
abundant modification that may involve processes including gene
transcription, signal transduction, DNA repair and mRNA splicing.
Studies have recently linked this modification to carcinogenesis
and metastasis (5).
2. Biological characteristics of PRMTs
Epigenetics represents a wide range of gene
expression changes (6), the
mechanisms of which include DNA methylation or demethylation,
histone methylation or demethylation, histone acetylation or
deacetylation and non-coding RNA. The processes of gene
transcription or silencing, signal transduction, DNA repair and DNA
replication are mediated by chromatin composed of DNA and histones
(H2A, H2B, H3 and H4) (7,8). As a type of epigenetic process,
histone methylation is one of the most common post-translational
modifications of histone, which is usually related to
transcriptional activation or inhibition of downstream genes
(9). The methylation of arginine
may be catalyzed by PRMTs (10).
Arginine methylation is a process in which the
nitrogen atom of arginine in a polypeptide is modified by a methyl
group in a reaction catalyzed by nine PRMT enzymes (11). PRMTs catalyze the transfer of a
methyl group from S-adenosyl methionine to the terminal guanidine
nitrogen atom of arginine to produce methyl arginine and
S-adenosine homocysteine (12).
PRMTs catalyze arginine to form three different
amino acids: Monomethyl-arginine (MMA), asymmetric dimethylarginine
(ADMA) and symmetric dimethyl-arginine (SDMA) (13). ADMA is formed by connecting two
methyl groups to one terminal nitrogen atom, SDMA is formed by
connecting one methyl group to each guanidine nitrogen atom at the
end of the arginine and MMA is formed by connecting a single methyl
group to a terminal nitrogen atom (14). In addition, PRMTs catalyze
non-histone substrates, e.g., PRMT1 methylates RNA-binding proteins
(eukaryotic translation initiation factor 4A, infected cell protein
27 and polyadenylate binding protein 1) and transcription factors
Twist1, TATA-box-binding protein-associated factor 15, CCAAT
enhancer-binding protein α, runt-related transcription factor 1 and
transcription factors (7).
Based on the different catalytic products, PRMTs may
be divided into three types: Type I PRMTs are involved in the
catalytic synthesis of MMA and ADMA, and include PRMT1-3, PRMT6,
PRMT8 and arginine methyltransferase 1 (CARM1 or PRMT4); type II
PRMTs are involved in the catalytic synthesis of MMA and SDMA, and
include PRMT5, PRMT9; and type III PRMTs are involved in the
catalytic synthesis of MMA, which is mainly limited to the
production of methyl methacrylate by PRMT7 (10,15).
PRMTs participate in numerous cellular processes as
epigenetic regulators. For instance, PRMT2 was reported to be
overexpressed in several cancer types, such as breast cancer and
glioblastoma, as well as during cellular processes, such as
inflammatory response, Wnt signaling, cell growth and apoptosis
(16). PRMT5 catalyzes the
symmetrical dimethylation of histones and arginine residues in
numerous non-histone proteins, thereby regulating RNA splicing and
suppressing gene transcription (17). PRMTs are also closely related to
various diseases. PRMT3 was observed to be involved in the
accumulation of liver triglycerides dependent on liver X receptors
(LXRs) in vivo and in processes of CVD (18). PRMT7 has a key role in the
self-methylation-induced epithelial-mesenchymal transition process
and promotes the migration and invasive behavior of breast cancer
cells (19). In recent years,
studies have focused on the role of PRMTs in CVD. The following
will introduce the role of PRMTs in the occurrence and development
of CVDs; these roles are outlined below based on the relationship
between PRMTs, cholesterol metabolism and CVD. As presented in
Table I, to clarify the roles of
PRMT1-PRMT6 in CVDease, the characteristics, mediating factors and
related roles of each PRMT are listed.
 | Table IRole of PRMT1-PRMT7 in CVD. |
Table I
Role of PRMT1-PRMT7 in CVD.
| PRMT enzyme | Type | Arginine
methylation | Mediating
factors | Role in CVD | (Refs.) |
|---|
| PRMT1 | Ⅰ | MMA and ADMA | CaMKII | Inhibits myocardial
hypertrophy by inhibiting CaMKII | (10,15,49) |
| PRMT1 | Ⅰ | MMA and ADMA | PIP2 | Prevention of heart
failure by reducing IKs and extending the duration of ventricular
action potentials | (10,15,50) |
| PRMT2 | Ⅰ | MMA and ADMA | LXRs | Promotion of RCT by
increasing the expression of ABCA1 and ABCG1 | (10,15,22,28) |
| PRMT3 | Ⅰ | MMA and ADMA | LXRs | Promotion of RCT by
increasing the expression of ABCA1 and ABCG1 | (10,15,30,33,46) |
| | | | H4 and ox-LDL | Induction of the
occurrence of AS by inhibiting the production of intracellular
NO | (7,10,15,28,44,46,47) |
| PRMT4 | Ⅰ | MMA and ADMA | BCL2-associated
X | Evasion of
cardiomyocyte apoptosis by modulating Bax | (10,15,52) |
| PRMT5 | Ⅱ | MMA and SDMA | SREBP1 | Promotion of the
expression of genes associated with cholesterol biosynthesis | (10,15,36,37,39) |
| PRMT5 | Ⅱ | MMA and SDMA | GATA4 | Inhibition of
cardiomyocyte cell masting by inhibiting the transcriptional
activity of GATA4 | (10,15,57-59,61) |
| §PRMT5 | Ⅱ | MMA and SDMA | O-GlcN | Prevention of DCM
by inhibition of protein O-GlcN acylation | (10,15,53) |
| PRMT6 | Ⅰ | MMA and ADMA | H3R2Me2a | Enhancement of the
expression of atrial natriuretic peptide | (10,15,63-65) |
| PRMT7 | Ⅲ | MMA | β-catenin | Inhibition of the
Wnt/β-catenin pathway by regulating methylation of β-catenin and
thus myocardial hypertrophy | (10,15,67,68) |
3. PRMTs and cholesterol metabolism
PRMT2 promotes cholesterol efflux
PRMT2, also known as heterologous ribonucleoprotein
methyltransferase 1, is a key member of PRMTs and is located on
chromosome 21q22.3(20). PRMT2 is
widely present in various tissues of the human body, exhibiting
high expression in various tissues, including the blood vessels,
heart and nervous system. It mainly functions as an auxiliary
activator that interacts with a variety of nuclear proteins and
participates in RNA metabolism and transcriptional regulation, such
as in group protein methylation (21).
ATP-binding cassette transporter A1 (ABCA1) and
ABCG1 are members of the ABC superfamily, which are involved in
cholesterol metabolism, removing excess cholesterol from cells and
preventing AS (22). It has an
important role in mediating the transmembrane transport of
cholesterol and proteins by consuming ATP. Li et al
(23) indicated that overexpression
of PRMT2 significantly increased the expression of ABCA1 and
reduced cholesterol levels in macrophages induced by oxidized
low-density lipoprotein (ox-LDL), thereby affecting the efflux of
cholesterol and degradation of macrophage-derived foam cells. The
mechanism of these effects are related to inhibition of ox-LDL in
RAW 264.7 macrophages by PRMTs, which are involved in forming foam
cells induced by cholesterol efflux mediated by ABCA1 and
ABCG1.
