1. Introduction
The aging population combined with the burden of
various risk factors, including atrial fibrillation, hypertension,
diabetes, hyperlipidemia, smoking, lack of physical activity,
unhealthy diets, abdominal obesity and alcohol consumption, all
lead to an increased lifelong risk of stroke (1,2).
Ischemic stroke is a result of either permanent or transient
regional reduction in brain blood supply in the brain, resulting in
motor difficulties, such as in movement or speech. Through advances
in pharmacological and mechanical thrombolysis, significant
progress has been made in the field of therapeutic interventions
for ischemic stroke. However, these methods are only effective
during a narrow window of time (1,2). To
the best of our knowledge, the pathogenesis of ischemic stroke
remains unclear, meaning that further studies are needed to
identify valuable therapeutic targets for improving treatment
methods. The identification of genes associated with the
progression of ischemic stroke is currently being studied to
identify relevant targets (1,2).
Ferroptosis is caused by an increase in
iron-dependent toxic lipid reactive oxygen species (ROS) levels,
particularly when the oxidation of membrane polyunsaturated fatty
acids (PUFAs) cannot be performed due to the inactivation of lipid
hydroperoxide glutathione (GSH) peroxidase 4 (GPX4) (3,4).
Ferroptosis is a regulated process that differs from apoptosis and
other forms of non-apoptotic cell death, which are typically
caspase-dependent (5). In
addition, it has been found to be associated with neuronal cell
death during a stroke (4).
Upregulated mutant p53 was first discovered in
cancer. Subsequent research has demonstrated other functions of p53
and it is now considered to be the most important tumor suppressor,
known as 'guardian of the genome (3). p53 is essential for growth and
development; mice treated with a p53 inhibitor or that had p53
expression knocked out were found to have manifestations of
extracerebral malformation, spina bifida, ocular abnormalities,
embryonic brain malformations and other developmental issues
(6). The earlier the loss of genes
regulating p53, such as mouse double minute (MDM)2 and MDM4, the
more serious the phenotypic abnormalities (7). Physiologically, ubiquitination and E3
ubiquitin ligases strictly controls p53 and keeps its functional
levels low during cell and embryonic development (8). Therefore, p53 is controlled precisely
under physiological condition and it regulates growth and
development.
Recently, it has been found that p53 is involved in
the pathogenesis of ischemic stroke and the ferroptotic signaling
pathway (9). As a transcription
factor, p53 can directly activate or inhibit the transcription of a
long list of genes, several of which serve a key role in
ferroptosis (6). In the present
review, the primary signaling pathways and relationship between
p53, ferroptosis and ischemic stroke are discussed.
2. Potential pathogenesis and treatment
status of cerebral ischemia injury
Ischemia stroke is the result of
ischemia/reperfusion (I/R) or ischemia in the brain, in which the
damaged region of the brain can be divided into two following
different regions: Ischemic core region and the penumbra region
(10). In the core area, if the
cerebral blood flow decreases below the end-stage depolarization
threshold, it would irreversibly destroy the structure and function
of neurons residing here (11).
The penumbra area refers to the neuronal regions around the
infarcted core, which maintains its structural integrity but has
impaired neuronal function due to the decrease in cerebral blood
flow (12,13). Compared with those in the core
area, neurons with normal structure and activity in the penumbra
area can be preserved if treated in the appropriate window of time.
This area can maintain a potential viability for 16-48 h, which
provides a treatment window for clinical intervention (11). Therefore, this is a potential
therapeutic target for the treatment of acute ischemic brain injury
in a clinical setting.
It has been previously reported through experimental
data that the recovery of the blood supply will not restore the
function of injured neurons, but instead aggravate the damage, in a
phenomenon known as cerebral I/R injury (14). This typically occurs in various
blood flow occlusion conditions in the clinic, such as
cardiopulmonary resuscitation, which causes delayed/prolonged
neuronal damage, impairing the function of the central nervous
system (15). To the best of our
knowledge, the underlying mechanism remains unclear, although
several theories have been proposed (2). The cessation of arterial blood flow
leads to hypoxia in neurons, which in turn disrupts electron
transport chain function in the mitochondria due to the loss of
oxygen and glucose supply. This results in reduced ATP production,
promoting anaerobic metabolism and dysfunctional
Na+-K+-ATPase and Ca2+-ATPase pump
activity (4). A series of
intracellular signaling cascades are activated as a result of
decreased ATP and antioxidant levels. Insufficient blood supply
results in irreversible injury and necrosis in the core area, but
the peri-infarcted area is potentially salvageable. Following the
reperfusion phase, blood flow to the ischemic tissue is restored,
which also restores the oxygen supply (14). However, this results in increased
production of ROS, coupled with an insufficient quantity of
antioxidants due to the cellular dysfunction caused by ischemia
(14). Therefore, the reperfusion
continues to aggravate the oxidative stress (2), promoting endothelial dysfunction, DNA
damage and a local inflammatory cascade. This cascade of
inflammation then activates the microglia in the brain, increasing
the permeability of the blood-brain barrier (BBB) and infiltration
by peripheral immune cells (2),
further aggravating the injury.
Patients who have experienced an ischemic stroke are
typically treated with thrombolytic therapy [tissue plasminogen
activator (tPA)] within 4.5 h of the onset of stroke symptoms
(4,10,16,17).
tPA treatment outside of this specific time window can result in a
hemorrhagic performance, which causes additive but unnecessary
damage to the brain (10). Other
treatment strategies, including thrombectomy and preventive drug
treatments, such as the early application of blood pressure- and
cholesterol-lowering drugs, have also been used for treating
ischemic stroke (16). Since a
second stroke frequently occurs immediately after the initial
stroke, timely treatment is of importance for reducing the depth of
disabilities caused by the initial and/or secondary stroke
(16). Patients with disabilities
caused by ischemic strokes, such as hemiparesis, facial paresis,
dysarthria, unconsciousness, language and speech disorders or
impaired vision, have a significantly reduced quality of life
(16). Several factors, such as
diet, smoking habits, high blood pressure and diabetes, have been
shown to increase the risk of stroke (18). In recent years, the incidence rate
of stroke and the prevalence of younger patients with stroke have
both increased (10). Therefore,
adequate control of manageable risk factors, such as diabetes and
hypertension, can prevent ischemic stroke in high-risk groups
(19). In addition, further
research into the accurate mechanism of ischemic stroke can
facilitate the identification of novel avenues for the management
of this disease.
3. Ferroptosis
Features of ferroptosis
A primary cause of ferroptosis is the increase in
iron-dependent toxic lipid ROS levels through the peroxidation of
PUFAs, which is caused by the dysregulation of the endogenous
antioxidant network (5,20-23).
Another key condition for ferroptosis is the overload of
Fe2+ in cells (5,17,21,24).
These two aforementioned elements are hypothesized to induce
irreversible lipid damage and increased membrane permeability.
During mammalian development, caspase-dependent
apoptosis is the most common method of regulated cell death
(4,25). Ferroptosis was initially identified
in oncogenic Ras-expressing human foreskin fibroblast cell cells
treated with the synthetic small molecule erastin or RAS-selective
lethal 3 (RSL3), which did not induce caspase activation but was
significantly reversed by specific antioxidants and iron chelators
(26). These results suggest that
the molecular mechanism of ferroptosis differed from that of
classical apoptosis.
Intracellular ATP consumption, caspases and lysosome
activation are not necessary for ferroptosis, in stark contrast to
apoptosis, necrosis and autophagy (27). The characteristics and
morphological features of ferroptosis can be used to identify
ferroptotic cells. Typically, ferroptotic mitochondria will exhibit
disordered cristae and increases in mitochondrial membrane density
(4,22,28,29).
By contrast, nuclear changes, such as nuclear condensation or
chromatin marginalization and condensation, do not occur in
ferroptosis (30). Instead, lipid
peroxidation is observed, such that the condition of the cells
after treatment with iron chelators or lipid peroxidation
inhibitors can be used to determine whether ferroptosis is
occurring (26). Recently,
antibodies against transferrin (TF) receptor 1 (TfR1), including
3F3 iron body membrane antibody, have also been used to detect
ferroptosis (31).
