1. Overview of CAPN3
Discovery
In 1989, Sorimachi et al (1) discovered a molecular clone named
p94 that encodes a novel calcium-dependent protease comprising 821
amino acid residues with a relative molecular mass of 94 kDa. In
2013, p94 was renamed calpain 3 (CAPN3) (2), or calcium-activated neutral
protease, by the American Association of Experimental Biology. In
humans, CAPN3 is located in the chromosomal region 15q15.1-q21.1
(3). Mainly expressed
specifically in skeletal muscle (1), the mRNA of CAPN3 has also been
detected in the early embryonic heart, but it will gradually
disappear from the ventricular area, and finally, only the
transcript of CAPN3 is present (4,5).
In addition, CAPN3 is also expressed in the cytoplasm and nucleus
of neuron-like PC12 cells, as well as in rat astrocytes and in the
brain of Microcebus (6,7).
Structure
So far, the calpain family has 16 members, including
classic calpains and non-classical calpains (summarized in Fig. 1). As a classic calpain, CAPN3
exhibits a large subunit containing four domains (I-IV). Domain I
is distinctly different from the other domains. Domain II is a
conserved cysteine protease domain comprising protease core domain
1 (PC1) and PC2 that confer protease activity. Domain III, also
known as the calpain-type β-sandwich domain (CBSW or C2L), is
responsible for protein structural changes upon CAPN3 activation.
Domain IV, or the penta-EF-hand (E, E-helix; F, F-helix) (PEF)
domain, mainly participates in calcium ion binding and CAPN3
homodimerization. The sequences of domain II and IV in CAPN3 are
homologous with those in other calpains (1).
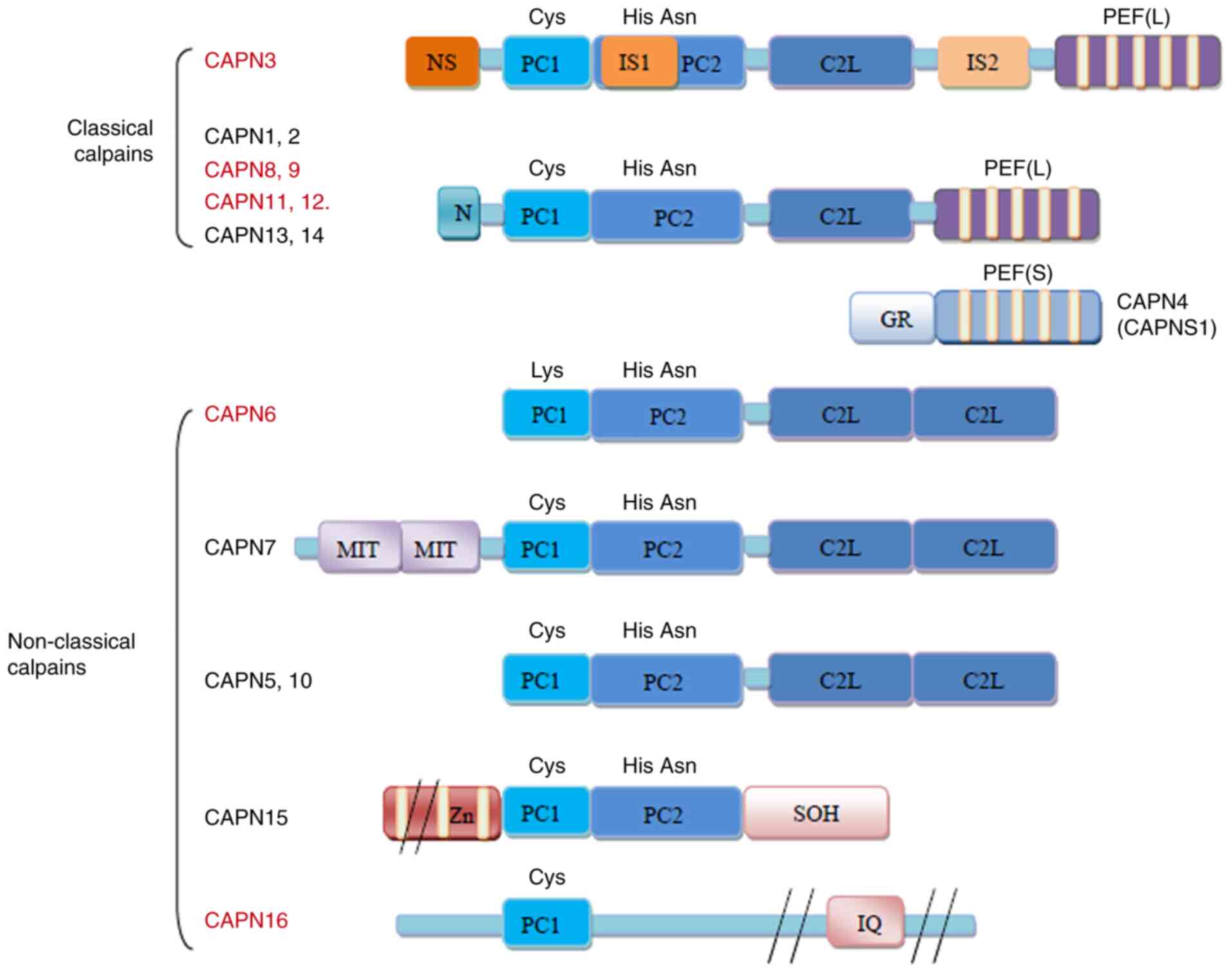 | Figure 1Structure of the human calpains. The
calpains presented in red are predominantly expressed in specific
tissues or organs, while those in black are more widely expressed.
The major difference in the structure of CAPN3 is that it contains
three additional insertion sequences, namely NS at the N-terminus,
IS1 of PC2 and IS2 between CBSW/C2L and PEF (L). Small subunits are
not present in CAPN3. CAPN3, calpain 3; GR, Gly-rich domain; MIT,
microtubule-interacting and transport motif; Zn, zinc-finger motif;
SOH, small optic lobes product homology domain; IQ,
calmodulin-interacting motif; NS, N-terminus; IS1, insertion
sequence 1; PC1, protease core domain 1; PEF, penta-EF-hand (E,
E-helix; F, F-helix); CBSW/C2L, calpain-type β-sandwich. |
In particular, CAPN3 contains three additional
insertion sequences, namely the N-terminus (NS), insertion sequence
1 (IS1) of PC2 and IS2 between CBSW/C2L and PEF (L) (8). IS1, which interrupts the protease
core, must be cleaved in order to be activated for substrate
binding (9). The PEF domain is
where four Ca2+ bind at positions 1, 2, 3 and 5 of
EF-hand to promote CAPN3 homodimerization. In calpain, EF5 is used
to form dimers independently of Ca2+ (10,11). In CAPN3, the addition of a CBSW
domain at the NS enhances its trimer-forming properties. Therefore,
CAPN3 actually forms a homotrimer. Although PEF domain deletion has
been observed to abolish trimer formation, insertion of the
CAPN3-specific sequences NS, IS1 and IS2 had no impact (12). Small subunits are not present in
CAPN3 (1) (summarized in
Fig. 1).
Various splice variants
There are numerous different splice variants of
CAPN3 (summarized in Fig. 2),
such as Lp82 that is expressed in the cytoplasm of the lens, cortex
and nuclear fibers of rodents (lack of exons 6, 15 and 16)
(13,14), Lp85 (Lp82+IS3) that is expressed
in the lens (15), Rt88
(Lp82+IS1) that is expressed in the retina and numerous other
isoenzymes (with deleted exons 6, 15 and 16) that are expressed in
the embryonic muscles (16,17). The cDNAs of CAPN3 variants are
highly conserved and the differences in their expression patterns
may be related to the function of each variant in specific tissues
or different species (18). The
IS1, IS2 and NS sequences that are either inserted or deleted in
CAPN3 variants endow CAPN3 with numerous characteristics that are
different from those of the original CAPN3.
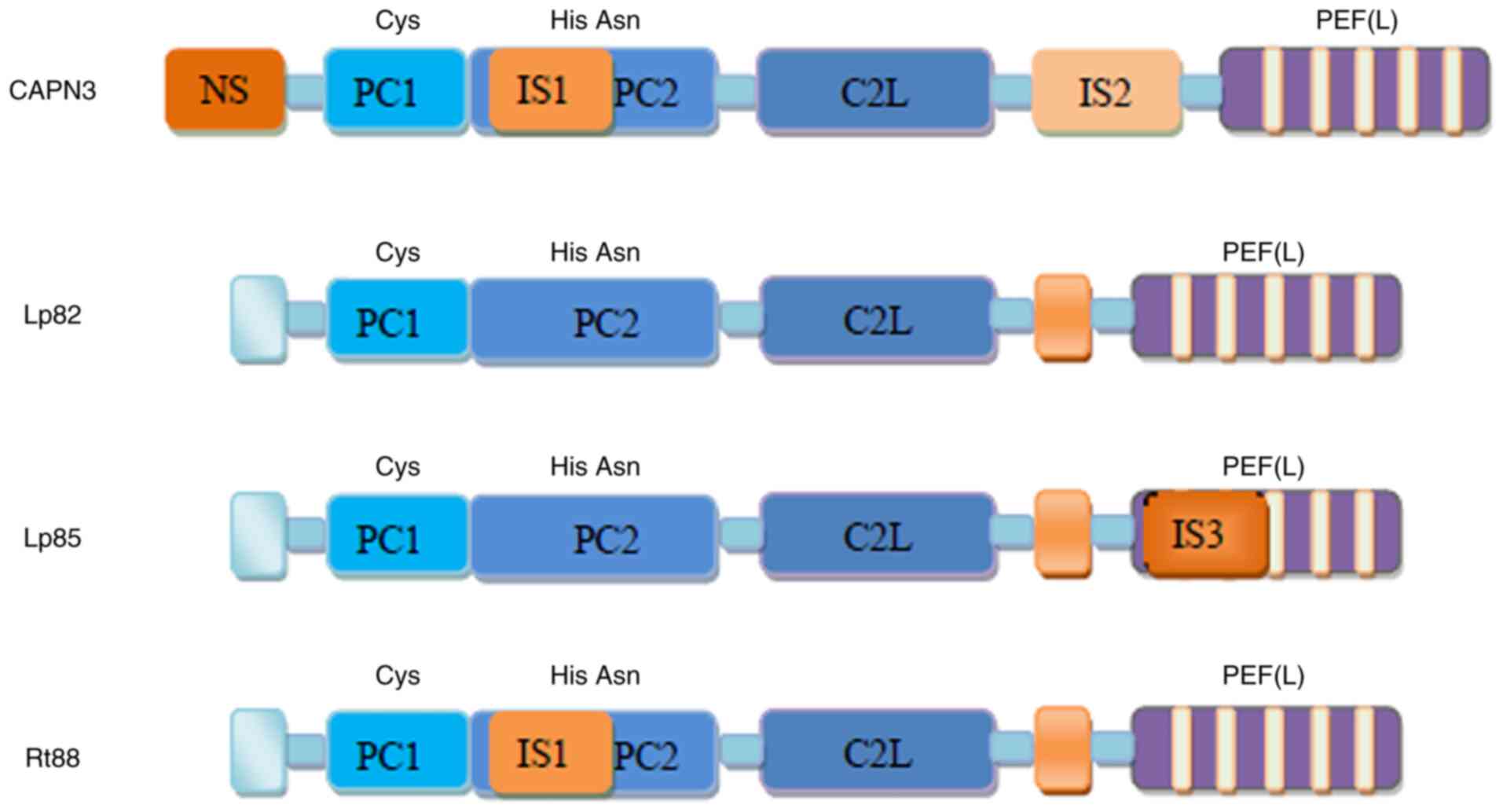 | Figure 2Representative CAPN3 isoforms
generated by alternative splicing of CAPN3. Lp82, lack of exons 6,
15 and 16; Lp85, Lp82+IS3; Rt88, Lp82+IS1. IS3, insertion sequence
3; CAPN3, calpain 3; PEF, penta-EF-hand (E, E-helix; F, F-helix);
NS, N-terminus; IS1, insertion sequence 1; PC1, protease core
domain 1; C2L, calpain-type β-sandwich. |
2. CAPN3 activation and inhibition
Platform element for inhibition of autolytic
degradation (PLEIAD), also known as SUMO-interacting
motif-containing protein 1/chromosome 5 open reading frame 25, is a
multi-SUMO protein that is mainly located in the cytoplasm and
occasionally in the sarcomere I zone. Studies have indicated that
PLEIAD is a novel CAPN3 binding protein. The C-terminal N2A titin
of PLEIAD, which is highly conserved in vertebrates, is able to
inhibit the activity of CAPN3. The N-terminus of PLEIAD is able to
bind to co-transcription factor C-terminal binding protein 1
(CTBP1) and CTBP1 is proteolyzed and functionally modified in COS7
cells expressing CAPN3. Other than CAPN3 inhibition, PLEIAD also
has a role in CAPN3 substrate recruitment. Hence, PLEIAD is a novel
CAPN3 regulator (19).