Glomest first proposed the concept of reverse
cholesterol transport (RCT), which is the only means by which the
body is able to excrete excess cholesterol (24). Studies have indicated that RCT
dysfunction is involved in the occurrence and development of AS;
determining the molecular regulatory mechanism of RCT may provide
new ideas for the prevention and treatment of CVD (25). ABCA1 has a key role in RCT by
mediating the transfer of intracellular cholesterol to
apolipoprotein A1 (ApoA1). The accumulation of cholesterol in
macrophages may increase the expression of ABCA1 and cooperate with
ABCG1 to increase the outflow of cholesterol and promote the
formation of RCT (26), which is
meaningful for reducing accumulated cholesterol in the blood vessel
wall (27). As an important
mediator of ABCA1 and potential regulator of LXR, PRMT2 regulates
LXR-mediated ABCA1 expression and ABCA1-dependent cholesterol
efflux (28). Overexpression of
PRMT2 enhances ABCA1 gene expression, whereas ABCA1 gene expression
decreases upon PRMT2 depletion. In PRMT2 knockout macrophages,
which exhibit defects in ABCA1 upregulation and cholesterol efflux
to ApoA1, AS is more likely to occur because the cells cannot
effectively drain cholesterol. Therefore, it is essential to
understand the role of PRMT2 in LXR-mediated AS.
Cardiac hypertrophy is a common sign of CVD, which
refers to two changes of cardiac chamber dilatation or cardiac wall
hypertrophy, collectively referred to as cardiac hypertrophy. It is
important for the differential diagnosis and prognosis of CVD to
correctly determine the location and degree of cardiac
hypertrophy.
While cardiac hypertrophy may have numerous causes,
hypertensive heart disease and coronary AS heart disease are the
most common ones. The elevated cholesterol caused by abnormal
cholesterol metabolism and excretion increases the incidence of
coronary heart disease and markedly increases the occurrence of AS
(18).
PRMT2 is closely related to the occurrence and
development of AS (23). Studies
have indicated that macrophages of mice lacking PRMT2 are more
prone to AS due to their inability to efficiently excrete
cholesterol (28). The influence of
cholesterol efflux in macrophage-derived foam cells by PRMT2
overexpression inhibits the deposition of cholesterol in
macrophages and the formation of foam cells, which is considered to
be an important pathway against AS (23). Severe hypercholesterolemia leads to
stenosis of the lumen, resulting in persistent damage, which in
turn leads to systemic AS, resulting in myocardial ischemia and
coronary heart disease. In severe cases, acute myocardial
infarction aggravates CVD, which causes the heart to enlarge.
PRMT3 regulates cholesterol synthesis
and efflux
The unique ‘zinc finger’ structure at the N-terminus
of PRMT3 distinguishes it from other arginine methyltransferases,
indicating possible unique functions. PRMT3 has an important role
in several biological processes in the body. In recent years, PRMT3
has been considered a new target for anti-AS and CVD treatment.
Studies are required to explore the role of PRMT3 in these
diseases.
LXRs, which are members of the nuclear receptor
superfamily, are cholesterol receptors in the body. These receptors
promote cholesterol efflux by increasing the ABC transporter
subtype and control processes such as absorption, metabolism,
transport and decomposition. LXR is essential for cholesterol
metabolism in the liver and macrophages and is considered an
important target for treating disorders related to cholesterol
metabolism (29). PRMT3 is
considered a specific coactivator of LXR-mediated cholesterol
metabolism and LXR transcription cofactor. PRMT3 regulates
LXR-mediated cholesterol metabolism by upregulating the expression
of ABCA1 and ABCG1, thereby maintaining cholesterol homeostasis in
macrophages (30).
Kim et al (31) indicated that inhibiting the function
of PRMT3 prevents the accumulation of triglycerides in hepatocytes,
destroys the ability of LXR to stimulate adipogenesis and relieves
the driving effect of LXR on cholesterol metabolism and
LXR-mediated fatty acid and cholesterol metabolism. As a common
high-efficiency inhibitor of PRMT3, SGC707 reduces LXR activity by
inhibiting PRMT3, which weakens the degree of liver steatosis.
Chronic treatment of hyperlipidemia ApoE-knockout mice with the
allosteric PRMT3 inhibitor SGC707 effectively reduced the degree of
hepatic steatosis and significantly reduced the expression level of
lipoprotein lipase mRNA in adipocytes (32). This finding indicates that PRMT3 is
an intermediate factor that strongly and selectively affects the
activity of LXR in hepatocytes. The direct combination of LXR
signals strengthens transcriptional assistance and regulates liver
adipogenesis. Overexpression of PRMT3 may increase the expression
of lipogenic proteins, whereas PRMT3 silencing and PRMT3 knockout
in mouse embryonic fibroblast cell lines was indicated to reduce
the expression of lipogenic proteins (33).
PRMT3 is considered a novel target in human AS and
CVD. PRMT3 has a key role in cholesterol-driven pathologies, such
as AS (30). The accumulation of
systemic cholesterol and increased susceptibility to AS caused by
PRMT3 may increase the burden on the heart, affecting normal blood
circulation. This may lead to hardening of the arteries, promoting
the deposition of cholesterol and fat in the arterial intima and
forming AS plaques. The degree of stenosis increases significantly
and promotes the formation of AS, which is also considered to be
the pathological basis of coronary heart disease. It may lead to
the occurrence of cardiac hypertrophy or aggravate the original
degree of cardiac hypertrophy, which is not conducive to the
control and treatment of heart disease.
PRMT5 promotes cholesterol
biosynthesis and metabolism
PRMT5 is the main type II protein arginine
methyltransferase that catalyzes the symmetrical transfer of two
methyl groups to histones and functions by binding to specific
proteins in the nucleus and cytoplasm (34). PRMT5 controls a variety of metabolic
pathways, including the metabolism of cholesterol, fatty acids,
other lipids and amino acids but exerts its strongest impact on
cholesterol metabolism. Studies have indicated that the expression
of PRMT5 in activated T-cells is necessary for the expression of
cholesterol biosynthesis- and metabolism-related genes (35). The expression of cholesterol
metabolism-related genes in PRMT5-knockout T cells was indicated to
be reduced by ~42%, whereas in T-helper cells lacking PRMT5, ~75%
of the cholesterol biosynthesis pathway was inhibited (36). These results indicate that PRMT5
controls T-cell cholesterol metabolism and regulates the expression
of various enzymes involved in the cholesterol pathway.
Sterol-regulatory element-binding proteins (SREBPs)
are essential for maintaining the balance between protein and lipid
biosynthesis, and SREBP transcription factors regulate lipid and
sterol biosynthesis (37). Three
subtypes of SREBP have been identified in mammals. Among them,
SREBP1 is mainly involved in regulating fatty acid, triglyceride
and phospholipid synthesis genes, and SREBP2 is involved in
activating cholesterol synthesis genes and enhancing cholesterol
biosynthesis (38).
Studies have indicated that PRMT5 enhances the
stability of SREBP1 by promoting the expression of SREBP1 and
cholesterol biosynthetic pathway-related enzymes, thereby
regulating cholesterol synthesis (39). PRMT5 is overlapped and co-purified
with SREBP to increase its transcriptional and enzymatic activity
and interacts with SREBP1 in an enzyme activity-dependent manner,
which includes arginine methylation modification and inhibition of
proteasome enzymatic degradation. PRMT5 inhibits the
phosphorylation and proteasomal degradation of SREBP1 by
methylating it at R3231, stabilizing the cleavage nucleus form of
SREBP1 in T cells, increasing the synthesis of new fat and
promoting the expression of genes related to cholesterol
biosynthesis (36).
Of note, mechanistic studies have indicated that
PRMT5 is involved in lipid metabolism reprogramming and tumor
growth and metastasis through sirtuin 7 (SIRT7)-mediated
deglycosylation of PRMT5 K387(40).
SIRT7 acts as an eraser for PRMT5 succinylation by mediating PRMT5
K387 deacetylation and inducing SREBP1a arginine methylation,
promoting PRMT5-mep50 complex formation and increasing cholesterol,
fatty acid and triglyceride biosynthesis in cells. PRMT5 regulates
fatty acid metabolism and lipid droplet biosynthesis in white
adipose tissue and realizes the reprogramming of lipid metabolism;
a normal lipid metabolism is important for the regulation and
abnormality of cholesterol metabolism (39).
In summary, as outlined in Fig. 1, PRMTs regulate ABC levels and RCT
generation by mediating the expression of LXP and SREBP,
controlling cholesterol excretion and degradation and participating
in the dynamic balance of cholesterol regulation.