Ferroptosis has been observed in several processes,
including the inhibition of different types of human cancer
(32-34),
tissue I/R injury (24,25,33,35-37),
neurodegenerative diseases (21,24,25,33,36,38,39),
intracerebral hemorrhage (40) and
both the innate and adaptive immune responses (30,41,42).
In addition, ferroptosis can form part of the process downstream of
different molecular signaling pathways (43). Recent studies have also confirmed
that it is involved in solute carrier family 7 member 11
(SLC7A11)/GSH/GPX4 signaling pathways and can be controlled by
specific factors, such as p53, in cells in the central nervous
system (43).
Iron metabolism
In mammalian cells, iron is normally bound and
absorbed by various transporters or receptors, through non-heme and
heme-dependent absorption pathways. Fe2+ is involved in
the regulation of various cellular processes, including oxygen
transport, cell proliferation, cell division, energy production and
DNA synthesis. In addition, Fe2+ can also serve as a key
co-factor in regulating various activities, such as cell size,
inflammatory response and cell death (36).
In the non-heme-dependent iron absorption pathway,
extracellular iron (Fe3+) binds to TF/TFR1 on the cell
membrane, resulting in membrane infiltration and localization in
the specialized endosomes formed (4). Fe3+ in the endosomes is
then released from TF and reduced to Fe2+ by the ferric
reductase steam 3, which is specifically located in endosomes
(24,25,27).
Fe2+ can be released into the labile iron pool (LIP) in
the cytoplasm through the endosomal membrane by solute carrier
family 11 member 2 (SLC11A2) (44). When the iron load exceeds the
carrying capacity of TF in certain pathological states,
Fe3+ are present as non-TF-bound iron, Fe3+
is reduced to Fe2+ by ferrireductases on the cell
surface or released cellular reductants and Fe2+ is
moved into cells through transmembrane transporters, such as
SLC11A2(44). In the
heme-dependent iron absorption pathway, Fe2+ present in
hemoglobin or heme in plasma is internalized into endosomes after
binding to various transporters, such as Feline leukemia virus
subgroup C receptor heme transporter 2, solute carrier family 48
member 1 and solute carrier family 46 member 1(45). In cells, heme treated with
cytoplasmic heme oxygenase 1 (HO-1), releases Fe2+ into
the labile iron pool.
Solute carrier family 40 member 1 (SLC40A1) is the
only known iron export protein in mammalian cells (45). Iron oxidases ceruloplasmin (CP),
hephaestin (HEPH) and HEPH like 1 (bacteriophage like 1) can all
regulate iron balance through SLC40A1-dependent iron output
(45,46). In addition, there is evidence that
overexpression of SLC40A1 can improve ferroptosis through reduction
of the level of Fe2+ in cells and knockdown of SLC40A1
can promote ferroptosis through the accumulation of Fe2+
(47). The overexpression of
SLC40A1 could reduce the levels of Fe2+ in cells, which
could reduce the risk of ferroptosis; however, Fe2+ will
accumulate in cells when SLC40A1 is knocked-down, which contributes
to ferroptosis (45).
Ferritin is an iron storage protein that can store
70-80% of the internalized iron ions (4,28);
it is primarily localized in the cytoplasm but can also be found in
the mitochondria. Ferritin can be divided into two subtypes: H type
and L type. The H subtype, also known as ferritin heavy chain 1
(FTH1), oxidizes Fe2+ into Fe3+. By contrast,
the L subtype, also known as ferritin light chain (FTL),
contributes to iron nucleation and mineralization (35).
In the central nervous system, iron ions need to be
transferred from the circulating blood to the brain parenchyma
through TF/TFR at the endothelial layer of the BBB (28). Since iron ions can accept and
donate electrons, pathological iron accumulation leads to oxidative
damage or even cell death (45).
Excessive Fe2+ can react with hydrogen peroxide
(H2O2) and organic peroxide to produce
hydroxyl radicals (HO•), which can attack DNA, proteins
and lipid membranes to disrupt cellular function and cause neuronal
death. In addition, lipid alkoxy radicals can be produced, which
are the primary sources of ROS produced by iron metabolism through
the Fenton reaction (27,30,48-51).
The increase in ROS levels not only reduces GSH consumption but
also reacts with PUFAs at the lipid membrane to induce lipid
peroxidation (52). Neuronal
membranes are rich in cholesterol and PUFAs, which are readily
oxidized by ROS. This renders neuronal membranes highly vulnerable
to damage, because superoxide dismutase and GPX activity in the
brain is decreased and the neurons there cannot clear ROS
adequately following an ischemic stroke (53).
Alterations in iron metabolism, including iron
deficiency and iron overload, can adversely affect the central
nervous system (33). Iron
deficiency can reduce the activity of cytochrome oxidase in the
brain, which can lead to developmental disorders in the hippocampus
and prefrontal lobe, such as motor development and cognitive memory
impairment (4). Intracellular iron
accumulation is generally caused by iron export impairments, where
increases in the brain iron load can increase sensitivity to
ferroptosis and aggravate brain injury (49). The middle cerebral artery occlusion
(MCAO) animal model has revealed increased iron levels in the
affected brain hemisphere (10).
Treatment with lipostatin-1 or ferristatin-1 can protect against
neurotoxicity induced by glutamate, reduce the size of the
infarcted area and reduce the degree of brain edema and behavioral
disorders caused by cerebral ischemic injury (33,54).
Ferroptosis observed during the MCAO model caused by
iron metabolism disorders can be managed by iron inhibitors, such
as lipstatin-1 and iron statin-1, which are both used in research
settings. However, they are not yet approved for human use
(55).
Lipid metabolism
Fatty acids are not only a source of energy but are
also important precursors of all bio-membrane lipids (56). Acetyl coenzyme A in the cytoplasm
is typically catalyzed to malonyl coenzyme A by the key enzyme
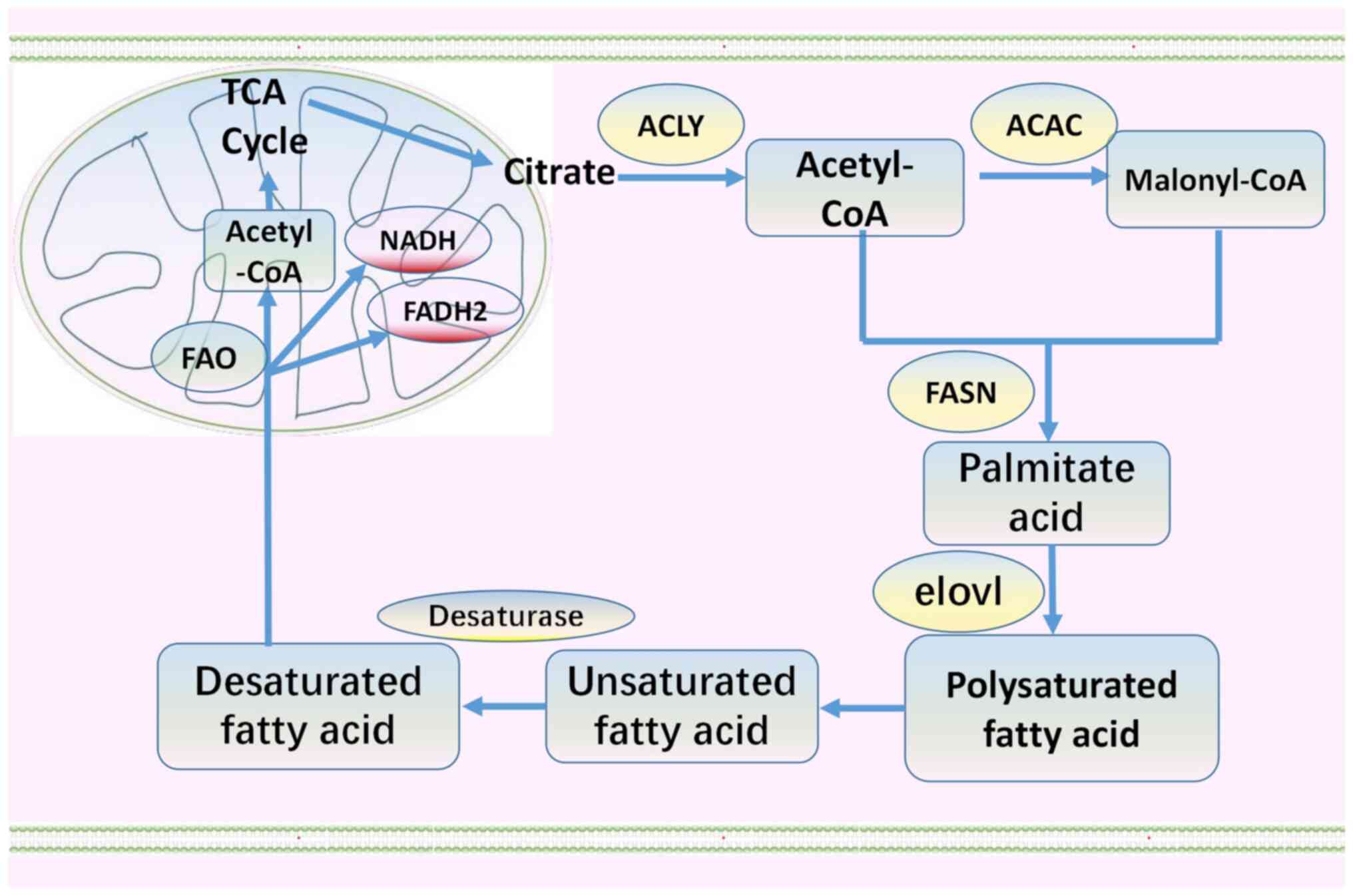
acetyl coenzyme A carboxylase (Fig.