Calmodulin (CaM), a known calcium signal transducer,
is able to bind CAPN3 at two sites located in the C2L domain to
activate CAPN3 autolysis. CaM may also promote CAPN3-mediated
cleavage of substrate titin in vivo. Therefore, CaM is the
first positive regulator of CAPN3 autolysis (20,21).
3. Features of CAPN3
Autolysis
Since CAPN3 contains specific sequences NS, IS1 and
IS2 that are not present in other calpains or proteases, CAPN3
exhibits unique characteristics. For instance, the complete
autolysis of CAPN3 is rapid and is independent of Ca2+
activation (22). CAPN3 is so
far the only intracellular enzyme that relies on Na+
activation.
The half-life of CAPN3 in vitro is <10 min
and its fast autolysis rate is attributed to the presence of IS1
and IS2 (23). Under normal
physiological conditions, CAPN3 undergoes intramolecular
dissolution, producing a nick with its IS1 sequence. As autolysis
continues, CAPN3 begins to hydrolyze exogenous substrates. The IS1
sequence then forms numerous small fragments and the remaining part
constitutes the two achiral Ca2+ binding sites in the
active center (24,25). Subsequent proteolysis of IS2
eventually inactivates CAPN3 (26). In the presence of IS1, the
autolysis of CAPN3 is unable to be prevented even by calpain
inhibitors. Binding between N2A connectin fragment and CAPN3 has
been observed to inhibit IS2 proteolysis and subsequent
dissociation of CAPN3 fragments (27).
Compared with other classical calpains, due to the
presence of the PC2, CBSW and PEF domains, the required
Ca2+ concentration for CAPN3 autolysis is only 0.1 mM
with a normal physiological Na+ concentration in cells.
The required Ca2+ concentration for CAPN3 activation is
lower than that for other classical calpains (28,29).
In addition, CAPN3 may be activated by
Na+ in vitro at a required Na+
concentration of 100 mM. Calcium binding site 1 (CBS-1) in PC1 is
the binding site for Na+. In the absence of IS1 and IS2,
CAPN3 loses its Na+ dependency (30). Therefore, the dependence of CAPN3
to Na+ requires synergy between CBS-1, IS1 and IS2.
Regain of activity after
inactivation
The rapid and continuous autolysis nature and the
instability feature of CAPN3 allow CAPN3 to regulate its own
activity (summarized in Fig. 3).
Studies have indicated that intramolecular complementation (iMOC)
between the two autolytic fragments of CAPN3 (N and C terminus)
enables CAPN3 to regain the activity of the active core protease
domain (31). Under appropriate
conditions, the use of autolyzed fragments containing CAPN3
wild-type amino acid sequence through iMOC has been indicated to
enable two different CAPN3 mutant molecules to amend each other, so
that part of the CAPN3 mutant activity is restored (31,32).
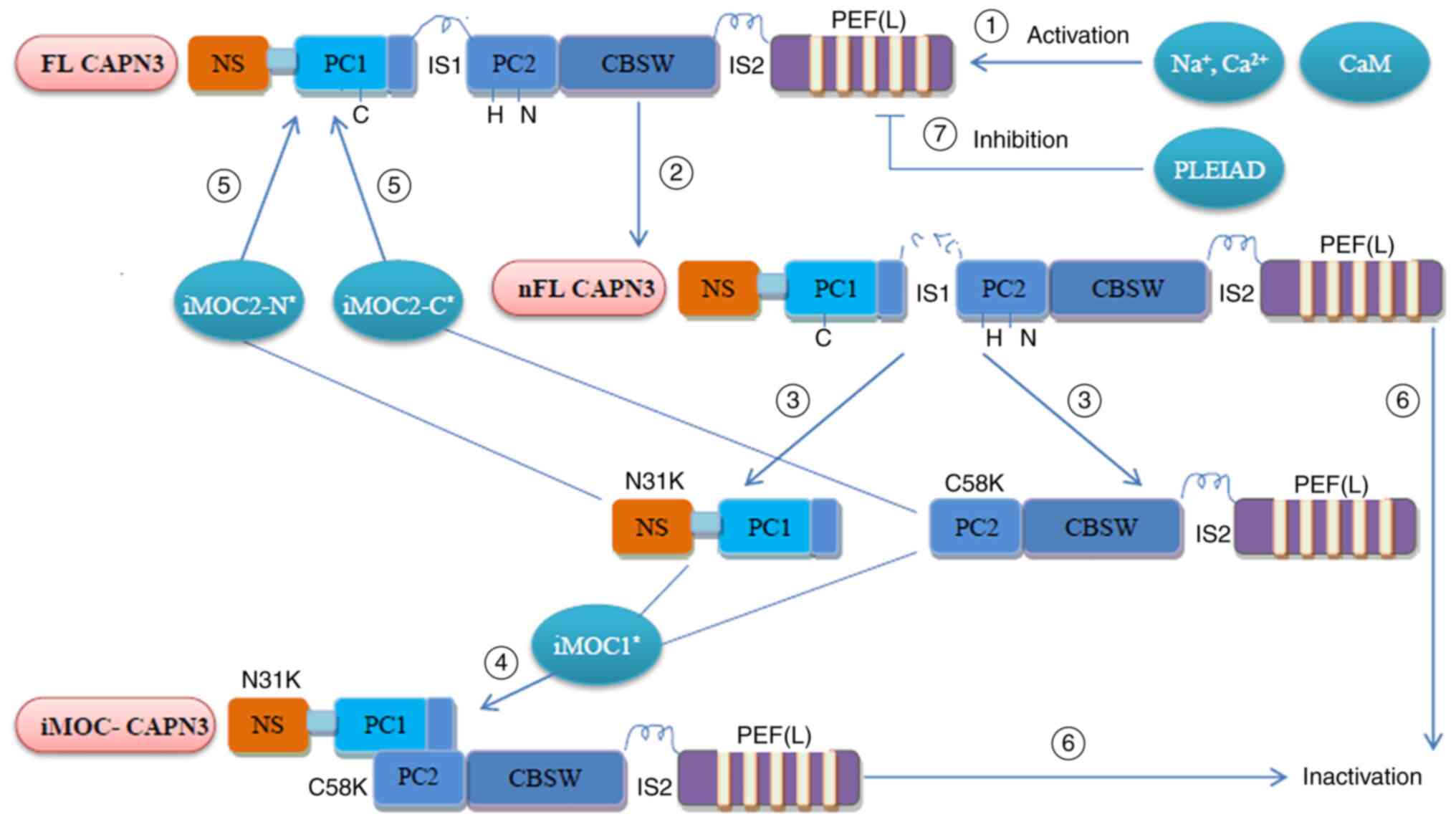 | Figure 3Regulation of CAPN3 activity. ①
Translated FL-CAPN3 (FL-CAPN3) is activated by physiological
Na+/Ca2+ and CaM; ② FL-CAPN3 undergoes
intramolecular dissolution and the IS1 sequence produces a nick
(but not dissociated), which is nFL-CAPN3; ③ N31K and C58K are
dissociated from each other; ④ N31K and C58K were recombined into
active CAPN3 (iMOC-CAPN3) by iMOC1, and its location and function
were completely different from FL/nFL-CAPN3; ⑤ N31K/C58K and
full-length CAPN3 reorganization by iMOC2-N/C to restore partial
activity; ⑥ The IS2 and other sites in nFL-CAPN3 undergo further
autolysis and the recombined iMOC-CAPN3 enters the next round of
autolysis, and will eventually be inactivated; ⑦ PLEIAD is able to
bind to FL-CAPN3 and inhibit CAPN3 activity through its C-terminus.
FL-CAPN3, full-length calpain 3; nFL-CAPN3, nick full-length CAPN3;
PEF, penta-EF-hand (E, E-helix; F, F-helix); NS, N-terminus; IS1,
insertion sequence 1; PC1, protease core domain 1; CBSW,
calpain-type β-sandwich; iMOC, intramolecular complementation;
PLEIAD, platform element for inhibition of autolytic degradation;
CaM, calmodulin. |
4. Physiological function
Calpain is a 'regulatory protease' that requires
Ca2+ for function in cells. It processes substrates
through limited and specific proteolysis to facilitate
Ca2+ signal transduction and regulate various protein
functions in cells (33). CAPN3
is the only muscle-specific member of the calpain family. It is
expressed in the myofibril, cytoplasm and triad components of
skeletal muscle (34). In
resting human skeletal muscle, most (87%) CAPN3 is expressed in
myofibrils and only a small proportion (<10%) exists in an
autolyzed state (35). The
biological function of CAPN3 is closely related to its location in
the cell. Nuclear localized CAPN3 is related to cell survival
(7,36), while CAPN3 localized in the
cytoplasm may be related to the control of cell motility or
skeletal plasticity (37,38).
CAPN3 has both proteolytic and non-proteolytic functions.
CAPN3 promotes calcium release of
skeletal muscle fibers and calcium uptake of sarcoplasmic
reticulum
The triad is the structural component of the muscle
responsible for calcium transport and excitation-contraction
coupling. Aldolase is also present as one of the components in
muscles rich in triads. Glycolytic enzyme aldolase A (AldoA) is the
binding partner of CAPN3. CAPN3 is able to degrade AldoA, but AldoA
is not the body substrate of CAPN3. Aldolase and CAPN3 are able to
interact with ryanodine receptor (RyR) to form the main calcium
release channel. Compared with the wild-type, the levels of AldoA
and RyR associated with the triplet in CAPN3-deficient muscle are
decreased; hence, CAPN3 helps maintain the integrity of the triplet
in skeletal muscle, which then promotes the release of calcium from
muscle fibers (39,40).
The Na+-Ca2+ exchanger isoform
3 (NCX3) is also expressed in the triad of skeletal muscle. A
number of studies have indicated that NCX3 is associated with the
increased Ca2+ content in the sarcoplasmic reticulum.
CAPN3 is able to increase the activity of NCX3, but only the
NCX3-AC variant, which is mainly expressed in the skeletal muscle,
is sensitive to calpain. Increased intracellular levels of
Ca2+ ([Ca2+]i) or [Na+]i
significantly increase cellular Ca2+ uptake through the
reverse mode of NCX3 ingestion. Exercise causes
excitation-contraction uncoupling, NCX3 increases the uptake of
Ca2+ and supplements Ca2+ in the sarcoplasmic
reticulum (41).
CAPN3 promotes muscle formation and
muscle remodeling
CAPN3 mRNA is expressed at high levels in muscles
(42). CAPN3 is distributed in
the amorphous plaques of myoblasts, the area close to the myotube
nucleus, the adhesion plaques and stress fibrous structures of
myotubes and the filaments of fibroblasts (43). CAPN3 is located in the Z line and
N2 line regions of the sarcomere by binding to its chaperone
protein titin (33). The most
characteristic function of CAPN3 is that it is located in the
sarcomere. CAPN3 is an important protein required for muscle
formation and a prerequisite for maintaining normal muscle function
(42).