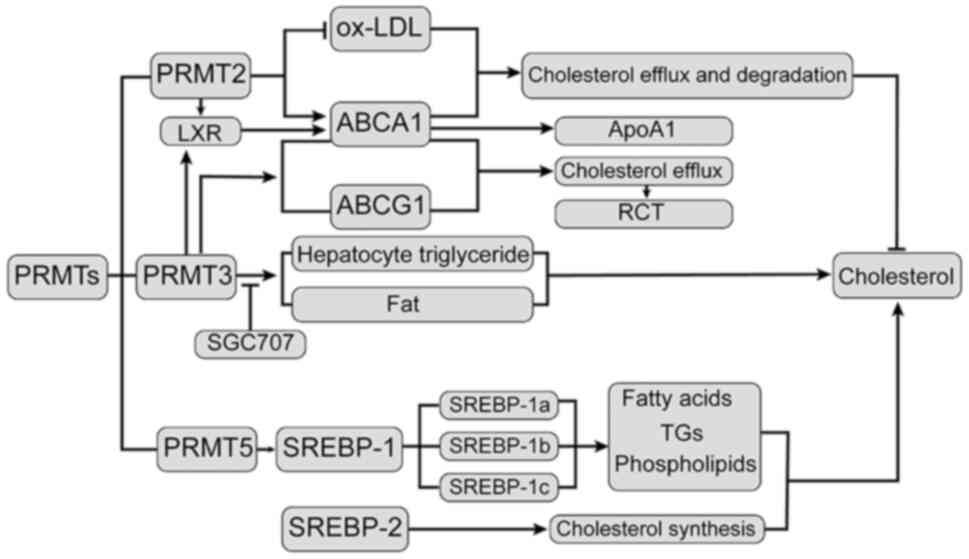 | Figure 1Relationship between PRMTs and
cholesterol. Overexpression of PRMT2 lowers ox-LDL-induced
macrophage cholesterol levels, significantly increasing the
expression of ABCA1 and thus affecting cholesterol efflux and
degradation. LXR is a cholesterol receptor in the body that
promotes cholesterol outflow by increasing ABCA1 and controlling
cholesterol absorption, metabolism, transport and decomposition.
SGC707 is a common phosphorylation PRMT3 high-efficiency inhibitor;
through inhibition of PRMT3 to reduce LXR activity, it may prevent
the accumulation of liver cell TGs and destroy the capacity of LXR
to stimulate fat production. PRMT5 interacts with SREBP1 in an
enzyme-based manner, involving the regulation of fatty acids, TGs
and phospholipids to regulate cholesterol synthesis. SREBP2 is
involved in activating the cholesterol synthesis gene to enhance
the biosynthesis of cholesterol. PRMT2, protein arginine
methyltransferase 2; ox-LDL, oxidized low-density lipoprotein; LXR,
liver X receptor; SREBP1, sterol-regulatory element binding protein
1; TGs, triglycerides; Apo, apolipoprotein; ABCA1, ATP binding
cassette subfamily A member 1. |
4. Relationship between PRMT family and
AS
AS is the most common CVD and is characterized by
the formation of AS or fibrous plaques in the vascular intima,
which involves the large and middle arteries and leads to ischemic
changes in the corresponding organs. Factors such as miRNAs,
autophagy and epigenetic modifications may be associated with AS by
affecting macrophage polarization (39). In recent years, numerous studies
have indicated that PRMT2 and PRMT3 are closely associated with
AS.
Relationship between PRMT2 and AS
After vascular injury factor intervention, PRMT2
knockout mice exhibited abnormal proliferation of vascular cells
and intimal hyperplasia (41),
leading to vascular endothelial dysfunction, which is a potential
factor for AS. It has been reported that bone marrow-derived
macrophages in mice lacking PRMT2 exhibit reduced cholesterol
efflux, suggesting that PRMT2 is involved in the formation of foam
cells and occurrence of AS (28).
Li et al (23) indicated
that overexpression of PRMT2 inhibited the formation of foam cells
in RAW264.7 macrophages induced by ox-LDL. This mechanism may be
related to an increase in ABCA1 expression and ABCA1-mediated
cholesterol efflux.
Relationship between PRMT3 and AS
Chen et al (42) determined that the mRNA expression
level of PRMT3 in the myocardial tissues of patients with AS was
two-fold higher than that in healthy controls. The common carotid
intima-media thickness is a measurement index of subclinical AS and
variations in the PRMT3 gene are related to changes in this
thickness (30). ADMA is a
methylation-modified product of PRMT3. Doğan et al (43) indicated that ADMA is a potential
factor for hypervolemia and AS in patients undergoing hemodialysis.
These studies demonstrated that PRMT3 is closely related to AS by
regulating the common carotid intima-media thickness and ADMA.
Small changes in plasma ADMA may cause large changes
in cells, thereby altering the production of nitric oxide (NO) and
leading to the development of CVDs (44). The impact of PRMT3 on AS may involve
the following mechanisms: ADMA and ox-LDL molecules may inhibit NO
production under the regulation of PRMT3, which is an important
cause of AS. PRMT3 transfers a methyl group to arginine residues of
histones and other proteins, resulting in asymmetric ADMA (30). ADMA is an endogenous competitive
inhibitor of epidermal NO synthase and is a favorable predictor of
CVD in patients with end-stage renal disease (45). Among patients with chronic kidney
disease, the inhibitory effect of ADMA on NO synthesis is enhanced
(46), which increases the risk of
AS. In addition, ox-LDL molecules induce AS and enable monocytes to
enter endothelial cells by binding to lectin-like ox-LDL receptor-1
(LOX-1), promoting foam cell formation and reducing intracellular
NO concentrations (46).
PRMT3 is a cofactor for LXR transcription (31). As a transcription factor, LXR
induces the expression of genes involved in cholesterol transport
and efflux and prevents AS by promoting reverse cholesterol
transport, reducing the accumulation of cholesterol in macrophages
and preventing the formation of foam cells (28,47).
Hoekstra et al (30)
experimentally demonstrated that PRMT3 is an auxiliary activator
that regulates the lipogenicity of LXR. Furthermore, PRMT3 may
affect cholesterol metabolism and increase susceptibility to AS.
The allosteric PRMT3 inhibitor SGC707 did not alter the
susceptibility to AS, but SGC707 may affect the ADMA/NO system,
revealing a correlation between PRMT3 and AS.
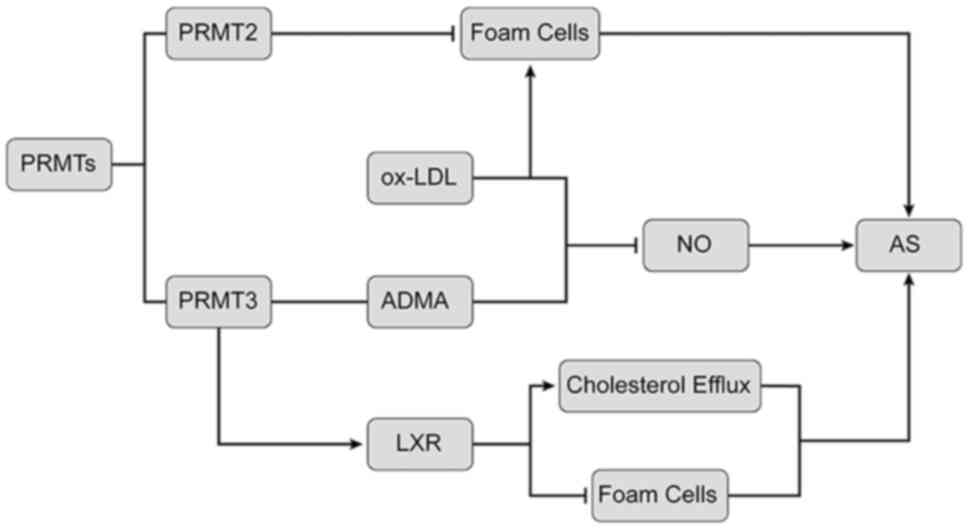
In summary, as illustrated in Fig. 2, PRMT2 inhibits ox-LDL-induced foam
cell formation. PRMT3 is closely related to AS by regulating the
expression of arginine methylation products and LOX-1.