1). Subsequently, through fatty acid synthase, malonyl CoA and
acetyl CoA are condensed to produce the 16-carbon fatty acid
palmitate (C16:0; Fig. 1)
(33,45). C16:0 is then extended by the elovl
fatty acid elongation enzyme and desaturated by fatty acid
desaturase (Fig. 1). Fatty acids
are catabolized by fatty acid oxidation in the mitochondria
(Fig. 1). This process produces
acetyl CoA, NADH and FADH2. Acetyl CoA then enters the Krebs cycle,
whilst NADH and FADH2 enter the electron transport chain to produce
ATP (45,53,57).
Fatty acids are stored in the form of lipid droplets, which can
buffer and store excess lipids. The formation of lipid droplets
prevents palmitic acid-induced lipotoxicity by separating damaged
membranes. Therefore, increasing lipid storage through the
formation of lipid droplets can limit ferroptosis. In addition,
monitoring the dynamic balance between lipid droplet formation and
degradation is important for evaluating the progress of ferroptosis
(57).
Under pathological conditions, ROS, including
superoxide anions, H2O2 and OH•,
are formed due to the incomplete reduction of oxygen (41,42).
These oxidants can attack the carbon-carbon double bonds of lipids,
especially PUFAs, to cause lipid peroxidation (45). Free PUFAs from neuronal membranes
are particularly sensitive to lipid peroxidation; they can be
esterified into membrane phospholipids and oxidized to transmit
ferroptotic signals (42).
Acyl-Coenzyme A (Acyl-CoA) synthase long-chain family member 4
(ACSL4) and lysophosphatidylcholinyl transferase 3 participate in
the biosynthesis of phosphatidylethanolamine, activate PUFA and
regulate the transmembrane properties of PUFA upstream of
iron/lipid signaling transduction (28,36,52,58).
Lipids can also be oxidized directly by oxygenase. Under these
conditions, lipid peroxidation occurs, which alters the electric
potential, fluidity and permeability of membranes, resulting in an
imbalance in osmotic potential and membrane breakage, and
ultimately ferroptosis (59).
Abnormal lipid metabolism is an important cause of
ferroptosis (4,53). Ferrostatin-1 has been shown to
reduce the infarct size in the brain of a mouse MCAO model of
stroke, by inhibiting the accumulation of ROS induced by glutamate
in neurons (33). Small molecule
lipoxygenase inhibitors can also capture free radicals and produce
antioxidants, which inhibit the production of ROS and lipid
peroxidation to block ferroptosis in neurons (33,52,60).
Ferroptosis observed during MCAO induced by ROS
accumulation as a result of lipid metabolism disorders can be
directly regulated by the small molecule inhibitor of ACSL4, which
prevents the accumulation of lipid ROS (45,61).
This includes natural products tricystin C and synthetic
thiazolidinediones, such as rosiglitazone, which can reduce the
load of PUFA (61).
Participants in the ferroptosis
signaling pathway
Substrate-specific subunit SLC7A11 and auxiliary
regulatory subunit SLC3A2 form the main pump of the cysteine
glutamate antiporter (system xc-), which imports cystine into cells
whilst pumping out glutamate (4,36,38,43,49,62).
This cycle is driven by high concentrations of intracellular
glutamate but does not require ATP (27). Erastin, sulfasalazine, sorafenib,
RAS-selective lethal 3 and related molecular proteins such as p53
can all target system xc- to inhibit ferroptosis (23,33,63).
In addition, it has been reported that the deficiency of SLC7A11
can lead to liver injury as a result of ferroptosis caused by an
overload of iron (62).
Cystine is reduced to cysteine by GSH and
thioredoxin reductase 1. Cysteine combines with glutamic forms GSH,
which can in turn bind to cytotoxic lipid peroxides (64). Glutamic acid cysteine ligase (GCL)
is comprised of a catalytic subunit and a modified subunit; it is
the rate-limiting enzyme for the de novo synthesis of GSH by
combining glutamic acid, cysteine and glycine in cells (49). However, cysteine content is low
across the whole cell and is therefore considered to be a limiting
factor in GSH synthesis. Therefore, the availability of cystine is
important for maintaining the levels of GSH for preventing
ferroptosis (30,49,65).
RAS-selective lethal 3 can also inhibit the
GSH-dependent enzyme GPX4. GPX4 is an isozyme member of the GPX
family (4). Compared with other
GPX members, GPX4 is the only known key cellular enzyme involved in
the regulation of ferroptosis, which can directly reduce lipid
peroxide to non-toxic alcohols (L-OH) in the membrane to prevent
ferroptosis during lipid oxidation (22,23,25,33,36,39,49,59,65,66).
GPX4-knockout mice have been shown to exhibit ferroptosis in the
brain and early embryonic lethality. The absence of GPX4 in the
mouse forebrain neurons can promote ferroptosis and lead to
cognitive impairments and neurodegeneration (63). GPX4 selenocysteine, the active site
of GPX4, can form a catalytic quadruplex with tryptophan, glutamine
and asparagine to catalyze the reduction of lipid hydroperoxide to
inhibit ferroptosis (42). ROS
inhibitors, such as ferrostatin-1 and liproxstatin-1, in addition
to GPX4 promoters, such as dopamine and selenium, have all been
reported to be effective in preventing ferroptosis in various
animal models (49,67).
PUFA is converted to acyl-coA by the fatty
acid-activating enzyme ACSL4. Acyl-CoA is involved in the lipid
peroxidation of membrane phospholipids (24,65).
After a series of complex dynamic processes, PUFA lipid peroxide
(L-OOH) is converted to oxidized glutathione and non-toxic alcohol
(L-OH) (23,25,27,30).
In cells, L-OOH can be oxidized by Fe2+ to produce
highly active alkoxy radicals (L-O•) (41). Under physiological conditions,
L-OOH and L-OH levels are held in equilibrium due to the activity
of GPX4. When GPX4 is absent or becomes inactivated, L-OOH
accumulates above physiological levels, resulting in the increased
production of L-O• and cell membrane damage (50).
Coenzyme Q (CoQ)
CoQ with 10 isoprene units at the tail of its side
chain is called CoQ10. The main function of CoQ10 is to transfer
electrons in the mitochondrial electron transport chain. In
addition, the reduced form of CoQ10, namely panthenol, is an
effective lipophilic antioxidant. CoQ10 is an endogenous inhibitor
of ferroptosis, which can neutralize free radicals produced during
the I/R damage process (33).