Muscle formation
CAPN3 is mostly inactive in muscles. It may be
activated by autolysis in the active site to destroy the
cytoskeleton of actin. By lysing several endogenous proteins, the
sarcomere and sarcomere components are lysed; this may enhance the
adaptability of muscle cells to external and/or internal stimuli.
Titin and filamin C, substrates of CAPN3, are co-localized in
multiple locations within the cytoskeleton structure in the body
(38). Therefore, CAPN3 is a
muscle cytoskeleton regulator.
As a scaffold protein, CAPN3 may have a role during
the early stages of myogenesis. Muscle-specific filamin C (FLNC) is
a candidate substrate of CAPN3. The C-terminus of FLNC binds to the
cytoplasmic domain of δ- and γ-glycans. CAPN3 cleaves the
C-terminus of FLNC in living cells in order to eliminate the
interaction between FLNC and δ- and γ-glycans, while the
FLNC-sarcoglycan interaction may have a regulatory effect on
CAPN-mediated myoblast fusion (44). M-cadherin has a role in the
fusion of myoblasts. CAPN3 is able to cut M-cadherin and β-catenin.
In the absence of CAPN3, M-cadherin and β-catenin accumulate
abnormally on the myometrial membrane, while myoblasts and myotubes
continue to fuse, which may inhibit muscle differentiation steps,
such as integrin complex body rearrangement and sarcomere assembly,
inhibiting the formation of sarcomere (45). Therefore, down-regulation of
CAPN3 leads to a decrease in the number of muscle cells and in the
size of myotubes formed (43).
Muscle remodeling
During the development of myoblasts into fully
differentiated myotubes in vitro, a number of 'reserve
cells' are maintained. These reserve cells are closely related to
satellite cells responsible for muscle regeneration. The level of
endogenous CAPN3 mRNA expressed in the reserve cells is higher than
that of proliferating myoblasts. CAPN3 is able to decrease the
transcriptional activity of MyoD, a key myogenic regulator, through
proteolysis in a manner that is independent of the
ubiquitin-proteasome degradation pathway, participate in the
establishment of the reserve cell bank and promote the renewal of
satellite cell compartments (46). The expression of CAPN3 mRNA after
muscle injury (denervation-devasculaization) is related to the
muscle wet weight ratio. CAPN3 is upregulated from day 7 to 14 in
order to promote satellite cell renewal by inhibiting cell
differentiation. CAPN3 is decreased significantly from day 14 to 28
in order to promote myoblast differentiation in L6 cells and
enhance the recovery function. Isoforms lacking exon 6 dominate the
early regeneration process (47,48).
Substrates of CAPN3 are divided into two major
functional categories: Metabolic substrates and myofibrils,
including myosin light chain 1 (MLC1). CAPN3 has a proteolytic
effect on MLC1 in vitro (49). Among these substrates, there are
three E3 SUMO ligases belonging to the protein inhibitor of
activated states (PIAS) family. CAPN3 is able to cleave PIAS
protein and negatively regulate the activity of PIAS3 sumoylase. In
the muscle tissue of patients with limb-girdle muscular dystrophy
(MD) type 2A (LGMD2A), SUMO2 is dysregulated (50). Therefore, CAPN3, through
fast-acting one-way proteolytic switch, has a significant role in
muscle remodeling. CAPN3 also has a role upstream of the
ubiquitin-proteasome pathway by targeting ubiquitination and
proteasome degradation. Increased expression of CAPN3 has been
reported to enhance ubiquitination and promote sarcomere remodeling
by cutting and releasing myofibrillar proteins (51).
In addition, in the triplet, CAPN3 co-localizes with
calmodulin kinase IIβ (CaMKIIβ). CAPN3 and CaMKIIβ have a role in
gene regulation during adaptive endurance exercise. Muscles of
CAPN3 knockout mice (C3KO) have been reported to exhibit decreased
triplet integrity and weakened CaMKIIβ signaling. After atrophy
induction, it has been indicated that C3KO muscles were unable to
activate CaMKIIβ signaling and inducible heat shock protein 70, and
that the expression of cell stress-related genes remained
unchanged; hence the inflammatory response required to promote
muscle recovery was absent. Meanwhile, C3KO muscles have been
indicated to exhibit decreased immune cell infiltration and
decreased expression of myogenic genes (34). In patients with severe LGMD2A,
recent muscle regeneration determined by the number of neonatal
myosin heavy chain/vimentin-positive fibers was significantly
decreased. Compared with those in patients with residual CAPN3, the
signs of abnormal regeneration of spiral fibers were highly
enhanced in patients with complete CAPN3 deficiency (52). Therefore, during sarcomere
remodeling, CAPN3 is necessary for the muscle regeneration
process.
Promotion of neurodegenerative
processes
The IκBα/nuclear factor-κB (NF-κB) signaling pathway
is related to cell apoptosis and it regulates the process of
neurodegeneration by direct alteration of gene expression in
neurons. Loss of CAPN3 proteolytic activity has been observed to
severely interfere with the IκBα/NF-κB pathway (53). On the contrary, when CAPN3 is
activated, the IκBα/CAPN3 complex is formed. Increased levels of
calpain-dependent IκBα cleavage products in the nucleus
subsequently stimulate the activation of nuclear CAPN3-like
proteases in neurons and calpain-dependent cell death. The
proteolysis of IκBα, which activates the IκBα/NF-κB pathway,
promotes neurodegenerative processes (7).
A role in astrocyte plasticity and/or
motility
Most astrocytes in the brains of rats and Microcebus
co-express glial fibrillary acidic protein (GFAP), which is an
ubiquitous target of calpain in vitro (54). GFAP is necessary for the movement
of astrocytes (55). CAPN3 is
located in the cytoplasm of astrocytes, where GFAP is closely
related to CAPN3. In addition, cells expressing GFAP and CAPN3 are
particularly common in the sub-ventricular zone (SVZ) and SVZ
astrocytes are actually the progenitor cells of stem cells that
produce new neurons (56).
Therefore, CAPN3 may have a role in astrocyte motility or
cytoskeletal plasticity (6).
5. CAPN3 in diseases
Since calpain participates in various physiological
processes in cells, dysregulation of CAPN3 may cause different
diseases (Table I), such as MD,
myositis or epilepsy.
 | Table ICAPN3 protein and genes in different
diseases. |
Table I
CAPN3 protein and genes in different
diseases.
A, MD
|
|---|
| Disease | Protein
expressiona | Gene
mutationa | (Refs.) |
|---|
| Limb-girdle MD type
2A | Normal or low
expression | Mutation | (59-69) |
| Limb-girdle MD type
2B | Low expression | Mutation | (86,87) |
| Limb-girdle MD type
2J | Low expression | - | (89,90) |
| Tibial MD | Low expression | - | (89,90) |
| Duchenne MD | Low expression | - | (91) |
| Facioscapulohumeral
MD | Overexpression | Mutation | (92-94) |
| Ullrich congenital
MD | Low expression | - | (95) |
|
B, Other diseases
|
| Disease | Protein
expression | Gene mutation | (Refs.) |
|
| Idiopathic
eosinophilic myositis | - | Mutation | (96) |
| Inclusion body
myositis | Low expression | - | (97,98) |
| Rhabdomyolysis
syndrome | Low expression | - | (99,100) |
| Melanoma | Overexpression | - | (101-106) |
| Epilepsy | - | Mutation | (107,108) |
| Alzheimer's
disease | Uncertain | - | (111) |
| Diabetes | Uncertain | - | (112,113) |
| Cardiovascular
diseases | - | Mutation | (114,115) |
| Vitiligo | Overexpression | - | (116) |
| Age-related
cataract | Overexpression | - | (117) |
Muscular diseases
MD
MD is a group of muscular diseases caused by genetic
factors characterized by progressive muscle weakness and
degeneration of muscles that govern exercise. A common pathological
feature is muscle atrophy accompanied by hyperplasia of fibrous
tissue and adipose tissue. MD has a high degree of genetic
heterogeneity characterized by autosomal dominant or recessive
inheritance of genes (57).
CAPN3 is closely related to the occurrence of a variety of MD.
LGMD2A
LGMD2A is the most commonly diagnosed type of LGMD,
accounting for 30-40% of all LGMD cases (58). LGMD2A is caused by a recessive
mutation in CAPN3. So far, >500 mutation sites have been
reported. Different pathogenic CAPN3 mutations include missense
mutations, frameshift mutations, nonsense mutations,
deletions/insertions, splice site mutations and single mutations.
Among them, missense mutations that are distributed along the
entire CAPN3 gene are most common (59-64). These mutations have been observed
to weaken CAPN3 proteolytic activity by affecting the protein
inter-domain interactions, decrease CAPN3 autocatalytic activity by
lowering its affinity towards Ca2+, cause complete or
partial loss of CAPN3 protein (62,65-67) or result in changes of other
characteristics of CAPN3. For instance, D705G and R448H mutations
affect the ability and stability of CAPN3 to bind titin in
vitro (68,69). These mutations have been reported
to cause skeletal muscle Ca2+ imbalance (70,71), abnormal muscle adaptation
(72,73), sarcomere disorder (74,75), oxidative damage (23,76), mitochondrial abnormality
(42,77) and impaired muscle regeneration
(77,78) (summarized in Fig. 4), which then lead to
inflammation, necrosis, fibrosis, atrophy and progressive muscle
degeneration. All these complications are the characteristics of
LGMD2A.