5. PRMTs and heart failure
Heart failure is the leading cause of
hospitalization and death among the elderly worldwide (48). Heart failure is a chronic disease
that generally starts with ventricular hypertrophy and its main
feature is an increase in cell size associated with increased
ventricular remodeling and fibrosis. In the chapter below, the role
of PRMTs in the pathogenesis of heart failure is introduced.
PRMT1 and heart failure
PRMTs are overexpressed in numerous types of cancer
(4); however, the role of PRMTs in
heart disease has remained largely elusive. It has been indicated
that calmodulin-dependent protein kinase II (CaMKII) disorder is
closely related to myocardial hypertrophy and heart failure.
However, the mechanisms regulating CaMKII activity are not fully
understood (49). PRMT1 is
essential for preventing excessive activation of CaMKII in the
heart. For instance, cardiac PRMT1-negative mice rapidly developed
dilated cardiomyopathy (DCM) and heart failure within two months,
accompanied by cardiomyocyte hypertrophy and fibrosis. In isolated
cardiomyocytes, loss of PRMT1 may cause a hypertrophic response
with increased expression of remodeling genes. In the heart or
cardiomyocytes deficient in PRMT1, the level of active CaMKII was
significantly increased. PRMT1 interacts with and methylates CaMKII
at arginine residues 9 and 275, resulting in CaMKII inhibition.
Pharmacological inhibition of CaMKII restores contractile function
in PRMT1-deficient mice (49).
Therefore, PRMT1 deactivates CaMKII and maintains normal heart
function, which is important for developing drugs to treat heart
failure.
In addition, PRMT1 may regulate slow delayed
rectifier potassium current (IKs) activity by regulating the
affinity of phosphatidylinositol 4,5-bisphosphate (PIP2), a
membrane lipid of the IKs channel. IKs, assembled by the
pore-forming KCNQ1α subunit and the auxiliary KCN1β subunit, is
crucial for the late repolarization of cardiac action potentials.
The activity of IKs may be modulated by the regulation of PIP2
availability or PIP2 affinity of IKs. The combination of PIP2 and
KCNQ1 may serve to stabilize the channel in the open state. PRMT1
may regulate the IKs channel function by inducing arginine
methylation of the KCNQ1 subunit in the IKs channel. Studies on the
recombinant IKs channel have shown that in 293T cells containing
human KCNQ1 subunits, inhibition of PRMT1 reduces the methylation
of KCNQ1, thereby reducing the binding of PIP2 to KCNQ1 and
inducing a decrease in IKs activity. This indicates that PRMT1 is
necessary for the binding of KCNQ1 to PIP2. Recombinant IKs channel
studies have indicated that inhibition of PRMT1 reduces the
methylation of KCNQ1 in 293T cells containing human KCNQ1 and KCNE1
subunits, thereby reducing the binding of PIP2 to KCNQ1 and
inducing a decrease in IKs (50).
The PRMT1 function is necessary for the channel to be combined with
PIP2. Inhibition of PRMT1 prolonged the duration of ventricular
action potentials by reducing IKs. PRMT1-mediated regulation of
cardiac IKS activity may be a key target for preventing excessive
prolongation of the time course and arrhythmias in patients with
heart failure (50).
PRMT4 and heart failure
PRMT4 is a type I protein arginine methyltransferase
involved in a variety of cell biological processes. Myocardial
infarction is caused by irreversible myocardial necrosis induced by
a sharp decrease in blood flow to a certain area of the heart.
Heart remodeling after myocardial infarction eventually leads to
heart failure. Previous studies have mostly focused on the
involvement of PRMT4 in the occurrence and development of various
tumors, but its role in cardiomyocytes remains unclear. PRMT4
expression is significantly reduced in the ischemic myocardium and
hypoxic cardiomyocytes, and overexpression of PRMT4 aggravates
cardiac remodeling after myocardial infarction (51). PRMT4 overexpression promotes
hypoxia-induced cardiomyocyte apoptosis, whereas inhibition of
PRMT4 expression allows cardiomyocytes to avoid apoptosis by
regulating the apoptosis-related protein Bax (52). It was also indicated that loss of
PRMT4 increased Notch1-mediated podocyte apoptosis through the
PRMT4-protein kinase AMP-activated catalytic subunit α1-Notch1-CB1R
signaling axis (52). In summary,
hypoxia-induced upregulation of PRMT4 promotes cardiomyocyte
apoptosis and aggravates cardiac remodeling after myocardial
infarction. However, the precise underlying mechanisms require
further investigation. Of note, this discovery may provide new
ideas for the treatment of myocardial infarction by inhibiting
PRMT4 expression.
In Fig. 3, the role
of PRMTs in heart failure is summarized. PRMT1 maintains the normal
function of the heart by inactivating CaMKII. Hypoxia-induced PRMT4
overexpression upregulates cardiomyocyte apoptosis, whereas the
following PRMT5 interacts with GATA4 to inhibit its role in
promoting hypertrophy gene expression, leading to heart
failure.
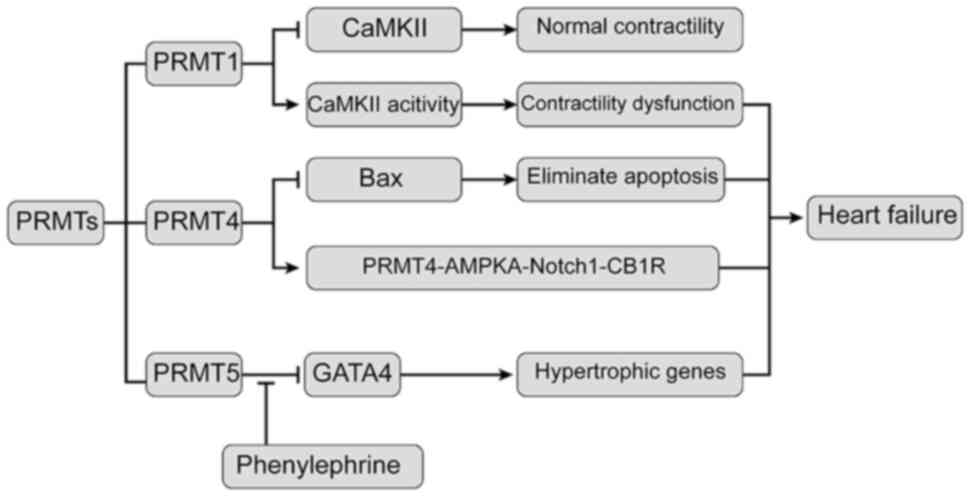 | Figure 3Relationship between PRMTs and heart
failure. PRMT1 interacts with and methylates CaMKII at arginine
residues 9 and 275, resulting in inhibition of CaMKII. Inhibition
of PRMT4 expression allows cardiomyocytes to eliminate apoptosis by
regulating the apoptosis-related protein Bax. PRMT4 increases
Notch1-mediated podocyte apoptosis through the
PRMT4-AMPKa-Notch1-CB1R signaling axis. PRMT5 leads to methylation
of arginine at sites 229, 265 and 317 of GATA4, resulting in
inhibition of transcriptional activity of GATA4. In addition, PRMT5
is transferred from the nucleus to the cytoplasm under
phenylepinephrine stimulation. In this way, PRMT5 reduces the
inhibition of GATA4 activity in the nucleus and leads to the
expression of hypertrophic genes in the cardiomyocytes. At last,
these above mechanisms lead to heart failure. CAMKII,
calmodulin-dependent protein kinase II; PRMT1, protein arginine
methyltransferase 1; AMPKa, protein kinase AMP-activated catalytic
subunit α1; GATA4, GATA binding protein 4. |
PRMT5 and heart failure
DCM is a complex myocardial disease characterized by
left ventricular dilatation and systolic dysfunction, and it
usually affects middle-aged males. Its occurrence is related to
viral infection, family inheritance and cellular immunity. The main
symptom of DCM is congestive heart failure.