Iron and ferrous chelators, such as deferoxamine
(DFO), VK-28, deferiprone, minocycline, nitrilotriacetic acid,
ethylenediaminetetraacetic acid and clioquinol, are effective
inhibitors of ferroptosis, as previously shown in several different
animal models (30,38); they deplete Fe2+ in the
LIP to prevent iron-dependent lipid peroxidation to inhibit
ferroptosis (49). Ferroxidase is
another type of iron metabolism inhibitor, such as CP, which
oxidizes the toxic Fe2+ to the less toxic
Fe3+ to inhibit ferroptosis (49). Inhibition of TF in vitro can
block ferroptosis, whereas TF supplementation can restore this
process (49). In cancer cells,
silencing of the expression of key genes associated with iron
metabolism such as Transferrin Receptor (TFRC) has been shown to
reduce iron uptake and susceptibility to ferritin disease (45). However, the function of the lipid
oxygenase family is iron-dependent (23,61,68).
Overall, iron is an indispensable factor in
ferroptosis. Any molecules or factors that can modulate the
unstable LIP in cells can either reduce or promote ferroptosis
(33,44). TF, TFR1, ferroportin (SLC11A3) and
HO-1 have all been found to weaken ferroptosis (69).
However, other factors, such as the transcription
factors nuclear factor erythroid 2-related factor 2 and p53, have
also been reported to regulate ferroptosis by transcriptionally
inducing the expression of GPX4, SLC7A11 and HO-1(3).
4. p53
In total, p53 has six major protein domains and is
negatively regulated by two homologous proteins, MDM2 and MDM4
(5,43,70,71).
A total of two intrinsically disordered N-terminal
domains, tRNA-specific adenosine deaminase (TAD)1 and TAD2, form
the binding sites of its negative regulator MDM2 (43,72,73).
Abrogation of TAD1 can inhibit the p53 response, whilst abrogation
of TAD2 does not notably alter p53 function, since p53 can continue
to induce the expression of target genes in addition to retaining
the ability to induce cell cycle arrest and apoptosis (74,75).
By contrast, deletion of both TAD1 and TAD2 results in the complete
abrogation of p53 function (74).
The proline-rich domain, also known as the polyproline region of
the PXXP repeat sequence (P stands for proline, whilst X can be any
amino acid), is of great significance for the stability of p53 and
for p53-mediated apoptosis (74).
Deletion of this region results in the nuclear export of p53, which
then becomes prone to ubiquitination and degradation mediated by
MDM2(76). Residues 102-292 in the
central core region of p53 contain the DNA binding domain (DBD) of
p53, which allows p53 to function as a transcription factor in a
sequence-specific manner, by recognizing the p53 response element
(77). The tetramerization domain
(TD) allows four p53 proteins to form a tetramer, facilitating the
acquisition of the appropriate protein conformation when bound with
DNA for sequence recognition (78). p53 as a tetramer can typically bind
to the target gene element or interact with other proteins. TD has
also been shown to be necessary for the post-translational
modifications of p53, such as phosphorylation and ubiquitination
(78). Only after
post-translational modification, namely acetylation and
phosphorylation, can the intrinsically disordered C-terminal
regulatory domain (CTD) change from an inactive conformation to an
active conformation, after which it binds with the DBD to exert its
function (79,80). This process requires the p53
cofactor P300 to participate in its acetylation or phosphorylation
(79,80). The CTD also contains nuclear export
and localization signals, which regulates intracellular location
(79,80).
Each domain of p53 has unique properties and
contributes to the overall function of p53 (79,80).
These domains not only interact with other proteins and regulate
signal transduction, but can also be affected by extensive
post-translational modifications to regulate protein stability,
turnover and cell localization (3).
p53 is mainly regulated by two negative regulators,
MDM2 and MDM4, which are homologous proteins. p53, MDM4 and MDM2
constitute a highly dynamic regulatory core, consisting of complex
positive and negative feedback loops, ensuring the precise
regulation of p53 under physiological conditions and rapid
responses to stress (1,70,81).
MDM2 is an oncogene that encodes an E3 ubiquitin ligase. p53
oligomerizes to form a tetramer, which then regulates cell
proliferation, apoptosis, cell cycle arrest, DNA repair and
metabolism upstream of oxidative stress, aging, autophagy and
ferroptosis (82). MDM4 is another
important negative regulator of p53. Although MDM4 does not have E3
ligase activity and does not directly control p53, it does form a
heterodimer with MDM2, stabilizing MDM2 E3 ligase activity and
promoting MDM2-mediated ubiquitination and p53 degradation
(73). In addition, p53
transcription can also induce MDM2 and MDM4, forming a negative
feedback pathway to strictly regulate p53 activity. Exogenous
MDM2-MDM4 dimers are maintained at a low level under physiological
conditions, reducing promotion of p53, which reduces the amount of
p53. However, with the increase in the half-life of the p53 protein
in cells, it can regulate the subsequent cell response by binding
with p53 response elements of its target genes (80). When the expression of MDM2, a
negative regulator of p53, was specifically ablated in the central
neuron system, the mice developed hydrated brain malformations
between embryonic days 12 and 5 due to apoptosis. By contrast, the
deletion of MDM4 expression, another negative regulator of p53,
resulted in multiple brain malformations between embryonic days 17
and 5 due to cell cycle arrest and apoptosis (73). Both phenotypes can be completely
rescued by p53 deletion. In general, p53 is involved in apoptosis,
DNA repair, cell cycle arrest, DNA replication stress response,
autophagy, ferritin-based diseases (such as neuroferritinopathy),
the pathological process of cancer inhibition, anti-infection,
immune response, tissue I/R injury, neurodegenerative diseases
(81), maternal reproduction,
development and aging (43,83,84).
5. p53-mediated regulation of iron and lipid
metabolism involved in ferroptosis
p53-mediated iron metabolism
p53 can directly activate the expression of iron
sensors, such as iron regulatory hormone, to regulate the
intracellular iron pool. p53 has been previously shown to
upregulate hepcidin antimicrobial peptide (HAMP), which encodes
ferritin (3), through a putative
p53-responsive element, to reduce iron export (3). In addition, p53 can directly
transcribe the mitochondrial iron-binding protein XN (frataxin) to
regulate mitochondrial iron homeostasis (3). Furthermore, p53 can mediate the
expression of iron oxidoreductase (FDXR) to form a FDXR/p53 loop to
upregulate FDXR, which prevents mitochondrial iron overload, whilst
FDXR deficiency inhibits p53 mRNA translation (43). p53 can also upregulate the
translation of fth1 mRNA and abrogates the stability of TF
mRNA, which results in increased cellular iron storage and reduced
cellular iron import. Ferritin is a protein, composed of both light
(FTL) and heavy chain (FTH1) subunits, which can store iron ions.
When FTH is upregulated, the balance between FTH1 and FTL is broken
and ferritin cannot maintain its balance. Cellular iron storage is
increasing and the iron ions are balanced inside and outside the
cells. Cells will reduce the import of iron ions when there are
enough iron ions inside the cell. Functionally, the p53 protein can
transactivate ferritin directly in response to ferroptosis and
inflammation (43). Ferritin can
reduce serum iron by chelation in reticuloendothelial macrophages
(43). In addition, the heme-p53
interaction can regulate iron metabolism and ferroptosis. p53 was
also found to be associated with hypoxia-inducible factor 1α to
increase p53 protein stability and expression levels during iron
deficiency (73). Iron overload
can reduce the levels and function of the p53 protein. Iron
porphyrin heme directly binds to p53 to interfere with p53-DNA
interactions, which promotes the nuclear export and degradation of
p53(73). The MDM2 antagonist,
nutlin-3, has been previously shown to delay the occurrence of
ferroptosis in liver and liver cancer cells in a p53-dependent
manner, to promote cell survival under metabolic stress (44).
p53 regulates lipid metabolism
p53 is also involved in the transcription of genes
associated with lipid metabolism through various mechanisms
(6); it can bind directly to the
promoter region of the transcription factor for sterol regulatory
element-binding transcription factor 1 to inhibit its expression,
which in turn regulates the expression of a group of genes involved
in lipid metabolism (5). p53 also
regulates the transcription of two enzymes involved in fatty acid
oxidation, namely carnitine palmitoyl transferase 1C and
phosphatidylate phosphatase, which regulates the transport of
activated fatty acids to the mitochondria to enhance the oxidation
of fatty acids in cells (5,85).
p53 also promotes the transcription of malonyl-CoA decarboxylase,
which catalyzes intracellular fatty acid oxygenation to prevent
lipid accumulation in cells (5).