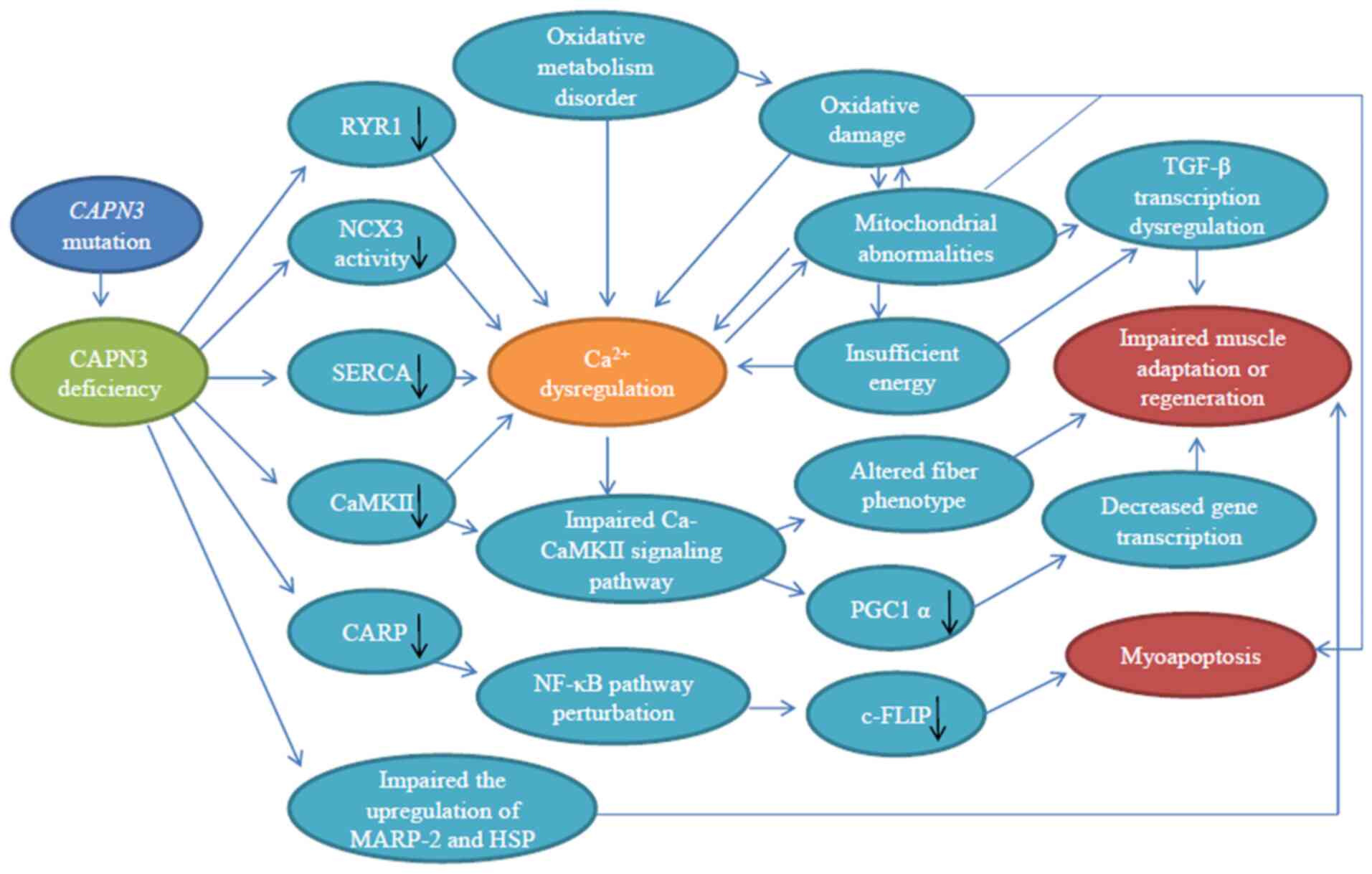 | Figure 4Schematic diagram of the pathogenic
mechanism of CAPN3 deficiency. CAPN3 deficiency results in reduced
levels of RyR1, SERCA, CaMKII and CARP, decreased NCX3 activity and
impaired upregulation of MARP-2 and HSP, causing Ca2+
imbalance and compromise Ca-CaMKII and NF-κB downstream signaling
pathways, which may lead to impaired gene transcription,
mitochondrial abnormalities, oxidative damage and altered fiber
phenotype. This eventually leads to abnormal muscle adaptation,
myocyte apoptosis and impaired regeneration. Among these
mechanisms, there are multiple feedback loops. Black arrows
indicate a decline. CAPN3, calpain 3; RyR, ryanodine receptor;
NCX3, Na+-Ca2+ exchanger isoform 3; SERCA,
sarco-endoplasmic reticulum Ca2+-ATPase; Ca-CaMKII,
Ca2+/calmodulin-dependent protein kinase II; CARP,
cardiac myotonic repeat protein; MARP-2, muscle ankyrin repeat
protein 2; HSP, heat shock protein; PGC1α, peroxisome
proliferator-activated receptor-γ coactlvator-1α; c-FLIP, cell
FLICE inhibitor protein. |
Dysregulation of Ca2+ in the skeletal
muscles is a potential event of MD, including LGMD2A (71,79,80). As one of the constituents of the
triplet, the absence of CAPN3, which destroys the triplet
integrity, decreases the release of Ca2+ in the skeletal
muscles. CAPN3 deficiency also leads to degradation and dysfunction
of skeletal muscle sarco-endoplasmic reticulum
Ca2+-ATPase 1 and 2 proteins, resulting in impaired
Ca2+ homeostasis in human myotubes (79) and NCX3 dysfunction, causing
impaired reticular Ca2+ storage (41).
CAPN3 knockout studies have indicated that
Ca2+/calmodulin-dependent protein kinase II
(Ca-CaMKII)-mediated signal transduction is impaired (81), which not only decreases the slow
muscle fiber phenotype and the fast muscle fiber phenotype
(82,83), but also lowers the level of p38
MAPK activation. Eventually, the level of peroxisome
proliferator-activated receptor-γ coactivator-1α (PGC1α; a
transcriptional co-regulator that coordinates muscle adaptive
response) is decreased, leading to the decreased levels of
transcription of genes involved in muscle adaptation (40). In addition, the loss of CAPN3
protease activity has been observed to impair the upregulation of
muscle ankyrin repeat protein 2 and hsp, causing stress and muscle
degeneration in skeletal muscles. Exercise has been indicated to
exacerbate this change (73). In
the end, long-term failure of muscles to adapt and reshape causes
the occurrence of LGMD2A.
In addition, a multi-protein complex comprising
CAPN3 and cardiac myotonic repeat protein (CARP) is present in the
N2A region of the sarcomeric protein titin. CAPN3 regulates CARP
subcellular localization by cleaving the N-terminus of CARP. The
higher the activity of CAPN3, the more important is the retention
of CARP's sarcomere. Overexpression of CARP decreases the DNA
binding activity of NF-κB p65, a factor that exhibits
anti-apoptotic effects. CAPN3 is unable to decrease the inhibitory
effect of CARP on NF-κB, which may decrease the survival rate of
muscle cells (84). In addition,
the activity of anti-apoptotic factor cell FLICE inhibitor protein
(c-FLIP) depends on the NF-κB pathway in normal muscle cells. CAPN3
is involved in regulating the expression of c-FLIP. CAPN3-dependent
IκBα is expressed after NF-κB activation (75). In the muscle cells of patients
with LGMD2A, NF-κB is activated under cytokine induction in the
absence of CAPN3, IκBα accumulation prevents nuclear translocation
of NF-κB, the NF-κB pathway is dysregulated, c-FLIP expression is
downregulated, and finally, cell apoptosis and muscle shrinking
occur (74,75).
CAPN3 deficiency may also weaken the antioxidant
defense mechanism of skeletal muscles in LGMD2A patients [super-
oxide dismutase (SOD)-1 and nuclear factor erythroid-2-related
factor 2, but not SOD-2], as well as increase lipid peroxidation
and protein ubiquitination, causing obvious oxidative damage and
redox imbalance (76).
Muscles of C3KO induced by cardiotoxin have too many
lobulated fibers belonging to the type of oxidative metabolism,
increased connective tissue, insufficient muscle energy and no
increase in mitochondrial content, PGC-1α and ATP synthase subunit
δ transcripts, resulting in significant TGF-β transcription level
increases with microRNA dysregulation, and the radial growth of
muscle fibers is weakened, Akt/mTOR complex 1 signaling is
disturbed, this signal is uncoupled from protein synthesis, and in
C3KO myoblast cultures, myotube fusion is defective (77), so that healthy sarcomere cannot
be reconstructed. This is the pathogenesis of abnormal muscle
regeneration in patients with LGMD2A.
Although the symptoms of most patients with LGMD2A
are usually uniform, studies have indicated that, compared with
homozygous missense mutations, compound heterozygous mutations
(such as pG222R/pR748Q) have a compensatory effect and that the
presence of 'molecular complementation' may rescue a certain amount
of the proteolytic activity of CAPN3, resulting in an exceptionally
benign phenotype (32).
Therefore, the severity and progression of the disease are
dependent on different gene mutations.
LGMD2B
Mutations in dysferlin (DYSF) are responsible for
LGMD2B. DYSF is absent or minimally expressed in the muscle tissues
of patients (85). DYSF is
thought to have a role in membrane repair. CAPN3 and recombinant
desmoyokin (AHNAK; a protein involved in subsarcolemmal
cytoarchitecture and membrane repair) coexist in the DYSF protein
complex. In skeletal muscle cells with normal CAPN3 expression
activity, the expression of AHNAK is decreased; conversely, in
cells without CAPN3 expression, the expression of ANHAK is
increased. Furthermore, cleaved fragments of AHNAK by CAPN3 lose
their affinity for DYSF in skeletal muscles. Therefore, CAPN3 is a
modulator of the DYSF protein complex in skeletal muscles (86). The expression of CAPN3 in the
skeletal muscles of patients with LGMD2B is decreased. Further
analysis has indicated missense mutations in CAPN3 that change the
amino acids of CAPN3 into the amino acids present in CAPN1 and
CAPN2 (87). Therefore, CAPN3 is
associated with the onset of LGMD2B, but the specific mechanism
exerted by CAPN3 in LGMD2B requires further exploration.
LGMD2J and tibial MD (TMD)
Titin C-terminal mutations in the M-band of striated
muscles may cause LGMD2J and TMD (88). CAPN3 binds titin at the
C-terminus and uses it as a substrate in vitro. There are
several CAPN3 cleavage sites in the C-terminus of titin. Hydrolysis
of titin at the C-terminus by CAPN3 may have an important role in
normal muscles (89). Titin
C-terminal mutations lead to the loss of binding site for CAPN3 and
also to the lack of CAPN3 in muscle (89,90). Therefore, CAPN3 is involved in
the pathogenesis of LGMD2J and TMD.
Duchenne MD (DMD)
DMD is a lethal X-linked muscle disease caused by
defective expression of cytoskeletal protein dystrophin (Dp)427.
Certain retinal neurons express Dp427 and/or the short subtype of
dystrophin. Therefore, patients with DMD may also experience
specific visual defects. A study that analyzed whether the lack of
Dp427 affects the late development of retina in mdx mice (the most
in-depth study of DMD animal models) indicated that, compared with
that of age-matched wild-type mice, the expression of genes E18-P5
and P5 that encode proteins related to retinal development and
synaptogenesis, including CAPN3, is transiently decreased in mdx
mice (91).
Facioscapulohumeral MD (FSHD)
FSHD is a muscle disease related to the loss of
heterozygous D4Z4 on chromosome 4q35. In previously reported
atypical cases, DNA analysis of patients indicated that the loss of
4q35 is associated with heterozygous CAPN3 mutations (92,93). Overexpression of FSHD region gene
1 (FRG1) may disrupt muscle development and cause FSHD-like
phenotypes. FSHD may also be related to splicing. FRG1 is related
to RNA-binding fox-1 homolog 1 (Rbfox1) RNA and may reduce its
stability. Rbfox1 is down-regulated in mice with high FRG1
expression and in patients with FSHD; on the contrary, CAPN3
subtypes lacking exon 6 (CAPN3 E6) were increased. Rbfox1 decreased
and CAPN3 E6 overexpression inhibited muscle differentiation.
Therefore, FSHD is caused by FRG1 overexpression, decreased Rbfox1
expression and high CAPN3 protein expression through misplaced
splicing (94).
Ullrich congenital MD (UCMD)
UCMD is a common MD caused by abnormality of COL6A2
that leads to a defect in collagen VI. UCMD is characterized by
unequal muscle fiber size and loss of muscle mass, as well as
hyperplasia of connective tissue and adipose tissue. Studies have
indicated that the expression of CAPN3 mRNA is decreased in
patients with UCMD and that the downregulation of CAPN3 is related
to altered nuclear immunolocalization of NF-κB. The weakening of
CAPN3 and NF-κB signal transduction may cause muscle cell reduction
and mass loss (95), but the
exact mechanism remains to be elucidated.
Other diseases
Idiopathic eosinophilic myositis
(IEM)
EM is frequently related to parasitic infections,
systemic diseases, drugs or L-tryptophan intake. When these causes
can be excluded, the pathology is IEM. A study has indicated that
CAPN3 is a candidate gene associated with IEM, which has an
autosomal recessive inheritance pattern. Idiopathic patients have
the following characteristics: i) EM in the first decade; ii)
elevated serum creatine phosphokinase levels (isolated or no
corresponding weakness); and iii) increased peripheral blood
eosinophils (96); its
pathogenesis is related to mutation of CAPN3. Further studies that
focus on the physiological functions of CAPN3 in skeletal muscles,
particularly analyses of CAPN3 protein and gene in patients, are
required to better understand IEM.
Inclusion body myositis (IBM)
IBM is the most commonly diagnosed primary myopathy
in the elderly (97). Disorders
of calcium homeostasis in cardiomyocytes may exacerbate factors
that mediate IBM muscle degeneration. Immune-mediated membrane
damage and/or abnormal accumulation of proteins may be the cause of
calcium disorders in patients with IBM. Analysis of muscles in
patients with IBM indicated that insufficient expression of CAPN3
mRNA and protein, which is conducive to proper calcium homeostasis
and increased abundance of two CAPN3 substrates (97,98). The pathogenesis of IBM remains to
be clarified and the role of CAPN3 in IBM remains to be
explored.