Cardiac protein O-GlcNAcylation, as a key enzyme for
dynamic regulation, has been detected in various cardiac diseases.
Studies suggested that O-GlcNAcylation has an important
physiological role in maintaining cardiac function and
O-GlcNAcylation dysregulation has been indicated to be closely
related to cardiac disease. Overexpression of O-GlcNAcylation
contributes to DCM formation and premature death (53). Reducing elevated O-GlcNAcylation may
be a potential treatment for human DCM.
As a novel protein O-GlcNAcylation regulator, PRMT5
mainly regulates O-GlcNAcylation by regulating the alternative
splicing of O-GlcNAcase mRNA, symmetrically dimethylating
downstream targets including RNA splicing proteins (34). Mechanistic studies have indicated
that PRMT5 is able to prevent DCM by inhibiting protein O-GlcN
acylation (53). In human DCM
samples, PRMT5 expression was significantly downregulated (36). The absence of PRMT5 causes abnormal
increases in myocardial O-GlcNAcylation levels, aggravates
myocardial injury and further reduces the ejection fraction and
systolic function, leading to cardiac dysfunction and triggers
DCM.
There is a further mechanism leading to DCM.
N6-methyladenosine (m6A) is the most abundant
internal epigenetic modification on eukaryotic mRNAs (54). As the sole nuclear m6A
reader, YTH domain containing 1 (YTHDC1) may be related to DCM.
Mice with YTHDC1 deficiency developed obvious left ventricular
chamber enlargement and severe systolic dysfunction, which are
typical manifestations of DCM (55). Titin is a sarcomeric protein that
determines the structure and biomechanical properties of striated
muscle and its defect is directly associated with DCM (56). According to a study by Gao et
al (55), Titin is the direct
target of YTHDC1 and knockout of YTHDC1 in heart resulted in
abnormal splicing of Titin, contributing to disarray of sarcomere
structures in the cardiomyocytes, which is a typical feature of
DCM. To conclude, depletion of YTHDC1 induces DCM by abnormal
splicing of Titin and DCM is a common cause of heart failure
(55). However, whether PRMTs are
involved in the regulation of YTHDC1 depletion remains elusive and
further research is anticipated to explore this novel
mechanism.
6. PRMTs and myocardial hypertrophy
PRMT5 and cardiac hypertrophy
PRMT5 is a protein arginine methyltransferase that
catalyzes the symmetric dimethylation of arginine residues on the
target protein. It participates in numerous important cellular
processes, ranging from gene expression regulation to cell
proliferation and differentiation. PRMT5 is highly expressed in the
heart; however, its functional role in the heart remains elusive.
Among the six GATA transcription factors in vertebrates, GATA4-6
are expressed in the heart and regulate the expression of numerous
heart-specific genes, such as β-myosin heavy chain, cardiac
troponin C, cardiac troponin I, atrial natriuretic peptide and
brain natriuretic peptide (57,58).
GATA4 is important for promoting the development of cardiac
hypertrophy. For instance, overexpression of GATA4 induces
cardiomyocyte hypertrophy in cultured cardiomyocytes and mouse
hearts (59), whereas inhibiting
the activity of GATA4 results in the suppression of hypertrophic
gene expression in cardiomyocytes (60). PRMT5 specifically interacts with
GATA4 in cardiomyocytes. This interaction leads to methylation of
arginine at positions 229, 265 and 317 of GATA4 to result in
inhibition of the transcriptional activity of GATA4. In addition,
PRMT5 was indicated to be transferred from the nucleus to the
cytoplasm in response to phenylephrine stimulation. Thus, the
absence of PRMT5 decreases inhibition of GATA4 activity in the
nucleus, leading to the expression of cardiomyocyte mast genes
(61). These findings indicate that
PRMT5 is an important regulator of cardiac hypertrophy signaling
and suggest that strategies aimed at activating PRMT5 in the heart
are useful for preventing cardiac hypertrophy and heart failure
(62).
PRMT6 and cardiac hypertrophy
PRMT6 is the main methyltransferase that acts on
asymmetric dimethylation of histone H3 at arginine 2 (H3R2Me2a)
(63). Researchers have evaluated
the expression of PRMTs in the left ventricle of failing and
control hearts. The results indicated that in failing human hearts,
PRMT6 was significantly upregulated compared with that in control
hearts, which also occurs in the early stages of cardiac
hypertrophy in mouse hearts undergoing pressure-overload
hypertrophy induced by coarctation of transverse aortic
constriction and in neonatal rat ventricular myocytes stimulated
with the hypertrophy agonist phenylephrine (64). These changes are related to the
significant increase in H3R2Me2a and a decrease in trimethylation
of lysine 4 on histone H3 in neonatal rat ventricular myocytes and
in vivo (65). Of note,
overexpression of PRMT6 in neonatal rat ventricular myocytes
enhanced the expression of the hypertrophy marker atrial
natriuretic peptide. By contrast, silencing of PRMT6 reduced the
expression of atrial natriuretic peptide and the cell size,
indicating that PRMT6 is essential for the phenylephrine-mediated
hypertrophy response (64).
Therefore, PRMT6 is a key regulator of myocardial hypertrophy,
suggesting that H3R2Me2a is an important histone modification and
that PRMT6 mediates myocardial hypertrophy through differential
regulation of histone H3 arginine methylation.
In Fig. 4, the role
of PRMTs in cardiac hypertrophy is illustrated. PRMT1 and PRMT6 are
involved in the mediation of cardiac hypertrophy, while PRMT5
reduces the inhibition of GATA4 activity and leads to the
expression of hypertrophic genes in cardiomyocytes.
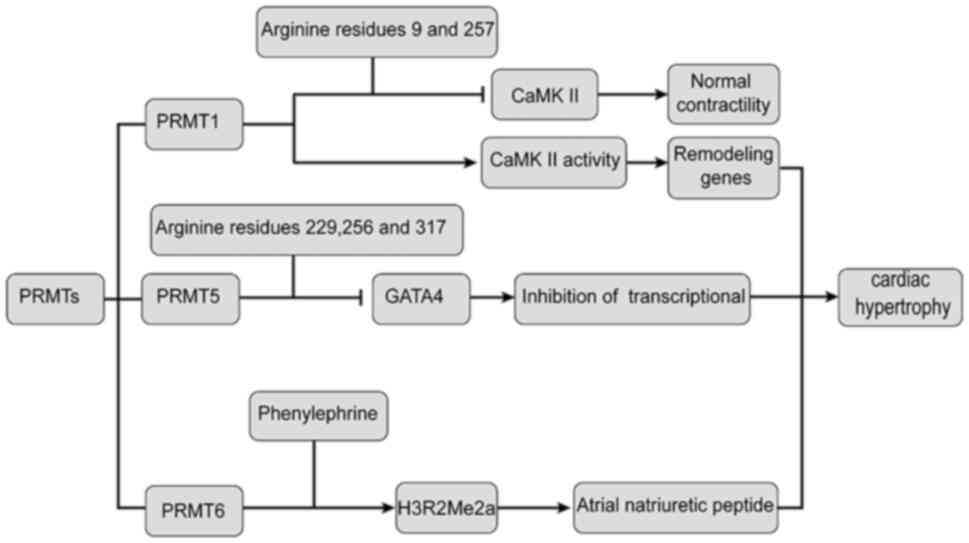 | Figure 4Relationship between PRMTs and
cardiac hypertrophy. PRMT1 interacts with CaMKII and is methylated
at arginine residues 9 and 275, leading to inhibition of CaMKII,
which may lead to hypertrophic gene expression. PRMT5 interacts
with GATA4, resulting in methylation at sites 229, 265 and 317 of
GATA4 arginine residues, thereby inhibiting the transcriptional
activity of GATA4, which may lead to the expression of hypertrophic
genes. PRMT6 mediates cardiac hypertrophy through differential
regulation of phenylephrine and asymmetric dimethylation of
H3R2Me2a. CAMKII, calmodulin-dependent protein kinase II; PRMT1,
protein arginine methyltransferase 1; AMPKa, protein kinase
AMP-activated catalytic subunit α1; GATA4, GATA binding protein 4;
H3R2Me2a, histone H3 at arginine 2. |
PRMT7 and cardiac hypertrophy
PRMT7 is a type III enzyme that catalyzes
monomethylation. PRMT7 is associated with biological processes,
such as regulating the proliferation of renal cell carcinoma by
regulating β-catenin activity (66). Mechanistic studies revealed that
PRMT7 suppresses the Wnt/β-catenin signaling pathway by methylation
of β-catenin at arginine residue 93, which is critical for the
regulation of β-catenin activity in the control of cardiac
hypertrophy and fibrosis (67).