In terms of protein-protein binding, glucose-6-phosphate
dehydrogenase can bind to p53 and is directly inhibited by p53,
leading to a decrease in NADPH production. p53 also serves a role
in lipid transport (5).
Apolipoprotein B (APOB) and APOB editing enzyme complex 1 have been
found to serve a role in atherosclerotic lipoproteins by the
regulation of p53 transcription (85).
6. p53-mediated ferroptosis
It has been found that, in H1299 cells with the p53
gene silenced and then treated with ROS, the cell activity remained
unchanged. However, when treated with ROS after p53 activation, 90%
of the cells died. This suggests that p53 activation reduced the
antioxidant capacity of these cells. In addition, after treatment
with the ferroptosis inhibitor fer-1, the cell death rate decreased
significantly. This also suggests that p53 serves an important role
in ferroptosis (3).
p53 has been previously found to promote
ferroptosis (36,43). p53 can downregulate the expression
of SLC7A11 (Fig. 2) (85). System xc- operates by transforming
cystine to intracellular cysteine. After the reaction between
cysteine and glutamate, glutathione is produced as the substrate of
GPX4 activity, which is required to inhibit ferroptosis (3). p53 can inhibit SLC7A11 to reduce
cystine uptake and intracellular glutathione production, which in
turn leads to an increase in intracellular ROS (5,36).
Ferroptosis is regulated by glutamine metabolism. During glutamine
catabolism, glutamine is first converted to glutamic acid by
glutaminase (GLS) 1 and GLS2, which is then converted into
α-ketoglutarate, an important substrate for the Kreb's cycle
(29). GLS2 is a hepatic
glutaminase present in the mitochondria. p53 induces GLS2
transcription, which in turn mediates oxygen consumption and ATP
production in cells (Fig. 2)
(5,85). GLS2 can also promote antioxidant
function by increasing the production of GSH and NADH in cells
(3,43). In male C57BL/6J mice, the knockdown
of GLS2 expression has been found to inhibit serum-dependent
ferroptosis induced by amino acid deprivation (3,85).
Spermidine/spermine N1 acetyltransferase 1 (SAT1) has also been
reported to be a direct p53 target, which catalyzes the acetylation
of spermidine and spermine, serving as a key enzyme for polyamine
catabolism (5,36,85).
SAT1 can be activated by nutlin-3, a small molecule MDM2 inhibitor,
in a p53-dependent manner, to promote the formation of
ROS-dependent lipid peroxides and render cells sensitive to
ferroptosis (43). In addition,
ROS-induced cell death can only be inhibited by ferriprotease-1 in
SAT1-overexpressing cells (86).
However, SAT1 did not affect the expression or activity of SLC7A11
and GPX4. Following SAT1 induction, the expression levels of
arachidonic acid (ALOX)15 lipoxygenase, a member of the
lipoxygenase family, increased (43). Its oxygenates PUFAs which is one of
the necessary conditions for ferroptosis (43). Ferroptosis induced by SAT1 can be
effectively blocked by the specific inhibitor of ALOX15, pd146176,
suggesting that ALOX15 is a mediator of p53 in the pathway between
SAT1 induction and ferroptosis (86). ALOX12 is another important positive
regulator of p53 in the lipoxygenase family to mediate ferroptosis.
ALOX12 inactivation can inhibit ROS stress-induced p53-mediated
ferroptosis, independent of GPX4 and ACSL4 activation (87). In conclusion, these observations
suggest that p53 serves an important role in regulating ferroptosis
by regulating the expression of its targets.
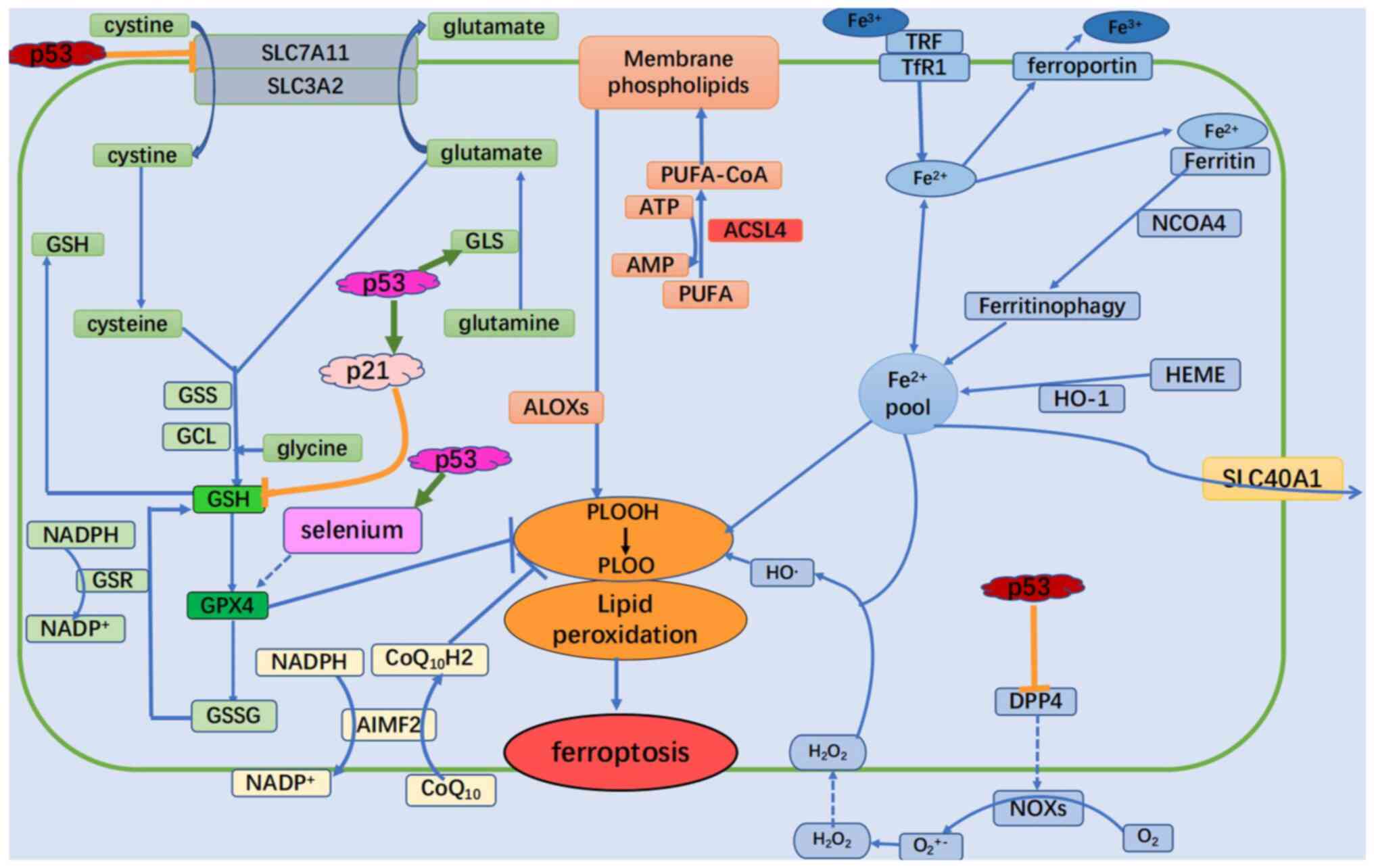 | Figure 2Summary points of previous studies on
ferroptosis signaling pathways that are potentially regulated by
p53. The dual role of p53 in the control of ferroptosis is shown.
p53 can enhance ferroptosis through the inhibition of SLC7A11
expression or the promotion of SAT1 and GLS2 expression. However,
p53 can also suppress ferroptosis through the inhibition of DPP4
activity or the induction of CDKN1A/p21 expression. GSS,
glutathione synthetase; GCL, glutamate-cysteine ligase; SLC,
soluble carrier family; GPX4, glutathione peroxidase 4; GSH,
glutathione; GSSG, oxidized glutathione; GSR, glutathione-disulfide
reductase; GLS, glutaminase; ALOXs, lipoxygenases; CoA, coenzyme A;
CoQ10, coenzyme Q10; CoQ10H2, ubiquinol; H2O2, hydrogen peroxide;
HO-1, heme oxygenase 1; NCOA4, nuclear receptor co-activator 4;
PUFA, polyunsaturated fatty acid; PUFA-CoA, PUFA-coenzyme; DPP4,
dipeptidyl peptidase 4; NOXs, NADPH oxidases; ACSL4, acyl-CoA
synthetase long chain family member 4; PLOOH, phospholipid
hydroperoxides; TRF, transferrin; TfR1, transferrin receptor. |
p53 can not only promote but also inhibit
ferroptosis. p21 is a major p53 target that inhibits glutathione
degradation, where its ability to induce cell cycle arrest and
aging allows it to respond to stress signals (Fig. 2) (3). p53 transactivates p21 to inhibit
glutathione degradation and promote GPX4 activity, which results in
the reduction of ROS accumulation from toxic lipids to inhibit
ferroptosis (88).