Rhabdomyolysis syndrome
Rhabdomyolysis syndrome is a medical emergency
caused by exposure to external triggers and may be related to
increased genetic susceptibility. Previously, two patients have
been diagnosed with rhabdomyolysis syndrome after consuming wild
quail meat. Analysis has indicated that the expression of CAPN3
protein in these two patients was decreased and that there was no
pathogenic mutation in the CAPN3 gene (99). Another study suggested that
certain patients with underlying genetic diseases (such as
recurrent episodes, a positive family history and high or
persistently increased CK levels) have clear genetic defects and
that CAPN3 may be related to increased genetic susceptibility to
rhabdomyolysis syndrome (100).
Melanoma
Numerous genes involved in intracellular calcium and
G protein signal transduction are highly expressed in melanoma,
with CAPN3 being one of the most highly expressed genes (101).
In the process of induction of irreversible growth
arrest and terminal differentiation, and subsequent apoptosis of
melanoma cells, the expression of CAPN3 variant 6 is inhibited, but
still maintained at a high level. After apoptotic injury, the high
expression of CAPN3 has no critical role in the regulation of
growth dynamics and/or cell viability. It has been suggested that
the upregulation of CAPN3 may not be a pathogenic factor, but is
related to the development of melanoma (102). Both hMp78 and hMp84 are
established new splice variants of CAPN3. They are localized in the
cytoplasm and in nucleoli. Compared with that in benign melanoma
cells, the expression of these variants is downregulated in
malignant melanoma cells. In A375 and HT-144 cells, hMp78 and hMp84
are over-expressed. In A375 cells, hMp84 exhibits catalytic
activity that induces p53 stability, regulates the expression of
certain genes related to p53 and oxidative stress and increases the
production of cellular reactive oxygen species, which then leads to
oxidative modification of phospholipids (F2-isoprostaglandin
formation) and DNA damage, and ultimately decreased cell
proliferation ability and cell death. In HT-144 cells, in addition
to p53 accumulation, the effects of hMp84 are consistent with those
observed in A375 cells. Therefore, it is thought that
downregulation of CAPN3 may contribute to the progression of
melanoma (103,104).
CAPN3 is also associated with the invasion of
melanoma cells (105). Compared
with that of M14C2/C4 cells with a low-invasive phenotype, the
growth rate of highly invasive M14C2/MK18 cells is more rapid. In
M14C2/MK18 cells, the expression of CAPN3 is downregulated.
Inhibition of CAPN3 activity in M14C2/C4 cells significantly
increases the invasiveness of M14C2/C4 cells, indicating that
downregulation of CAPN3 promotes malignant melanoma invasion
(106).
CAPN3 has a vital role in the development and
metastasis of melanoma, but the underlying mechanism and whether
the specific role of CAPN3 is related to tumor cell types require
further clarification.
Epilepsy
There has been a case report of CAPN3 mutation in a
pediatric case of LGMD with hereditary generalized epilepsy. It is
thought that CAPN3 mutation is related to the occurrence of
hereditary generalized epilepsy (107). Recently, members of a family
were diagnosed with generalized epilepsy and LGMD phenotypes. It
has been determined that patients with CAPN3 homozygous mutations
developed LGMD, while subjects with CAPN3 heterozygous mutations
developed epilepsy (108).
Hence, CAPN3 mutations may have a role in hereditary
generalized epilepsy.
Alzheimer's disease (AD)
AD is the most common type of dementia and its onset
and development are related to specific changes in DNA methylation
in affected brain regions (109,110). Patients with advanced AD
frequently lose the ability to recognize family members. The
fusiform gyrus (FUS) of the brain is important in facial
recognition. Studies have indicated that the expression levels of
CAPN3 and other four characteristic genes are abnormal in FUS and
that there are related changes in the DNA methylome profiles of
these genes (111). Compared
with that of currently used clinical standards, the level of
sensitivity of these related methylome profile changes is higher
and may effectively predict the prognosis of AD.
Diabetes
Obesity is an important factor in the development of
insulin resistance, which is the basis of type 2 diabetes. Studies
have indicated that the expression levels of CAPN3 in skeletal
muscle are positively correlated with carbohydrate oxidation but
negatively correlated with circulating glucose and insulin
concentrations, as well as body fat (112). Another study that performed
fasting (inducing insulin resistance) and refeeding (reversing
insulin) in healthy patients suggested that the expression of CAPN3
mRNA or protein is unaffected (113). Further studies are required to
explore the association between CAPN3 and diabetes.
Coronary artery disease (CAD)
Lipid homeostasis is closely related to
cardiovascular risk. Although hundreds of loci associated with
blood lipids and related cardiovascular traits have been
identified, there is only a small number of known genetic links
that explore long-term changes in blood lipids. A genotyping
analysis has revealed that there is a variant site
(P=1.2x10-4) in CAPN3 that is related to CAD (114). The integration variation,
haplotype and double ploidy of CAPN3 and FERM domain containing 5
genes are related to blood lipid variables (115). Therefore, CAPN3 is associated
with blood lipids and related cardiovascular diseases.
Vitiligo
Vitiligo is a disease that features skin
depigmentation. Its pathogenesis involves factors such as the
environment, genetics and biology of melanocytes. RNA sequencing
has identified CAPN3 as one of the five differentially expressed
genes, with the RNA levels of CAPN3 being significantly increased
(116). Further study of these
differentially expressed genes is required to understand the
pathogenesis and disease progression of vitiligo.
Age-related cataract
Age-related cataracts are related to degenerative
changes that slowly occur in old individuals. In certain cases,
vision is affected by lens opacity. Studies have suggested that
129α3Cx46-/- mice are able to form age-related
cataracts, while in the absence of CAPN3, the formation of
cataracts is delayed and the cataract appearance becomes more
diffuse and of the pulverulent type. Analysis has indicated that
CAPN3 is directly involved in the γ-crystallin cleavage pathway;
CAPN3 is therefore associated with the formation of age-related
cataracts in α3Cx46-/- mice (117). Since age-related cataract is
the leading cause of blindness worldwide, it is important to better
understand its pathogenesis.
6. Conclusions and future perspectives
CAPN3, a member of the calpain family, has certain
common features of calpains, but due to its special structure, it
has complex biological functions, including fast autolysis. To
better understand CAPN3, it is necessary to study the quaternary
structure, activity and natural substrates of CAPN3, particularly
by using full-length CAPN3 purified protein.
Although certain functions of CAPN3 have been
under-stood, due to its inherent instability, the molecular
mechanisms of its substrate or CAPN3 activation remain elusive. In
addition, iMOC may help regain the activity of the mutant CAPN3,
which in turn helps to partially alleviate LGMD2A caused by
missense mutations in the CAPN3 allele. Therefore, revealing the
physiological significance of iMOC and the molecular mechanisms of
CAPN3 is of great significance.
Existing CAPN3 and disease-related studies have
been helpful in explaining the pathogenesis of associated diseases,
which may facilitate the prediction of disease development and
prognosis, but there is a relatively large number of studies on
LGMD2A and only a small number of studies on other diseases; its
role in numerous diseases remains controversial and may be further
investigated. In terms of treatment, recombinant adeno-associated
virus (rAAV) that mediates CAPN3 gene transfer and autologous
induced pluripotent stem cells have been used to treat LGMD2A
(118,119), both of which have certain
curative effects, but with certain issues, including the potential
immune response caused by the introduction of rAAV or CAPN3, or the
persistence of genes. CAPN3-related diseases, particularly MD, are
genetic-related. The combination of genetics and biochemical
research will help to further clarify the pathogenic roles of this
unusual calpain molecule in order to provide a basis to facilitate
the development of precision gene and cell therapy in the
future.
In addition, current studies on CAPN3 mainly focus
on skeletal muscle. Although CAPN3 is a skeletal muscle-specific
calpain, is not limited to skeletal muscle due to its diverse
function and damage it causes in humans after mutation, which
requires to be further explored.
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
LC, FT, HG, XZ, XL and DX were involved in the
conceptualization of the study. LC, FT, XL and DX were involved in
software applications. LC, FT, HG, XZ, XL and DX provided the study
resources. LC and FT were involved in the writing and preparation
of the original draft, and in the writing, reviewing and editing of
the study. HG and XZ were involved in the processing of the
figures. XL and DX supervised the study. All authors read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Key R&D
Programme of China (grant nos. 2017YFA 0104201 and 2017YFA
0104200), the National Science Foundation of China (grant nos.
81330016, 82071353 and 82001593) and the Key R&D projects of
Science and Technology Department of Sichuan Province (grant no.
2020YFS 0104).
Abbreviations:
|
CAPN3
|
calpain 3
|
|
NS
|
N-terminus
|
|
IS1
|
insertion sequence 1
|
|
MD
|
muscular dystrophy
|
|
PC1
|
protease core domain 1
|
|
PEF
|
penta-EF-hand (E, E-helix
|
|
F
|
F-helix)
|
|
CBSW
|
calpain-type β-sandwich
|
|
PLEIAD
|
platform element for inhibition of
autolytic degradation
|
|
CTBP1
|
C-terminal binding protein 1
|
|
CaM
|
calmodulin
|
|
CBS-1
|
calcium binding site
|
|
iMOC
|
intramolecular complementation
|
|
AldoA
|
aldolase A
|
|
RyR
|
ryanodine receptor
|
|
NCX3
|
Na+-Ca2+
exchanger isoform 3
|
|
FLNC
|
filamin C
|
|
MLC1
|
myosin light chain 1
|
|
PIAS
|
protein inhibitor of activated
states
|
|
LGMD2A
|
limb-girdle muscular dystrophy type
2A
|
|
CaMKIIβ
|
calmodulin kinase IIβ
|
|
C3KO
|
CAPN3 knockout mice
|
|
NF-κB
|
nuclear factor κB
|
|
GFAP
|
glial fibrillary acidic protein
|
|
SVZ
|
sub-ventricular zone
|
|
Ca-CaMKII
|
Ca2+/CaM-dependent protein
kinase II
|
|
PGC1α
|
peroxisome proliferator-activated
receptor-γ coactivator-1α
|
|
CARP
|
cardiac myotonic repeat protein
|
|
c-FLIP
|
cell FLICE inhibitor protein
|
|
DYSF
|
dysferlin
|
|
TMD
|
tibial MD
|
|
DMD
|
Duchenne MD
|
|
FSHD
|
facioscapulohumeral MD
|
|
FRG1
|
FSHD region gene 1
|
|
CAPN3 E6-
|
CAPN3 subtypes lacking exon 6
|
|
UCMD
|
Ullrich congenital MD
|
|
IEM
|
idiopathic eosinophilic myositis
|
|
IBM
|
inclusion body myositis
|
|
AD
|
Alzheimer's disease
|
|
FUS
|
fusiform gyrus
|
|
CAD
|
coronary artery disease
|
|
AAV
|
adeno-associated virus
|
References
|
1
|
Sorimachi H, Imajoh-Ohmi S, Emori Y,
Kawasaki H, Ohno S, Minami Y and Suzuki K: Molecular cloning of a
novel mammalian calcium-dependent protease distinct from both m-
and mu-types. Specific expression of the mRNA in skeletal muscle. J
Biol Chem. 264:20106–20111. 1989. View Article : Google Scholar
|
|
2
|
Ono Y, Ojima K, Shinkai-Ouchi F, Hata S
and Sorimachi H: An eccentric calpain, CAPN3/p94/calpain-3.
Biochimie. 122:169–187. 2016. View Article : Google Scholar
|
|
3
|
Ohno S, Minoshima S, Kudoh J, Fukuyama R,
Shimizu Y, Ohmi-Imajoh S, Shimizu N and Suzuki K: Four genes for
the calpain family locate on four distinct human chromosomes.
Cytogenet Cell Genet. 53:225–229. 1990. View Article : Google Scholar
|
|
4
|
Fougerousse F, Durand M, Suel L, Pourquié
O, Delezoide AL, Romero NB, Abitbol M and Beckmann JS: Expression
of genes (CAPN3, SGCA, SGCB, and TTN) involved in progressive
muscular dystrophies during early human development. Genomics.