Accumulating evidence suggests a prominent role of
Wnt/β-catenin signaling in cardiac hypertrophy and myocardial
fibrosis. The importance of Wnt/β-catenin signaling in
cardiomyopathy is underscored by the fact that inhibition of
Wnt/β-catenin signaling inhibits cardiac remodeling,
post-infarction mortality and decreased cardiac function (68). Wnt/β-catenin signaling is regulated
at multiple levels through complex mechanisms (69). Upon binding of Wnt/β-catenin ligands
to frizzled receptor and co-receptor lipoprotein receptor-related
protein 5/6, a signaling cascade is initiated to activate β-catenin
by suppression of inhibitory phosphorylation, ubiquitination and
proteasomal degradation, resulting in stabilization and subsequent
nuclear translocation of β-catenin and induction of target gene
expression (70). Although the
precise regulation of β-catenin activity appears to be essential
for normal cardiac remodeling, the mechanism of β-catenin
inhibition is currently unknown.
7. Differences between PRMT1-PRMT6 in CVD
and their application in CVD treatment
In the above chapters, the link between PRMT1-PRMT6
and CVD was described. PRMT1 and PRMT2 are cardioprotective
enzymes. PRMT1 depletion leads to myocardial hypertrophy and heart
failure by creating an imbalance of CaMKII (49). In addition, PRMT1-mediated
regulation of cardiac IKs activity may be a target for preventing
excessive prolongation of the course of disease and arrhythmia in
patients with heart failure (50).
PRMT2 upregulates the expression of ABCA1 protein, inhibits foam
cell formation and resists AS, while ABCA1 mediates cholesterol
efflux (28). PRMT3-PRMT6 promote
the occurrence and development of heart disease. As a nuclear
cofactor, PRMT3 regulates LXR lipogenesis by upregulating the
expression of ABCA1 and ABCG1, thereby increasing the expression of
lipogenic proteins, resulting in systemic cholesterol accumulation
and increasing the susceptibility to AS (30,31,33).
PRMT4 is involved in myocardial infarction and its overexpression
promotes hypoxia-induced cardiomyocyte apoptosis by regulating the
apoptosis-related protein Bax (51,52).
PRMT5 and PRMT6 are involved in the occurrence of myocardial
hypertrophy. PRMT5 increases the synthesis of fat by enhancing the
stability of SREBP1 (36,39); At the same time, PRMT5 also prevents
myocardial hypertrophy and DCM by inhibiting the effect of GATA4
and inhibiting protein O-GlcN acylation (53,59,61),
respectively. Silencing of PRMT6 expression inhibits the
hypertrophy-promoting effect of phenylephrine and participates in
the formation of cardiac hypertrophy (64). PRMT7 inhibits cardiac remodeling,
post-infarction mortality and decreased cardiac function by
inhibiting the Wnt/β-catenin signaling pathway (67,68).
Therefore, increasing the activity of PRMT1 and PRMT2 or decreasing
the expression of PRMT3-PRMT6 may be potential therapeutic targets
for CVDs.
8. Conclusion
PRMT1-7 is involved in the occurrence of numerous
CVDs, such as AS, heart failure and myocardial hypertrophy, by
regulating the methylation of arginine residues, the activity of
LXR transcription factors and signal transduction enzymes. Other
PRMTs, such as PRMT8 and -9, have not been demonstrated to have a
role in CVD and require further analysis. The present study mainly
introduced the role of PRMT2, -3 and -5 in cholesterol metabolism
and reviewed the relationship between PRMT1 and maintenance of
normal cardiac function between PRMT4 and myocardial infarction,
between PRMT5-7 and myocardial hypertrophy, and between PRMT2 and
-3 and AS in vascular diseases. As an epigenetic species, the
differential expression of PRMT1-PRMT7 and their mediating roles in
CVD are complex, involving both enzyme activity-dependent and
possibly non-enzyme activity-dependent effects. Different ways of
action may be the reason why they have different roles in CVDs.
Although the role of PRMTs in CVD has been examined,
numerous issues remain to be addressed, such as how PRMT2
upregulates the expression of ABCA1 and the detailed underlying
mechanism. Furthermore, whether cardiovascular-related inhibitors
of PRMTs affect other organs remains elusive. In addition, PRMTs
may also affect the occurrence and development of CVDs in manners
that have not been identified. Further studies of PRMTs will reveal
their roles and mechanisms in CVDs, leading to the identification
of drug targets and improving their clinical applications.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 82060250),
the Guangxi Science and Technology Plan Project (grant no.
AD20238035), the Open Project of Guangxi Key Laboratory of Brain
and Cognitive Neuroscience (grant no. GKLBCN-20190105-02), the
Guangxi Zhuang Autonomous Region Students' Innovation and
Entrepreneurship Training Program of China (grant nos. 202010601050
and 202110601010), Hunan Provincial Health Commission Project
(grant no. 20201934) and the Natural Science Foundation of Hunan
Province (grant no. 2021JJ30611).
Availability of data and materials
Not applicable.
Authors' contributions
SiZ and CZ were involved in the conception and
design of this study. AH and FH performed the literature search
selection. AM and ShZ contributed to manuscript revision. ZW
coordinated the study and reviewed the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Lin WJ, Gary JD, Yang MC, Clarke S and
Herschman HR: The mammalian immediate-early TIS21 protein and the
leukemia-associated BTG1 protein interact with a protein-arginine
N-methyltransferase. J Biol Chem. 271:15034–15044. 1996.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang J, Tan GJ, Han LN, Bai YY, He M and
Liu HB: Novel biomarkers for cardiovascular risk prediction. J
Geriatr Cardiol. 14:135–150. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhao D, Liu J, Wang M, Zhang X and Zhou M:
Epidemiology of cardiovascular disease in China: Current features
and implications. Nat Rev Cardiol. 16:203–212. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jarrold J and Davies CC: PRMTs and
arginine methylation: Cancer's best-kept secret? Trends Mol Med.
25:993–1009. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yang Y and Bedford MT: Protein arginine
methyltransferases and cancer. Nat Rev Cancer. 13:37–50.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Zhang W, Song M, Qu J and Liu GH:
Epigenetic modifications in cardiovascular aging and diseases. Circ
Res. 123:773–786. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Al-Hamashi AA, Diaz K and Huang R:
Non-histone arginine methylation by protein arginine
methyltransferases. Curr Protein Pept Sci. 21:699–712.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Xu S, Pelisek J and Jin ZG:
Atherosclerosis is an epigenetic disease. Trends Endocrinol Metab.