7. p53-mediated ferroptosis in the MCAO
model
p53 activation is induced by various types of
cellular stress, including DNA damage, oncogene activation,
ribosomal stress or hypoxia. Ischemia and hypoxia caused by the
occurrence of brain MCAO provide the conditions for the activation
of p53. The brain contains the highest PUFA content, providing
abundant precursor material for lipid peroxidation. In addition,
the neurons in the brain are prone to downstream molecular events,
such as Ca2+ influx, which accumulates iron ions and
increases lipid ROS, increasing the risk of ferroptosis (89). After severe ischemic and hypoxic
brain injury, the accumulation of Fe2+ in the basal
ganglia, thalamus, periventricular and subcortical white matter
areas, increases lipid oxide levels and reduces GPX4 function,
supporting their involvement in ferroptosis (4,24,25,41,89).
The expression of ROS, GPX4, GSH, GSSH, SLC7A11, TFRC (31), FTH1 and FTL can be used as
indicators of ferroptosis (31). A
previous study reported that the use of ferroptosis inhibitors
significantly improve the prognosis of ischemic stroke (4).
p53 transcription can promote ferroptosis after
MCAO in the cerebrum. SLC7A11 expression promotes ferroptosis,
whilst in p53-deficient cells, SLC7A11 upregulation promotes
sensitivity to ferroptosis (70,90,91).
It has been previously shown that p53 can promote the nuclear
translocation of ubiquitin specific peptidase 7, a nuclear
deubiquitinase, to remove ubiquitin from histone 2B ubiquitination
(H2BUB) at lysine 120. This removes H2BUB from the SLC7A11
promoter, inhibiting SLC7A11 transcription (92). Ferroptotic cells in the MCAO model
may undergo irreversible cell death through this pathway (4). p53 regulation of SLC7A11 expression
can also regulate ALOX12-dependent lipid peroxidation to regulate
ferroptosis (87). Glutamine is
transformed into glutamic acid by GLS1 and GLS2 after entering the
cell (3). Glutamic acid serves two
important functions, acting as a precursor for GSH synthesis and as
an intermediate for its conversion to α-ketoglutarate (3). It is widely known that GSH synthesis
is a key process in ferroptosis. GSL2 expression could promote GSH
production to increase cellular antioxidant function (3). p53 can induce GLS2 transcription to
regulate glutamate levels in cells and then induce ferroptosis
(70). Glutamate can also be
exported from cells through SLC7A11 in exchange for extracellular
cystine (62). Therefore, SLC7A11
can mediate glutamate export, leading to reduction in intracellular
glutamate. Subsequently, SLC7A11 absorbs additional glutamine
through negative feedback and activates the glutamate enzymes to
increase glutamate production, leading to
SLC7A11/glutamine-dependent ferroptosis. Therefore, p53
transcription-mediated SLC7A11 activity can interact with
p53-mediated glutamine-dependent ferroptosis to jointly regulate
cell death (3). In addition, p53
transcription promotes the expression of spermidine/SAT1 to
regulate ALOX15-dependent lipid peroxidation and increase the
sensitivity of cells to ferroptosis (70).
p53 directly participates in the regulation of
HAMP, frataxin, FDXR, ferritin, heme and ferroptosis by regulating
iron metabolism (93). p53 also
regulates the expression of lipid metabolism genes and transporters
upstream of ferroptosis. Following cerebral ischemia and hypoxia,
the regulation of various genes regulating iron and lipid
metabolism is altered (53,56).
In cerebral ischemic reperfusion process, both iron and lipid
metabolism will change under the influence of regulators such as
p53. However, the mechanism underlying the regulation of iron and
lipid metabolism by p53 remains unclear and requires further
verification.
p53 can also inhibit ferroptosis (42). p53 can bind to dipeptidyl peptidase
4 (DDP4) in the nucleus, inhibiting the binding of DPP4 to NADPH
oxidase 1 to mediate the production of ROS in the cell membrane
(43). p53 also promotes the
expression of p21, which induces the production of GSH to inhibit
lipid peroxidation (3). The tumor
suppressor p21 is a primary p53 target that inhibits glutathione
degradation to inhibit ferroptosis (70,94).
Although p53 can inhibit ferroptosis, its value in the MCAO model
remains unclear.
8. Conclusions and future perspectives
The present review aimed to discuss the role of
p53-mediated ferroptosis in ischemic stroke. Ferroptosis is a
unique regulatory form of cell death that was first discovered in
2012 and involves a complex network of gene sets and signaling
pathways that is distinct from apoptosis, necrosis, autophagy and
oxygenation (42). p53 was first
found in cancer cells. However, p53 can act on numerous proteins
involved in ferroptotic signaling pathways and has been shown to
serve an important role in the progression of several nervous
system diseases. The iron overload that occurs during MCAO is
caused by several mechanisms, such as system xc-, GSH depletion,
GPX4 inactivity, lipoxygenase inactivation and/or intracellular
iron accumulation. These mechanisms can be either promoted or
inhibited by known inducers of ferroptosis (glutamate, erastine and
rsl3) and inhibitors (fer-1, lipstatin-1, DFO and vitamin E). Based
on the above, we hypothesized that p53-mediated ferroptosis is
potentially an important form of cell death in the MCAO model.
Further studies on ferroptosis will provide novel opportunities for
the diagnosis and treatment of ischemic stroke.
Although p53 has been found to act directly on
proteins involved in ferroptosis signaling, it is not clear if p53
interacts with each target protein. The majority of associated
previous studies have focused on the effect of ferroptosis on brain
function. However, to the best of our knowledge, relatively few
studies have been performed regarding brain immune infiltration,
immune cell activation and circulatory function following the
establishment of the MCAO model. Therefore, future research should
not only reveal the therapeutic effect of inhibiting ferroptosis in
brain cells, but should also pay attention to the comprehensive
treatment of all aspects of ischemic stroke.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 81870943).
Availability of data and materials
Not applicable.
Authors' contributions
SX and XL conceived the topic of review. YW was
responsible for reviewing and editing the manuscript. All authors
have read and approved the final manuscript. Data authentication is
not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang T, Wang H, Li Q, Fu J, Huang J and
Zhao Y: MALAT1 Activates the P53 signaling pathway by regulating
MDM2 to promote ischemic stroke. Cell Physiol Biochem.
50:2216–2228. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sun MS, Jin H, Sun X, Huang S, Zhang FL,
Guo ZN and Yang Y: Free radical damage in ischemia-reperfusion
injury: An obstacle in acute ischemic stroke after
revascularization therapy. Oxid Med Cell Longev.
2018(3804979)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kang R, Kroemer G and Tang D: The Tumor
Suppressor protein p53 and the ferroptosis network. Free Radic Biol
Med. 133:162–168. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bu ZQ, Yu HY, Wang J, He X, Cui YR, Feng
JC and Feng J: Emerging role of ferroptosis in the pathogenesis of
ischemic stroke: A new therapeutic target? ASN Neuro.
13(17590914211037505)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Liu J, Zhang C, Hu W and Feng Z: Tumor
suppressor p53 and metabolism. J Mol Cell Biol. 11:284–292.
2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Laubach K, Zhang J and Chen X: The p53
Family: A role in lipid and iron metabolism. Front Cell Dev Biol.
9(715974)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Filichia E, Shen H, Zhou X, Qi X, Jin K,
Greig N, Hoffer B and Luo Y: Forebrain neuronal specific ablation
of p53 gene provides protection in a cortical ischemic stroke
model. Neuroscience. 295:1–10. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hernández Borrero LJ and El-Deiry WS:
Tumor suppressor p53: Biology, signaling pathways, and therapeutic
targeting. Biochim Biophys Acta Rev Cancer.