48:145–156. 1998. View Article : Google Scholar
|
|
5
|
Fougerousse F, Anderson LV, Delezoide AL,
Suel L, Durand M and Beckmann JS: Calpain3 expression during human
cardiogenesis. Neuromuscul Disord. 10:251–256. 2000. View Article : Google Scholar
|
|
6
|
König N, Raynaud F, Feane H, Durand M,
Mestre-Francès N, Rossel M, Ouali A and Benyamin Y: Calpain 3 is
expressed in astrocytes of rat and Microcebus brain. J Chem
Neuroanat. 25:129–136. 2003. View Article : Google Scholar
|
|
7
|
Marcilhac A, Raynaud F, Clerc I and
Benyamin Y: Detection and localization of calpain 3-like protease
in a neuronal cell line: Possible regulation of apoptotic cell
death through degradation of nuclear IkappaBalpha. Int J Biochem
Cell Biol. 38:2128–2140. 2006. View Article : Google Scholar
|
|
8
|
McCartney CE, Ye Q, Campbell RL and Davies
PL: Insertion sequence 1 from calpain-3 is functional in calpain-2
as an internal propeptide. J Biol Chem. 293:17716–17730. 2018.
View Article : Google Scholar
|
|
9
|
Ye Q, Campbell RL and Davies PL:
Structures of human calpain-3 protease core with and without bound
inhibitor reveal mechanisms of calpain activation. J Biol Chem.
293:4056–4070. 2018. View Article : Google Scholar
|
|
10
|
Imajoh S, Kawasaki H and Suzuki K: The
COOH-terminal E-F hand structure of calcium-activated neutral
protease (CANP) is important for the association of subunits and
resulting proteolytic activity. J Biochem. 101:447–452. 1987.
View Article : Google Scholar
|
|
11
|
Partha SK, Ravulapalli R, Allingham JS,
Campbell RL and Davies PL: Crystal structure of calpain-3
penta-EF-hand (PEF) domain-a homodimerized PEF family member with
calcium bound at the fifth EF-hand. FEBS J. 281:3138–3149. 2014.
View Article : Google Scholar
|
|
12
|
Hata S, Doi N, Shinkai-Ouchi F and Ono Y:
A muscle-specific calpain, CAPN3, forms a homotrimer. Biochim
Biophys Acta Proteins Proteom. 1868:1404112020. View Article : Google Scholar
|
|
13
|
Ma H, Fukiage C, Azuma M and Shearer TR:
Cloning and expression of mRNA for calpain Lp82 from rat lens:
Splice variant of p94. Invest Ophthalmol Vis Sci. 39:454–461.
1998.
|
|
14
|
Ma H, Shih M, Fukiage C, Azuma M, Duncan
MK, Reed NA, Richard I, Beckmann JS and Shearer TR: Influence of
specific regions in Lp82 calpain on protein stability, activity,
and localization within lens. Invest Ophthalmol Vis Sci.
41:4232–4239. 2000.
|
|
15
|
Ma H, Shih M, Hata I, Fukiage C, Azuma M
and Shearer TR: Lp85 calpain is an enzymatically active
rodent-specific isozyme of lens Lp82. Curr Eye Res. 20:183–189.
2000. View Article : Google Scholar
|
|
16
|
Herasse M, Ono Y, Fougerousse F, Kimura E,
Stockholm D, Beley C, Montarras D, Pinset C, Sorimachi H, Suzuki K,
et al: Expression and functional characteristics of calpain 3
isoforms generated through tissue-specific transcriptional and
posttranscriptional events. Mol Cell Biol. 19:4047–4055. 1999.
View Article : Google Scholar
|
|
17
|
Azuma M, Fukiage C, Higashine M, Nakajima
T, Ma H and Shearer TR: Identification and characterization of a
retina-specific calpain (Rt88) from rat. Curr Eye Res. 21:710–720.
2000. View Article : Google Scholar
|
|
18
|
Nakajima T, Fukiage C, Azuma M, Ma H and
Shearer TR: Different expression patterns for ubiquitous calpains
and Capn3 splice variants in monkey ocular tissues. Biochim Biophys
Acta. 1519:55–64. 2001. View Article : Google Scholar
|
|
19
|
Ono Y, Iemura S, Novak SM, Doi N, Kitamura
F, Natsume T, Gregorio CC and Sorimachi H: PLEIAD/SIMC1/C5orf25, a
novel autolysis regulator for a skeletal-muscle-specific calpain,
CAPN3, scaffolds a CAPN3 substrate, CTBP1. J Mol Biol.
425:2955–2972. 2013. View Article : Google Scholar
|
|
20
|
Ermolova N, Kramerova I and Spencer MJ:
Autolytic activation of calpain 3 proteinase is facilitated by
calmodulin protein. J Biol Chem. 290:996–1004. 2015. View Article : Google Scholar
|
|
21
|
Ono Y and Sorimachi H: Calpains: An
elaborate proteolytic system. Biochim Biophys Acta. 1824:224–236.
2012. View Article : Google Scholar
|
|
22
|
Sorimachi H and Kawabata Y: Calpain and
pathology in view of structure-function relationships. Nihon
Yakurigaku Zasshi. 122:21–29. 2003.In Japanese. View Article : Google Scholar
|
|
23
|
Sorimachi H, Toyama-Sorimachi N, Saido TC,
Kawasaki H, Sugita H, Miyasaka M, Arahata K, Ishiura S and Suzuki
K: Muscle-specific calpain, p94, is degraded by autolysis
immediately after translation, resulting in disappearance from
muscle. J Biol Chem. 268:10593–10605. 1993. View Article : Google Scholar
|
|
24
|
Rey MA and Davies PL: The protease core of
the muscle-specific calpain, p94, undergoes Ca2+-dependent
intramolecular autolysis. FEBS Lett. 532:401–406. 2002. View Article : Google Scholar
|
|
25
|
Diaz BG, Moldoveanu T, Kuiper MJ, Campbell
RL and Davies PL: Insertion sequence 1 of muscle-specific calpain,
p94, acts as an internal propeptide. J Biol Chem. 279:27656–27666.
2004. View Article : Google Scholar
|
|
26
|
Fukiage C, Nakajima E, Ma H, Azuma M and
Shearer TR: Characterization and regulation of lens-specific
calpain Lp82. J Biol Chem. 277:20678–20685. 2002. View Article : Google Scholar
|
|
27
|
Ono Y, Torii F, Ojima K, Doi N, Yoshioka
K, Kawabata Y, Labeit D, Labeit S, Suzuki K, Abe K, et al:
Suppressed disassembly of autolyzing p94/CAPN3 by N2A
connectin/titin in a genetic reporter system. J Biol Chem.
281:18519–18531. 2006. View Article : Google Scholar
|
|
28
|
Ono Y, Hayashi C, Doi N, Tagami M and
Sorimachi H: The importance of conserved amino acid residues in p94
protease sub-domain IIb and the IS2 region for constitutive
autolysis. FEBS Lett. 582:691–698. 2008. View Article : Google Scholar
|
|
29
|
Hata S, Doi N, Kitamura F and Sorimachi H:
Stomach-specific calpain, nCL-2/calpain 8, is active without
calpain regulatory subunit and oligomerizes through C2-like
domains. J Biol Chem. 282:27847–27856. 2007. View Article : Google Scholar
|
|
30
|
Parr T, Sensky PL, Scothern GP, Bardsley
RG, Buttery PJ, Wood JD and Warkup C: Relationship between skeletal
muscle-specific calpain and tenderness of conditioned porcine
longissimus muscle. J Anim Sci. 77:661–668. 1999. View Article : Google Scholar
|
|
31
|
Ono Y, Shindo M, Doi N, Kitamura F,
Gregorio CC and Sorimachi H: The N- and C-terminal autolytic
fragments of CAPN3/p94/calpain-3 restore proteolytic activity by
inter- molecular complementation. Proc Natl Acad Sci USA.
111:E5527–E5536. 2014. View Article : Google Scholar
|
|
32
|
Sáenz A, Ono Y, Sorimachi H, Goicoechea M,
Leturcq F, Blázquez L, García-Bragado F, Marina A, Poza JJ,
Azpitarte M, et al: Does the severity of the LGMD2A phenotype in
compound heterozygotes depend on the combination of mutations?
Muscle Nerve. 44:710–714. 2011. View Article : Google Scholar
|
|
33
|
Ojima K, Ono Y, Hata S, Koyama S, Doi N
and Sorimachi H: Possible functions of p94 in connectin-mediated
signaling pathways in skeletal muscle cells. J Muscle Res Cell
Motil. 26:409–417. 2005. View Article : Google Scholar
|
|
34
|
Kramerova I, Torres JA, Eskin A, Nelson SF
and Spencer MJ: Calpain 3 and CaMKIIβ signaling are required to
induce HSP70 necessary for adaptive muscle growth after atrophy.
Hum Mol Genet. 27:1642–1653. 2018. View Article : Google Scholar
|
|
35
|
Murphy RM, Vissing K, Latchman H, Lamboley
C, McKenna MJ, Overgaard K and Lamb GD: Activation of skeletal
muscle calpain-3 by eccentric exercise in humans does not result in
its translocation to the nucleus or cytosol. J Appl Physiol (1985).
111:1448–1458. 2011. View Article : Google Scholar
|
|
36
|
Baghdiguian S, Martin M, Richard I, Pons
F, Astier C, Bourg N, Hay RT, Chemaly R, Halaby G, Loiselet J, et
al: Calpain 3 deficiency is associated with myonuclear apoptosis
and profound perturbation of the IkappaB alpha/NF-kappaB pathway in
limb-girdle muscular dystrophy type 2A. Nat Med. 5:5031999.
View Article : Google Scholar
|
|
37
|
Sorimachi H, Kinbara K, Kimura S,
Takahashi M, Ishiura S, Sasagawa N, Sorimachi N, Shimada H, Tagawa
K, Maruyama K, et al: Muscle-specific calpain, p94, responsible for
limb girdle muscular dystrophy type 2A, associates with connectin
through IS2, a p94-specific sequence. J Biol Chem. 270:31158–31162.
1995. View Article : Google Scholar
|
|
38
|
Taveau M, Bourg N, Sillon G, Roudaut C,
Bartoli M and Richard I: Calpain 3 is activated through autolysis
within the active site and lyses sarcomeric and sarcolemmal
components. Mol Cell Biol. 23:9127–9135. 2003. View Article : Google Scholar
|
|
39
|
Kramerova I, Kudryashova E, Wu B,
Ottenheijm C, Granzier H and Spencer MJ: Novel role of calpain-3 in
the triad-associated protein complex regulating calcium release in
skeletal muscle. Hum Mol Genet. 17:3271–3280. 2008. View Article : Google Scholar
|
|
40
|
Kramerova I, Ermolova N, Eskin A, Hevener
A, Quehenberger O, Armando AM, Haller R, Romain N, Nelson SF and
Spencer MJ: Failure to up-regulate transcription of genes necessary
for muscle adaptation underlies limb girdle muscular dystrophy 2A
(calpainopathy). Hum Mol Genet. 25:2194–2207. 2016. View Article : Google Scholar
|
|
41
|
Michel LY, Hoenderop JG and Bindels RJ:
Calpain-3-mediated regulation of the Na+-Ca2+
exchanger isoform 3. Pflugers Arch. 468:243–255. 2016. View Article : Google Scholar
|
|
42
|
Beckmann JS and Spencer M: Calpain 3, the
'gatekeeper' of proper sarcomere assembly, turnover and
maintenance. Neuromuscul Disord. 18:913–921. 2008. View Article : Google Scholar
|
|
43
|
de Andrade Rosa I, Corrêa S, Costa ML and
Mermelstein C: The scaffolding protein calpain-3 has multiple
distributions in embryonic chick muscle cells and it is essential
for the formation of muscle fibers. Tissue Cell. 67:1014362020.