29:739–742. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yang Y and Luan Y, Yuan RX and Luan Y:
Histone methylation related therapeutic challenge in cardiovascular
diseases. Front Cardiovasc Med Sep. 8(710053)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rakow S, Pullamsetti SS, Bauer UM and
Bouchard C: Assaying epigenome functions of PRMTs and their
substrates. Methods. 175:53–65. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Couto E Silva A, Wu CY, Citadin CT,
Clemons GA, Possoit HE, Grames MS, Lien CF, Minagar A, Lee RH,
Frankel A and Lin HW: Protein arginine methyltransferases in
cardiovascular and neuronal function. Mol Neurobiol. 57:1716–1732.
2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Larsen SC, Sylvestersen KB, Mund A, Lyon
D, Mullari M, Madsen MV, Daniel JA, Jensen LJ and Nielsen ML:
Proteome-wide analysis of arginine monomethylation reveals
widespread occurrence in human cells. Sci Signal.
9(rs9)2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Blanc RS and Richard S: Arginine
methylation: The coming of age. Mol Cell. 65:8–24. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Morales Y, Cáceres T, May K and Hevel JM:
Biochemistry and regulation of the protein arginine
methyltransferases (PRMTs). Arch Biochem Biophys. 590:138–152.
2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
vanLieshout TL and Ljubicic V: The
emergence of protein arginine methyltransferases in skeletal muscle
and metabolic disease. Am J Physiol Endocrinol Metab.
317:E1070–E1080. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hu G, Yan C, Xie P, Cao Y, Shao J and Ge
J: PRMT2 accelerates tumorigenesis of hepatocellular carcinoma by
activating Bcl2 via histone H3R8 methylation. Exp Cell Res.
394(112152)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hamard PJ, Santiago GE, Liu F, Karl DL,
Martinez C, Man N, Mookhtiar AK, Duffort S, Greenblatt S, Verdun RE
and Nimer SD: PRMT5 regulates DNA repair by controlling the
alternative splicing of histone-modifying enzymes. Cell Rep.
24:2643–2657. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nahon JE, Groeneveldt C, Geerling JJ, van
Eck M and Hoekstra M: Inhibition of protein arginine
methyltransferase 3 activity selectively impairs liver X
receptor-driven transcription of hepatic lipogenic genes in vivo.
Br J Pharmaco. 175:3175–3183. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Geng P, Zhang Y, Liu X, Zhang N, Liu Y,
Liu X, Lin C, Yan X, Li Z, Wang G, et al: Automethylation of
protein arginine methyltransferase 7 and its impact on breast
cancer progression. FASEB J. 31:2287–2300. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vhuiyan MI, Pak ML, Park MA, Thomas D,
Lakowski TM, Chalfant CE and Frankel A: PRMT2 interacts with
splicing factors and regulates the alternative splicing of BCL-X. J
Biochem. 162:17–25. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hou W, Nemitz S, Schopper S, Nielsen ML,
Kessels MM and Qualmann B: Arginine methylation by PRMT2 controls
the functions of the actin nucleator cobl. Dev Cell. 45:262–275.e8.
2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang S, Li L, Wang J, Zhang T, Ye T, Wang
S, Xing D and Chen W: Recent advances in the regulation of ABCA1
and ABCG1 by lncRNAs. Clin Chim Acta. 516:100–110. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li YY, Zhou SH, Chen SS, Zhong J and Wen
GB: PRMT2 inhibits the formation of foam cell induced by ox-LDL in
RAW 264.7 macrophage involving ABCA1 mediated cholesterol efflux.
Biochem Biophys Res Commun. 524:77–82. 2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Rohatgi A: Reverse cholesterol transport
and atherosclerosis. Arterioscler Thromb Vasc Biol. 39:2–4.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zaiou M and Bakillah A: Epigenetic
regulation of ATP-binding cassette protein A1 (ABCA1) gene
expression: A new Era to alleviate atherosclerotic cardiovascular
disease. Diseases. 6(34)2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang T, Zhao Y, You Z, Li X, Xiong M, Li H
and Yan N: Endoplasmic reticulum stress affects cholesterol
homeostasis by inhibiting LXR α expression in hepatocytes and
macrophages. Nutrients. 12(3088)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhang M, Zhao GJ, Yin K, Xia XD, Gong D,
Zhao ZW, Chen LY, Zheng XL, Tang XE and Tang CK: Apolipoprotein A-1
binding protein inhibits inflammatory signaling pathways by binding
to apolipoprotein A-1 in THP-1 macrophages. Circ J. 82:1396–1404.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hussein MA, Shrestha E, Ouimet M, Barrett
TJ, Leone S, Moore KJ, Hérault Y, Fisher EA and Garabedian MJ:
LXR-mediated ABCA1 expression and function are modulated by high
glucose and PRMT2. PLoS One. 10(e0135218)2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ramón-Vázquez A, de la Rosa JV, Tabraue C,
Lopez F, Díaz-Chico BN, Bosca L, Tontonoz P, Alemany S and
Castrillo A: Common and differential transcriptional actions of
nuclear receptors liver X receptors α and β in macrophages. Mol
Cell Biol. 39:e00376–18. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hoekstra M, Nahon JE, de Jong LM, Kröner
MJ, de Leeuw LR and Van Eck M: Inhibition of PRMT3 activity reduces
hepatic steatosis without altering atherosclerosis susceptibility
in ApoE knockout mice. Biochim Biophys Acta Mol Basis Dis.
1865:1402–1409. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kim DI, Park MJ, Lim SK, Park JI, Yoon KC,
Han HJ, Gustafsson JA, Lim JH and Park SH: PRMT3 regulates hepatic
lipogenesis through direct interaction with LXRα. Diabetes.
64:60–71. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
He PP, Jiang T, OuYang XP, Liang YQ, Zou
PQ, Wang Y, Shen QQ, Liao L and Zheng XL: Lipoprotein lipase:
Biosynthesis, regulatory factors, and its role in atherosclerosis
and other diseases. Clin Chim Acta. 480:126–137. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wu YT, Li JB, Lin HQ, Zhang GX, Hong CM,
Li M, Guo ZJ and Yang YB: Inhibition of miR-200b-3p alleviates
lipid accumulation and promotes cholesterol efflux by targeting
ABCA1 in macrophage-derived foam cells. Exp Ther Med.
22(831)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Vinet M, Suresh S, Maire V, Monchecourt C,
Némati F, Lesage L, Pierre F, Ye M, Lescure A, Brisson A, et al:
Protein arginine methyltransferase 5: A novel therapeutic target
for triple-negative breast cancers. Cancer Med. 8:2414–2428.
2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Webb LM, Sengupta S, Edell C,
Piedra-Quintero ZL, Amici SA, Miranda JN, Bevins M, Kennemer A,
Laliotis G, Tsichlis PN and Guerau-de-Arellano M: Protein arginine
methyltransferase 5 promotes cholesterol biosynthesis-mediated Th17
responses and autoimmunity. J Clin Invest. 130:1683–1698.
2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Shimano H and Sato R: SREBP-regulated
lipid metabolism: Convergent physiology-divergent pathophysiology.
Nat Rev Endocrinol. 13:710–730. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yang HX, Zhang M, Long SY, Tuo QH, Tian Y,
Chen JX, Zhang CP and Liao DF: Cholesterol in LDL receptor
recycling and degradation. Clin Chim Acta. 500:81–86.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kober DL, Xu S, Li S, Bajaj B, Liang G,
Rosenbaum DM and Radhakrishnan A: Identification of a degradation
signal at the carboxy terminus of SREBP2: A new role for this
domain in cholesterol homeostasis. Proc Natl Acad Sci USA.
117:28080–28091. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Liu L, Zhao X, Zhao L, Li J, Yang H, Zhu
Z, Liu J and Huang G: Arginine methylation of SREBP1a via PRMT5
promotes de novo lipogenesis and tumor growth. Cancer Res.