1876(188556)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhao J, Dong Y, Chen X, Xiao X, Tan B,
Chen G, Hu J, Qi D, Li X and Xie R: p53 inhibition protects against
neuronal ischemia/reperfusion injury by the p53/PRAS40/mTOR
pathway. Oxid Med Cell Longev. 2021(4729465)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Paul S and Candelario-Jalil E: Emerging
neuroprotective strategies for the treatment of ischemic stroke: An
overview of clinical and preclinical studies. Exp Neurol.
335(113518)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kanazawa M, Takahashi T, Ishikawa M,
Onodera O, Shimohata T and del Zoppo GJ: Angiogenesis in the
ischemic core: A potential treatment target? J Cereb Blood Flow
Metab. 39:753–769. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
del Zoppo GJ, Sharp FR, Heiss WD and
Albers GW: Heterogeneity in the penumbra. J Cereb Blood Flow Metab.
31:1836–1851. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sommer CJ: Ischemic stroke: Experimental
models and reality. Acta Neuropathol. 133:245–261. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Abrams D, MacLaren G, Lorusso R, Price S,
Yannopoulos D, Vercaemst L, Bělohlávek J, Taccone FS, Aissaoui N,
Shekar K, et al: Extracorporeal cardiopulmonary resuscitation in
adults: evidence and implications. Intensive Care Med. 48:1–15.
2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Barthels D and Das H: Current advances in
ischemic stroke research and therapies. Biochim Biophys Acta Mol
Basis Dis. 1866(165260)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P,
Gao G, Shi H, Chang S and Chang YZ: Mitochondrial ferritin
attenuates cerebral ischaemia/reperfusion injury by inhibiting
ferroptosis. Cell Death Dis. 12(447)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Przykaza L: Understanding the connection
between common stroke comorbidities, their associated inflammation,
and the course of the cerebral ischemia/reperfusion cascade. Front
Immunol. 12(782569)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Boehme AK, Esenwa C and Elkind MS: Stroke
risk factors, genetics, and prevention. Circ Res. 120:472–495.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Guo H, Zhu L, Tang P, Chen D, Li Y, Li J
and Bao C: Carthamin yellow improves cerebral ischemia-reperfusion
injury by attenuating inflammation and ferroptosis in rats. Int J
Mol Med. 47(52)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF,
Gao GY and Jiang JY: Inhibition of ferroptosis attenuates tissue
damage and improves long-term outcomes after traumatic brain injury
in mice. CNS Neurosci Ther. 25:465–475. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hirschhorn T and Stockwell BR: The
development of the concept of ferroptosis. Free Radic Biol Med.
133:130–143. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Magtanong L and Dixon SJ: Ferroptosis and
brain injury. Dev Neurosci. 40:382–395. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
She X, Lan B, Tian H and Tang B: Cross
talk between ferroptosis and cerebral ischemia. Front Neurosci.
14(776)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mao H, Zhao Y, Li H and Lei L: Ferroptosis
as an emerging target in inflammatory diseases. Prog Biophys Mol
Biol. 155:20–28. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hu X, Xu Y, Xu H, Jin C, Zhang H, Su H, Li
Y, Zhou K and Ni W: Progress in understanding ferroptosis and its
targeting for therapeutic benefits in traumatic brain and spinal
cord injuries. Front Cell Dev Biol. 9(705786)2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yan N and Zhang JJ: The emerging roles of
ferroptosis in vascular cognitive impairment. Front Neurosci.
13(811)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363 e3. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Han C, Liu Y, Dai R, Ismail N, Su W and Li
B: Ferroptosis and its potential role in human diseases. Front
Pharmacol. 11(239)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Feng H, Schorpp K, Jin J, Yozwiak CE,
Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka
JM, et al: Transferrin receptor is a specific ferroptosis marker.
Cell Rep. 30:3411–3423 e7. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hassannia B, Wiernicki B, Ingold I, Qu F,
Van Herck S, Tyurina YY, Bayır H, Abhari BA, Angeli JPF, Choi SM,
et al: Nano-targeted induction of dual ferroptotic mechanisms
eradicates high-risk neuroblastoma. J Clin Invest. 128:3341–3355.
2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, Redox Biology, and Disease. Cell. 171:273–285.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M,
Buchfelder M and Savaskan N: Nrf2-Keap1 pathway promotes cell
proliferation and diminishes ferroptosis. Oncogenesis.
6(e371)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lin W, Zhang T, Zheng J, Zhou Y, Lin Z and
Fu X: Ferroptosis is Involved in Hypoxic-ischemic brain damage in
neonatal rats. Neuroscience. 487:131–142. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11(88)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Guo P, Jin Z, Wu H, Li X, Ke J, Zhang Z
and Zhao Q: Effects of irisin on the dysfunction of blood-brain
barrier in rats after focal cerebral ischemia/reperfusion. Brain
Behav. 9(e01425)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Proneth B and Conrad M: Ferroptosis and
necroinflammation, a yet poorly explored link. Cell Death Differ.
26:14–24. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hambright WS, Fonseca RS, Chen L, Na R and
Ran Q: Ablation of ferroptosis regulator glutathione peroxidase 4
in forebrain neurons promotes cognitive impairment and
neurodegeneration. Redox Biol. 12:8–17. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li Q, Weiland A, Chen X, Lan X, Han X,
Durham F, Liu X, Wan J, Ziai WC, Hanley DF and Wang J:
Ultrastructural characteristics of neuronal death and white matter
injury in mouse brain tissues after intracerebral hemorrhage:
Coexistence of ferroptosis, autophagy, and necrosis. Front Neurol.
9(581)2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Bayir H, Anthonymuthu TS, Tyurina YY,
Patel SJ, Amoscato AA, Lamade AM, Yang Q, Vladimirov GK, Philpott
CC and Kagan VE: Achieving life through death: Redox biology of
lipid peroxidation in ferroptosis. Cell Chem Biol. 27:387–408.
2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Liu J, Zhang C, Wang J, Hu W and Feng Z:
The regulation of ferroptosis by tumor suppressor p53 and its
pathway. Int J Mol Sci. 21(8387)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Vogt AS, Arsiwala T, Mohsen M, Vogel M,
Manolova V and Bachmann MF: On iron metabolism and its regulation.
Int J Mol Sci. 22(4591)2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Nemeth E and Ganz T: Hepcidin-Ferroportin
interaction controls systemic iron homeostasis. Int J Mol Sci.
22(6493)2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Li J, Liu J, Xu Y, Wu R, Chen X, Song X,
Zeh H, Kang R, Klionsky DJ, Wang X and Tang D: Tumor heterogeneity
in autophagy-dependent ferroptosis. Autophagy. 17:3361–3374.
2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang Y, Khan S, Liu Y, Zhang R, Li H, Wu
G, Tang Z, Xue M and Yong WV: Modes of brain cell death following
intracerebral hemorrhage. Front Cell Neurosci.
16(799753)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zhou SY, Cui GZ, Yan XL, Wang X, Qu Y, Guo
ZN and Jin H: Mechanism of ferroptosis and its relationships with
other types of programmed cell death: insights for potential
interventions after intracerebral hemorrhage. Front Neurosci.
14(589042)2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Cepelak I, Dodig S and Dodig DC:
Ferroptosis: Regulated cell death. Arh Hig Rada Toksikol.
71:99–109. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Kerins MJ and Ooi A: The Roles of NRF2 in
modulating cellular iron homeostasis. Antioxid Redox Signal.
29:1756–1773. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lee JY, Kim WK, Bae KH, Lee SC and Lee EW:
Lipid metabolism and ferroptosis. Biology (Basel).
10(184)2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Wan J, Ren H and Wang J: Iron toxicity,
lipid peroxidation and ferroptosis after intracerebral haemorrhage.
Stroke Vasc Neurol. 4:92–95. 2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yan HF, Tuo QZ, Yin QZ and Lei P: The
pathological role of ferroptosis in ischemia/reperfusion-related
injury. Zool Res. 41:220–230. 2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Magtanong L, Ko PJ and Dixon SJ: Emerging
roles for lipids in non-apoptotic cell death. Cell Death Differ.