View Article : Google Scholar
|
|
44
|
Guyon JR, Kudryashova E, Potts A, Dalkilic
I, Brosius MA, Thompson TG, Beckmann JS, Kunkel LM and Spencer MJ:
Calpain 3 cleaves filamin C and regulates its ability to interact
with gamma- and delta-sarcoglycans. Muscle Nerve. 28:472–483. 2003.
View Article : Google Scholar
|
|
45
|
Kramerova I, Kudryashova E, Wu B and
Spencer MJ: Regulation of the M-cadherin-beta-catenin complex by
calpain 3 during terminal stages of myogenic differentiation. Mol
Cell Biol. 26:8437–8447. 2006. View Article : Google Scholar
|
|
46
|
Stuelsatz P, Pouzoulet F, Lamarre Y,
Dargelos E, Poussard S, Leibovitch S, Cottin P and Veschambre P:
Down-regulation of MyoD by calpain 3 promotes generation of reserve
cells in C2C12 myoblasts. J Biol Chem. 285:12670–12683. 2010.
View Article : Google Scholar
|
|
47
|
Stockholm D, Herasse M, Marchand S, Praud
C, Roudaut C, Richard I, Sebille A and Beckmann JS: Calpain 3 mRNA
expression in mice after denervation and during muscle
regeneration. Am J Physiol Cell Physiol. 280:C1561–C1569. 2001.
View Article : Google Scholar
|
|
48
|
Wu R, Yan Y, Yao J, Liu Y, Zhao J and Liu
M: Calpain 3 expression pattern during gastrocnemius muscle atrophy
and regeneration following sciatic nerve injury in rats. Int J Mol
Sci. 16:26927–26935. 2015. View Article : Google Scholar
|
|
49
|
Cohen N, Kudryashova E, Kramerova I,
Anderson LV, Beckmann JS, Bushby K and Spencer MJ: Identification
of putative in vivo substrates of calpain 3 by comparative
proteomics of overexpressing transgenic and nontransgenic mice.
Proteomics. 6:6075–6084. 2006. View Article : Google Scholar
|
|
50
|
de Morrée A, Lutje Hulsik D, Impagliazzo
A, van Haagen HH, de Galan P, van Remoortere A, 't Hoen PA, van
Ommen GB, Frants RR and van der Maarel SM: Calpain 3 is a
rapid-action, unidirectional proteolytic switch central to muscle
remodeling. PLoS One. 5:e119402010. View Article : Google Scholar
|
|
51
|
Kramerova I, Kudryashova E, Venkatraman G
and Spencer MJ: Calpain 3 participates in sarcomere remodeling by
acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet.
14:2125–2134. 2005. View Article : Google Scholar
|
|
52
|
Hauerslev S, Sveen ML, Duno M, Angelini C,
Vissing J and Krag TO: Calpain 3 is important for muscle
regeneration: Evidence from patients with limb girdle muscular
dystrophies. BMC Musculoskelet Disord. 13:432012. View Article : Google Scholar
|
|
53
|
Richard I, Roudaut C, Marchand S,
Baghdiguian S, Herasse M, Stockholm D, Ono Y, Suel L, Bourg N,
Sorimachi H, et al: Loss of calpain 3 proteolytic activity leads to
muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear
factor kappaB pathway perturbation in mice. J Cell Biol.
151:1583–1590. 2000. View Article : Google Scholar
|
|
54
|
DeArmond S, Fajardo M, Naughton S and Eng
L: Degradation of glial fibrillary acidic protein by a calcium
dependent proteinase: An electroblot study. Brain Res. 262:275–282.
1983. View Article : Google Scholar
|
|
55
|
Lepekhin EA, Eliasson C, Berthold CH,
Berezin V, Bock E and Pekny M: Intermediate filaments regulate
astrocyte motility. J Neurochem. 79:617–625. 2001. View Article : Google Scholar
|
|
56
|
Alvarez-Buylla A, Seri B and Doetsch F:
Identification of neural stem cells in the adult vertebrate brain.
Brain Res Bull. 57:751–758. 2002. View Article : Google Scholar
|
|
57
|
Schmidt WM, Uddin MH, Dysek S, Moser-Their
K, Pirker C, Höger H, Ambros IM, Ambros PF, Berger W and Bittner
RE: DNA damage, somatic aneuploidy, and malignant sarcoma
susceptibility in muscular dystrophies. PLoS Genet. 7:e10020422011.
View Article : Google Scholar
|
|
58
|
Oliveira Santos M, Ninitas P and Conceição
I: Severe limb-girdle muscular dystrophy 2A in two young siblings
from Guinea-Bissau associated with a novel null homozygous mutation
in CAPN3 gene. Neuromuscul Disord. 28:1003–1005. 2018. View Article : Google Scholar
|
|
59
|
Ono Y, Sorimachi H and Suzuki K: New
aspect of the research on limb-girdle muscular dystrophy 2A: A
molecular biologic and biochemical approach to pathology. Trends
Cardiovasc Med. 9:114–118. 1999. View Article : Google Scholar
|
|
60
|
Richard I, Roudaut C, Saenz A, Pogue R,
Grimbergen JE, Anderson LV, Beley C, Cobo AM, de Diego C, Eymard B,
et al: Calpainopathy-a survey of mutations and polymorphisms. Am J
Hum Genet. 64:1524–1540. 1999. View
Article : Google Scholar
|
|
61
|
Chae J, Minami N, Jin Y, Nakagawa M,
Murayama K, Igarashi F and Nonaka I: Calpain 3 gene mutations:
Genetic and clinico-pathologic findings in limb-girdle muscular
dystrophy. Neuromuscul Disord. 11:547–555. 2001. View Article : Google Scholar
|
|
62
|
Fanin M, Nascimbeni AC, Fulizio L,
Trevisan CP, Meznaric-Petrusa M and Angelini C: Loss of calpain-3
auto- catalytic activity in LGMD2A patients with normal protein
expression. Am J Pathol. 163:1929–1936. 2003. View Article : Google Scholar
|
|
63
|
Peddareddygari LR, Surgan V and Grewal RP:
Limb-girdle muscular dystrophy type 2A resulting from homozygous
G2338C transversion mutation in the calpain-3 gene. J Clin
Neuromuscul Dis. 12:62–65. 2010. View Article : Google Scholar
|
|
64
|
Perez F, Vital A, Martin-Negrier ML,
Ferrer X and Sole G: Diagnostic procedure of limb girdle muscular
dystrophies 2A or calpainopathies: French cohort from a
neuromuscular center (Bordeaux). Rev Neurol (Paris). 166:502–508.
2010.In French. View Article : Google Scholar
|
|
65
|
Fanin M, Fulizio L, Nascimbeni AC,
Spinazzi M, Piluso G, Ventriglia VM, Ruzza G, Siciliano G, Trevisan
CP, Politano L, et al: Molecular diagnosis in LGMD2A: Mutation
analysis or protein testing? Hum Mutat. 24:52–62. 2004. View Article : Google Scholar
|
|
66
|
Fanin M, Nascimbeni AC and Angelini C:
Screening of calpain-3 autolytic activity in LGMD muscle: A
functional map of CAPN3 gene mutations. J Med Genet. 44:38–43.
2007. View Article : Google Scholar
|
|
67
|
Pathak P, Sharma MC, Sarkar C, Jha P, Suri
V, Mohd H, Singh S, Bhatia R and Gulati S: Limb girdle muscular
dystrophy type 2A in India: A study based on semi-quantitative
protein analysis, with clinical and histopathological correlation.
Neurol India. 58:549–554. 2010. View Article : Google Scholar
|
|
68
|
Chrobáková T, Hermanová M, Kroupová I,
Vondrácek P, Maríková T, Mazanec R, Zámecník J, Stanek J, Havlová M
and Fajkusová L: Mutations in Czech LGMD2A patients revealed by
analysis of calpain3 mRNA and their phenotypic outcome. Neuromuscul
Disord. 14:659–665. 2004. View Article : Google Scholar
|
|
69
|
Ermolova N, Kudryashova E, DiFranco M,
Vergara J, Kramerova I and Spencer MJ: Pathogenity of some limb
girdle muscular dystrophy mutations can result from reduced
anchorage to myofibrils and altered stability of calpain 3. Hum Mol
Genet. 20:3331–3345. 2011. View Article : Google Scholar
|
|
70
|
Duguez S, Bartoli M and Richard I: Calpain
3: A key regulator of the sarcomere? FEBS J. 273:3427–3436. 2006.
View Article : Google Scholar
|
|
71
|
Groen EJ, Charlton R, Barresi R, Anderson
LV, Eagle M, Hudson J, Koref MS, Straub V and Bushby KM: Analysis
of the UK diagnostic strategy for limb girdle muscular dystrophy
2A. Brain. 130:3237–3249. 2007. View Article : Google Scholar
|
|
72
|
Chiannilkulchai N, Pasturaud P, Richard I,
Auffray C and Beckmann JS: A primary expression map of the
chromosome 15q15 region containing the recessive form of
limb-girdle muscular dystrophy (LGMD2A) gene. Hum Mol Genet.
4:717–725. 1995. View Article : Google Scholar
|
|
73
|
Ojima K, Kawabata Y, Nakao H, Nakao K, Doi
N, Kitamura F, Ono Y, Hata S, Suzuki H, Kawahara H, et al: Dynamic
distribution of muscle-specific calpain in mice has a key role in
physical-stress adaptation and is impaired in muscular dystrophy. J
Clin Invest. 120:2672–2683. 2010. View Article : Google Scholar
|
|
74
|
Baghdiguian S, Richard I, Martin M,
Coopman P, Beckmann JS, Mangeat P and Lefranc G: Pathophysiology of
limb girdle muscular dystrophy type 2A: Hypothesis and new insights
into the IkappaBalpha/NF-kappaB survival pathway in skeletal
muscle. J Mol Med (Berl). 79:254–261. 2001. View Article : Google Scholar
|
|
75
|
Benayoun B, Baghdiguian S, Lajmanovich A,
Bartoli M, Daniele N, Gicquel E, Bourg N, Raynaud F, Pasquier MA,
Suel L, et al: NF-kappaB-dependent expression of the anti-
apoptotic factor c-FLIP is regulated by calpain 3, the protein
involved in limb-girdle muscular dystrophy type 2A. FASEB J.
22:1521–1529. 2008. View Article : Google Scholar
|
|
76
|
Nilsson MI, Macneil LG, Kitaoka Y, Alqarni
F, Suri R, Akhtar M, Haikalis ME, Dhaliwal P, Saeed M and
Tarnopolsky MA: Redox state and mitochondrial respiratory chain
function in skeletal muscle of LGMD2A patients. PLoS One.
9:e1025492014. View Article : Google Scholar
|
|
77
|
Yalvac ME, Amornvit J, Braganza C, Chen L,
Hussain SA, Shontz KM, Montgomery CL, Flanigan KM, Lewis S and
Sahenk Z: Impaired regeneration in calpain-3 null muscle is
associated with perturbations in mTORC1 signaling and defective
mitochondrial biogenesis. Skelet Muscle. 7:272017. View Article : Google Scholar
|
|
78
|
Gallardo E, Saenz A and Illa I:
Limb-girdle muscular dystrophy 2A. Handb Clin Neurol. 101:97–110.
2011. View Article : Google Scholar
|
|
79
|
Toral-Ojeda I, Aldanondo G, Lasa-Elga r
resta J, Lasa-Fernández H, Fernández-Torrón R, López de Munain A
and Vallejo-Illarramendi A: Calpain 3 deficiency affects SERCA
expression and function in the skeletal muscle. Expert Rev Mol Med.
18:e72016. View Article : Google Scholar
|
|
80
|
Lasa-Elgarresta J, Mosqueira-Martín L,
Naldaiz-Gastesi N, Sáenz A, López de Munain A and
Vallejo-Illarramendi A: Calcium mechanisms in limb-girdle muscular
dystrophy with CAPN3 mutations. Int J Mol Sci. 20:45482019.