76:1260–1272. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yuan HF, Zhao M, Zhao LN, Yun HL, Yang G,
Geng Y, Wang YF, Zheng W, Yuan Y, Song TQ, et al: PRMT5 confers
lipid metabolism reprogramming, tumour growth and metastasis
depending on the SIRT7-mediated desuccinylation of PRMT5 K387 in
tumours. Acta Pharmacol Sin. 43:2373–2385. 2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yoshimoto T, Boehm M, Olive M, Crook MF,
San H, Langenickel T and Nabel EG: The arginine methyltransferase
PRMT2 binds RB and regulates E2F function. Exp Cell Res.
312:2040–2053. 2006.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chen X, Niroomand F, Liu Z, Zankl A, Katus
HA, Jahn L and Tiefenbacher CP: Expression of nitric oxide related
enzymes in coronary heart disease. Basic Res Cardiol. 101:346–353.
2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Doğan I, Eser B, Özkurt S, Yayar O, Özgür
B, Kayadibi H, Doğan T, Muşmul A and Soydan M: Serum ADMA,
endothelial dysfunction, and atherosclerosis in hypervolemic
hemodialysis patients. Turk J Med Sci. 48:1041–1047.
2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Alpoim PN, Sousa LP, Mota AP, Rios DR and
Dusse LM: Asymmetric dimethylarginine (ADMA) in cardiovascular and
renal disease. Clin Chim Acta. 440:36–39. 2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shafi T, Hostetter TH, Meyer TW, Hwang S,
Hai X, Melamed ML, Banerjee T, Coresh J and Powe NR: Serum
asymmetric and symmetric dimethylarginine and morbidity and
mortality in hemodialysis patients. Am J Kidney Dis. 70:48–58.
2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Dogan I, Dogan T, Yetim M, Kayadibi H,
Yilmaz MB, Eser B, Kalcik M and Karavelioglu Y: Relation of serum
ADMA, apelin-13 and LOX-1 levels with inflammatory and
echocardiographic parameters in hemodialysis patients. Ther Apher
Dial. 22:109–117. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ito A, Hong C, Oka K, Salazar JV, Diehl C,
Witztum JL, Diaz M, Castrillo A, Bensinger SJ, Chan L and Tontonoz
P: Cholesterol accumulation in CD11c+ immune cells is a causal and
targetable factor in autoimmune disease. Immunity. 45:1311–1326.
2016.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Butrous H and Hummel SL: Heart failure in
older adults. Can J Cardiol. 32:1140–1147. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Pyun JH, Kim HJ, Jeong MH, Ahn BY, Vuong
TA, Lee DI, Choi S, Koo SH, Cho H and Kang JS: Cardiac specific
PRMT1 ablation causes heart failure through CaMKII dysregulation.
Nat Commun. 9(5107)2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
An X, Lee J, Kim GH, Kim HJ, Pyo HJ, Kwon
I and Cho H: Modulation of IKs channel-PIP2
interaction by PRMT1 plays a critical role in the control of
cardiac repolarization. J Cell Physiol. 237:3069–3079.
2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wang Y, Ju C, Hu J, Huang K and Yang L:
PRMT4 overexpression aggravates cardiac remodeling following
myocardial infarction by promoting cardiomyocyte apoptosis. Biochem
Biophys Res Commun. 520:645–650. 2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li Z, Xu J, Song Y, Xin C, Liu L, Hou N,
Teng Y, Cheng X, Wang T, Yu Z, et al: PRMT5 prevents dilated
cardiomyopathy via suppression of protein O-GlcNAcylation. Circ
Res. 129:857–871. 2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kim D, Lim S, Park M, Choi J, Kim J, Han
H, Yoon K, Kim K, Lim J and Park S: Ubiquitination-dependent CARM1
degradation facilitates notch1-mediated podocyte apoptosis in
diabetic nephropathy. Cell Signal. 26:1774–1782. 2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Roundtree IA, Evans ME, Pan T and He C:
Dynamic RNA modifications in gene expression regulation. Cell.
169:1187–1200. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Gao S, Sun H, Chen K, Gu X, Chen H, Jiang
L, Chen L, Zhang S, Liu Y, Shi D, et al: Depletion of
m6A reader protein YTHDC1 induces dilated cardiomyopathy
by abnormal splicing of Titin. J Cell Mol Med. 25:10879–10891.
2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Roberts AM, Ware JS, Herman DS, Schafer S,
Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, et al:
Integrated allelic, transcriptional, and phenomic dissection of the
cardiac effects of titin truncations in health and disease. Sci
Transl Med. 7(270ra6)2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Gong Y, Zhang L, Zhang A, Chen X, Gao P
and Zeng Q: GATA4 inhibits cell differentiation and proliferation
in pancreatic cancer. PLoS One. 13(e0202449)2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Chang YM, Chang HH, Lin HJ, Tsai CC, Tsai
CT, Chang HN, Lin SL, PadmaViswanadha V, Chen RJ and Huang CY:
Inhibition of cardiac hypertrophy effects in d-galactose-induced
senescent hearts by alpinate oxyphyllae fructus treatment. Evid
Based Complement Alternat Med. 2017(2624384)2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Glenn DJ, Rahmutula D, Nishimoto M, Liang
F and Gardner DG: Atrial natriuretic peptide suppresses endothelin
gene expression and proliferation in cardiac fibroblasts through a
GATA4-dependent mechanism. Cardiovasc Res. 84:209–217.
2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhang G, Wang X, Bi X, Li C, Deng Y,
Al-Hashimi AA, Luo X, Gillette TG, Austin RC, Wang Y and Wang ZV:
GRP78 (glucose-regulated protein of 78 kDa) promotes cardiomyocyte
growth through activation of GATA4 (GATA-binding protein 4).
Hypertension. 73:390–398. 2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Chen M, Yi B and Sun J: Inhibition of
cardiomyocyte hypertrophy by protein arginine methyltransferase 5.
J Biol Chem. 289:24325–24335. 2014.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Cai S, Liu R, Wang P, Li J, Xie T, Wang M,
Cao Y, Li Z and Liu P: PRMT5 prevents cardiomyocyte hypertrophy via
symmetric demethylating HoxA9 and repressing HoxA9 expression.
Front Pharmacol. 11(600627)2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Bouchard C, Sahu P, Meixner M, Nötzold RR,
Rust MB, Kremmer E, Feederle R, Hart-Smith G, Finkernagel F,
Bartkuhn M, et al: Genomic location of PRMT6-dependent H3R2
methylation is linked to the transcriptional outcome of associated
genes. Cell Rep. 24:3339–3352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Raveendran VV, Al-Haffar K, Kunhi M,
Belhaj K, Al-Habeeb W, Al-Buraiki J, Eyjolsson A and Poizat C:
Protein arginine methyltransferase 6 mediates cardiac hypertrophy
by differential regulation of histone H3 arginine methylation.
Heliyon. 6(e03864)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Cheng D, Gao G, Di Lorenzo A, Jayne S,
Hottiger MO, Richard S and Bedford MT: Genetic evidence for partial
redundancy between the arginine methyltransferases CARM1 and PRMT6.
J Biol Chem. 295:17060–17070. 2020.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Liu F, Wan L, Zou H, Pan Z, Zhou W and Lu
X: PRMT7 promotes the growth of renal cell carcinoma through
modulating the β-catenin/C-MYC axis. Int J Biochem Cell Biol.
120(105686)2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Ahn BY, Jeong MH, Pyun JH, Jeong HJ, Vuong
TA, Bae JH, An S, Kim SK, Kim YK, Ryu D, et al: PRMT7 ablation in
cardiomyocytes causes cardiac hypertrophy and fibrosis through
β-catenin dysregulation. Cell Mol Life Sci. 79(99)2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Bergmann ME: WNT signaling in adult
cardiac hypertrophy and remodeling: Lessons learned from cardiac
development. Circ Res. 107:1198–1208. 2010.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Angers S and Moon RT: Proximal events in
Wnt signal transduction. Nat Rev Mol Cell Biol. 10:468–477.
2009.PubMed/NCBI View Article : Google Scholar
|
|
70
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009.PubMed/NCBI View Article : Google Scholar
|