23:1099–1109. 2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Bian X, Liu R, Meng Y, Xing D, Xu D and Lu
Z: Lipid metabolism and cancer. J Exp Med.
218(e20201606)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Cao Y, Li Y, He C, Yan F, Li JR, Xu HZ,
Zhuang JF, Zhou H, Peng YC, Fu XJ, et al: Selective ferroptosis
inhibitor liproxstatin-1 attenuates neurological deficits and
neuroinflammation after subarachnoid hemorrhage. Neurosci Bull.
37:535–549. 2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Zou Y and Schreiber SL: Progress in
understanding ferroptosis and challenges in its targeting for
therapeutic benefit. Cell Chem Biol. 27:463–471. 2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Karuppagounder SS, Alin L, Chen Y, Brand
D, Bourassa MW, Dietrich K, Wilkinson CM, Nadeau CA, Kumar A, Perry
S, et al: N-acetylcysteine targets 5 lipoxygenase-derived, toxic
lipids and can synergize with prostaglandin E2 to inhibit
ferroptosis and improve outcomes following hemorrhagic stroke in
mice. Ann Neurol. 84:854–872. 2018.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Shah R, Shchepinov MS and Pratt DA:
Resolving the role of lipoxygenases in the initiation and execution
of ferroptosis. ACS Cent Sci. 4:387–396. 2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620.
2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zhang Z, Yao Z, Wang L, Ding H, Shao J,
Chen A, Zhang F and Zheng S: Activation of ferritinophagy is
required for the RNA-binding protein ELAVL1/HuR to regulate
ferroptosis in hepatic stellate cells. Autophagy. 14:2083–2103.
2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Wenzel SE, Tyurina YY, Zhao J, St Croix
CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA,
Amoscato AA, et al: PEBP1 wardens ferroptosis by enabling
lipoxygenase generation of lipid death signals. Cell. 171:628–641,
e26. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wang GX, Tu HC, Dong Y, Skanderup AJ, Wang
Y, Takeda S, Ganesan YT, Han S, Liu H, Hsieh JJ and Cheng EH:
DeltaNp63 inhibits oxidative stress-induced cell death, including
ferroptosis, and cooperates with the BCL-2 family to promote
clonogenic survival. Cell Rep. 21:2926–2939. 2017.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Anthonymuthu TS, Kenny EM, Lamade AM,
Kagan VE and Bayir H: Oxidized phospholipid signaling in traumatic
brain injury. Free Radic Biol Med. 124:493–503. 2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Li Q, Han X, Lan X, Gao Y, Wan J, Durham
F, Cheng T, Yang J, Wang Z, Jiang C, et al: Inhibition of neuronal
ferroptosis protects hemorrhagic brain. JCI Insight.
2(e90777)2017.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Venkatesh D, O'Brien NA, Zandkarimi F,
Tong DR, Stokes ME, Dunn DE, Kengmana ES, Aron AT, Klein AM, Csuka
JM, et al: MDM2 and MDMX promote ferroptosis by PPARα-mediated
lipid remodeling. Genes Dev. 34:526–543. 2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Liu Y, Tavana O and Gu W: p53
modifications: Exquisite decorations of the powerful guardian. J
Mol Cell Biol. 11:564–577. 2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Ho T, Tan BX and Lane D: How the other
half lives: What p53 does when it is not being a transcription
factor. Int J Mol Sci. 21(13)2019.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Karni-Schmidt O, Lokshin M and Prives C:
The Roles of MDM2 and MDMX in Cancer. Annu Rev Pathol. 11:617–644.
2016.PubMed/NCBI View Article : Google Scholar
|
|
74
|
He F, Borcherds W, Song T, Wei X, Das M,
Chen L, Daughdrill GW and Chen J: Interaction between p53 N
terminus and core domain regulates specific and nonspecific DNA
binding. Proc Natl Acad Sci USA. 116:8859–8868. 2019.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Levine AJ: The many faces of p53:
Something for everyone. J Mol Cell Biol. 11:524–530.
2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Nagpal I and Yuan ZM: The basally
expressed p53-mediated homeostatic function. Front Cell Dev Biol.
9(775312)2021.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Lou J, Hao Y, Lin K, Lyu Y, Chen M, Wang
H, Zou D, Jiang X, Wang R, Jin D, et al: Circular RNA CDR1as
disrupts the p53/MDM2 complex to inhibit Gliomagenesis. Mol Cancer.
19(138)2020.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Gencel-Augusto J and Lozano G: p53
tetramerization: At the center of the dominant-negative effect of
mutant p53. Genes Dev. 34:1128–1146. 2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hamard PJ, Lukin DJ and Manfredi JJ: p53
basic C terminus regulates p53 functions through DNA binding
modulation of subset of target genes. J Biol Chem. 287:22397–22407.
2012.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Laptenko O, Shiff I, Freed-Pastor W,
Zupnick A, Mattia M, Freulich E, Shamir I, Kadouri N, Kahan T,
Manfredi J, et al: The p53 C terminus controls site-specific DNA
binding and promotes structural changes within the central DNA
binding domain. Mol Cell. 57:1034–1046. 2015.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Xiong Y, Zhang Y, Xiong S and
Williams-Villalobo AE: A glance of p53 functions in brain
development, neural stem cells, and brain cancer. Biology (Basel).
9(285)2020.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Kastenhuber ER and Lowe SW: Putting p53 in
Context. Cell. 170:1062–1078. 2017.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping
F, Huang W, Wu F, Zhang H and Zhang X: Inhibitor of
apoptosis-stimulating protein of p53 inhibits ferroptosis and
alleviates intestinal ischemia/reperfusion-induced acute lung
injury. Cell Death Differ. 27:2635–2650. 2020.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhao Y, Wu L, Yue X, Zhang C, Wang J, Li
J, Sun X, Zhu Y, Feng Z and Hu W: A polymorphism in the tumor
suppressor p53 affects aging and longevity in mouse models. Elife.
7(e34701)2018.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Gnanapradeepan K, Basu S, Barnoud T,
Budina-Kolomets A, Kung CP and Murphy ME: The p53 tumor suppressor
in the control of metabolism and ferroptosis. Front Endocrinol
(Lausanne). 9(124)2018.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Ou Y, Wang SJ, Li D, Chu B and Gu W:
Activation of SAT1 engages polyamine metabolism with p53-mediated
ferroptotic responses. Proc Natl Acad Sci USA. 113:E6806–E6812.
2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Chu B, Kon N, Chen D, Li T, Liu T, Jiang
L, Song S, Tavana O and Gu W: ALOX12 is required for p53-mediated
tumour suppression through a distinct ferroptosis pathway. Nat Cell
Biol. 21:579–591. 2019.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Venkatesh D, Stockwell BR and Prives C:
p21 can be a barrier to ferroptosis independent of p53. Aging
(Albany NY). 12:17800–17814. 2020.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kenny EM, Fidan E, Yang Q, Anthonymuthu
TS, New LA, Meyer EA, Wang H, Kochanek PM, Dixon CE, Kagan VE and
Bayir H: Ferroptosis contributes to neuronal death and functional
outcome after traumatic brain injury. Crit Care Med. 47:410–418.
2019.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Liu DS, Duong CP, Haupt S, Montgomery KG,
House CM, Azar WJ, Pearson HB, Fisher OM, Read M, Guerra GR, et al:
Inhibiting the system xC-/glutathione axis selectively
targets cancers with mutant-p53 accumulation. Nat Commun.
8(14844)2017.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Jiang L, Kon N, Li T..Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62.
2015.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Snyder NA and Silva GM: Deubiquitinating
enzymes (DUBs): Regulation, homeostasis, and oxidative stress
response. J Biol Chem. 297(101077)2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Shen J, Sheng X, Chang Z, Wu Q, Wang S,
Xuan Z, Li D, Wu Y, Shang Y, Kong X, et al: Iron metabolism
regulates p53 signaling through direct heme-p53 interaction and
modulation of p53 localization, stability, and function. Cell Rep.
7:180–193. 2014.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Chaudhary R and Lal A: Long noncoding RNAs
in the p53 network. Wiley Interdiscip Rev RNA. 8:10.1002/wrna.1410.
2017.PubMed/NCBI View Article : Google Scholar
|