View Article : Google Scholar
|
|
81
|
DiFranco M, Kramerova I, Vergara JL and
Spencer MJ: Attenuated Ca(2+) release in a mouse model of limb
girdle muscular dystrophy 2A. Skelet Muscle. 6:112016. View Article : Google Scholar
|
|
82
|
Kramerova I, Kudryashova E, Ermolova N,
Saenz A, Jaka O, López de Munain A and Spencer MJ: Impaired calcium
calmodulin kinase signaling and muscle adaptation response in the
absence of calpain 3. Hum Mol Genet. 21:3193–3204. 2012. View Article : Google Scholar
|
|
83
|
Liu J, Campagna J, John V, Damoiseaux R,
Mokhonova E, Becerra D, Meng H, McNally EM, Pyle AD, Kramerova I
and Spencer MJ: A small-molecule approach to restore A
slow-oxidative phenotype and defective CaMKIIβ signaling in limb
girdle muscular dystrophy. Cell Rep Med. 1:1001222020. View Article : Google Scholar
|
|
84
|
Laure L, Danièle N, Suel L, Marchand S,
Aubert S, Bourg N, Roudaut C, Duguez S, Bartoli M and Richard I: A
new pathway encompassing calpain 3 and its newly identified
substrate cardiac ankyrin repeat protein is involved in the
regulation of the nuclear factor-κB pathway in skeletal muscle.
FEBS J. 277:4322–4337. 2010. View Article : Google Scholar
|
|
85
|
Fanin M, Pegoraro E, Matsuda-Asada C,
Brown RH Jr and Angelini C: Calpain-3 and dysferlin protein
screening in patients with limb-girdle dystrophy and myopathy.
Neurology. 56:660–665. 2001. View Article : Google Scholar
|
|
86
|
Huang Y, de Morrée A, van Remoortere A,
Bushby K, Frants RR, den Dunnen JT and van der Maarel SM: Calpain 3
is a modulator of the dysferlin protein complex in skeletal muscle.
Hum Mol Genet. 17:1855–1866. 2008. View Article : Google Scholar
|
|
87
|
Anderson LV, Harrison RM, Pogue R,
Vafiadaki E, Pollitt C, Davison K, Moss JA, Keers S, Pyle A, Shaw
PJ, et al: Secondary reduction in calpain 3 expression in patients
with limb girdle muscular dystrophy type 2B and Miyoshi myopathy
(primary dysferlinopathies). Neuromuscul Disord. 10:553–559. 2000.
View Article : Google Scholar
|
|
88
|
Haravuori H, Vihola A, Straub V, Auranen
M, Richard I, Marchand S, Voit T, Labeit S, Somer H, Peltonen L, et
al: Secondary calpain3 deficiency in 2q-linked muscular dystrophy:
Titin is the candidate gene. Neurology. 56:869–877. 2001.
View Article : Google Scholar
|
|
89
|
Charton K, Sarparanta J, Vihola A, Milic
A, Jonson PH, Suel L, Luque H, Boumela I, Richard I and Udd B:
CAPN3-mediated processing of C-terminal titin replaced by
pathological cleavage in titinopathy. Hum Mol Genet. 24:3718–3731.
2015. View Article : Google Scholar
|
|
90
|
Charton K, Danièle N, Vihola A, Roudaut C,
Gicquel E, Monjaret F, Tarrade A, Sarparanta J, Udd B and Richard
I: Removal of the calpain 3 protease reverses the myopathology in a
mouse model for titinopathies. Hum Mol Genet. 19:4608–4624. 2010.
View Article : Google Scholar
|
|
91
|
Persiconi I, Cosmi F, Guadagno NA, Lupo G
and De Stefano ME: Dystrophin is required for the proper timing in
retinal histogenesis: A thorough investigation on the mdx mouse
model of duchenne muscular dystrophy. Front Neurosci. 14:7602020.
View Article : Google Scholar
|
|
92
|
Pastorello E, Cao M and Trevisan CP:
Atypical onset in a series of 122 cases with facioscapulohumeral
muscular dystrophy. Clin Neurol Neurosurg. 114:230–234. 2012.
View Article : Google Scholar
|
|
93
|
Sacconi S, Camaño P, de Greef JC, Lemmers
RJ, Salviati L, Boileau P, Lopez de Munain Arregui A, van der
Maarel SM and Desnuelle C: Patients with a phenotype consistent
with facioscapulohumeral muscular dystrophy display genetic and
epigenetic heterogeneity. J Med Genet. 49:41–46. 2012. View Article : Google Scholar
|
|
94
|
Pistoni M, Shiue L, Cline MS, Bortolanza
S, Neguembor MV, Xynos A, Ares M Jr and Gabellini D: Rbfox1
downregulation and altered calpain 3 splicing by FRG1 in a mouse
model of Facioscapulohumeral muscular dystrophy (FSHD). PLoS Genet.
9:e10031862013. View Article : Google Scholar
|
|
95
|
Paco S, Ferrer I, Jou C, Cusí V, Corbera
J, Torner F, Gualandi F, Sabatelli P, Orozco A, Gómez-Foix AM, et
al: Muscle fiber atrophy and regeneration coexist in collagen
VI-deficient human muscle: Role of calpain-3 and nuclear factor-κB
signaling. J Neuropathol Exp Neurol. 71:894–906. 2012. View Article : Google Scholar
|
|
96
|
Krahn M, Lopez de Munain A,
Streichenberger N, Bernard R, Pécheux C, Testard H, Pena-Segura JL,
Yoldi E, Cabello A, Romero NB, et al: CAPN3 mutations in patients
with idiopathic eosinophilic myositis. Ann Neurol. 59:905–911.
2006. View Article : Google Scholar
|
|
97
|
Parker KC, Kong SW, Walsh RJ, Bch
Salajegheh M, Moghadaszadeh B, Amato AA, Nazareno R, Lin YY,
Krastins B, et al Fast-twitch sarcomeric and glycolytic enzyme
protein loss in inclusion body myositis. Muscle Nerve. 39:739–753.
2009. View Article : Google Scholar
|
|
98
|
Amici DR, Pinal-Fernandez I, Mázala DA,
Lloyd TE, Corse AM, Christopher-Stine L, Mammen AL and Chin ER:
Calcium dysregulation, functional calpainopathy, and endoplasmic
reticulum stress in sporadic inclusion body myositis. Acta
Neuropathol Commun. 5:242017. View Article : Google Scholar
|
|
99
|
Musumeci O, Aguennouz M, Cagliani R, Comi
GP, Ciranni A, Rodolico C, Messina C, Vita G and Toscano A: Calpain
3 deficiency in Quail Eater's disease. Ann Neurol. 55:146–147.
2004. View Article : Google Scholar
|
|
100
|
Kruijt N, van den Bersselaar LR, Kamsteeg
EJ, Verbeeck W, Snoeck MMJ, Everaerd DS, Abdo WF, Jansen DRM,
Erasmus CE, Jungbluth H and Voermans NC: The etiology of
rhabdomyolysis: An interaction between genetic susceptibility and
external triggers. Eur J Neurol. 28:647–659. 2021. View Article : Google Scholar
|
|
101
|
Weeraratna AT, Becker D, Carr KM, Duray
PH, Rosenblatt KP, Yang S, Chen Y, Bittner M, Strausberg RL,
Riggins GJ, et al: Generation and analysis of melanoma SAGE
libraries: SAGE advice on the melanoma transcriptome. Oncogene.
23:2264–2274. 2004. View Article : Google Scholar
|
|
102
|
Huynh KM, Kim G, Kim DJ, Yang SJ, Park SM,
Yeom YI, Fisher PB and Kang D: Gene expression analysis of terminal
differentiation of human melanoma cells highlights global
reductions in cell cycle-associated genes. Gene. 433:32–39. 2009.
View Article : Google Scholar
|
|
103
|
Moretti D, Del Bello B, Cosci E, Biagioli
M, Miracco C and Maellaro E: Novel variants of muscle calpain 3
identified in human melanoma cells: Cisplatin-induced changes in
vitro and differential expression in melanocytic lesions.
Carcinogenesis. 30:960–967. 2009. View Article : Google Scholar
|
|
104
|
Moretti D, Del Bello B, Allavena G, Corti
A, Signorini C and Maellaro E: Calpain-3 impairs cell proliferation
and stimulates oxidative stress-mediated cell death in melanoma
cells. PLoS One. 10:e01172582015. View Article : Google Scholar
|
|
105
|
Rambow F, Job B, Petit V, Gesbert F,
Delmas V, Seberg H, Meurice G, Van Otterloo E, Dessen P, Robert C,
et al: New functional signatures for understanding melanoma biology
from tumor cell lineage-specific analysis. Cell Rep. 13:840–853.
2015. View Article : Google Scholar
|
|
106
|
Ruffini F, Tentori L, Dorio AS, Arcelli D,
D'Amati G, D'Atri S, Graziani G and Lacal PM: Platelet-derived
growth factor C and calpain-3 are modulators of human melanoma cell
invasiveness. Oncol Rep. 30:2887–2896. 2013. View Article : Google Scholar
|
|
107
|
Pizzanelli C, Mancuso M, Galli R, Choub A,
Fanin M, Nascimbeni AC, Siciliano G and Murri L: Epilepsy and limb
girdle muscular dystrophy type 2A: Double trouble, serendipitous
finding or new phenotype? Neurol Sci. 27:134–136. 2006. View Article : Google Scholar
|
|
108
|
Viloria-Alebesque A, Bellosta-Diago E,
Santos-Lasaosa S and Mauri-Llerda JÁ: Familial association of
genetic generalised epilepsy with limb-girdle muscular dystrophy
through a mutation in CAPN3. Epilepsy Behav Case Rep. 11:122–124.
2019. View Article : Google Scholar
|
|
109
|
Sanchez-Mut JV, Aso E, Panayotis N, Lott
I, Dierssen M, Rabano A, Urdinguio RG, Fernandez AF, Astudillo A,
Martin-Subero JI, et al: DNA methylation map of mouse and human
brain identifies target genes in Alzheimer's disease. Brain.
136:3018–3027. 2013. View Article : Google Scholar
|
|
110
|
De Jager P, Srivastava G, Lunnon K,
Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe
C, et al: Alzheimer's disease: Early alterations in brain DNA
methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci.
17:1156–1163. 2014. View Article : Google Scholar
|
|
111
|
Ma D, Fetahu IS, Wang M, Fang R, Li J, Liu
H, Gramyk T, Iwanicki I, Gu S, Xu W, et al: The fusiform gyrus
exhibits an epigenetic signature for Alzheimer's disease. Clin
Epigenetics. 12:1292020. View Article : Google Scholar
|
|
112
|
Walder K, McMillan J, Lapsys N, Kriketos
A, Trevaskis J, Civitarese A, Southon A, Zimmet P and Collier G:
Calpain 3 gene expression in skeletal muscle is associated with
body fat content and measures of insulin resistance. Int J Obes
Relat Metab Disord. 26:442–449. 2002. View Article : Google Scholar
|
|
113
|
Norton L, Parr T, Bardsley RG, Ye H and
Tsintzas K: Characterization of GLUT4 and calpain expression in
healthy human skeletal muscle during fasting and refeeding. Acta
Physiol (Oxf). 189:233–240. 2007. View Article : Google Scholar
|
|
114
|
Varga TV, Kurbasic A, Aine M, Eriksson P,
Ali A, Hindy G, Gustafsson S, Luan J, Shungin D, Chen Y, et al:
Novel genetic loci associated with long-term deterioration in blood
lipid concentrations and coronary artery disease in European
adults. Int J Epidemiol. 46:1211–1222. 2017.
|
|
115
|
Guo T, Yin RX, Pan L, Yang S, Miao L and
Huang F: Integrative variants, haplotypes and diplotypes of the
CAPN3 and FRMD5 genes and several environmental exposures associate
with serum lipid variables. Sci Rep. 7:451192017. View Article : Google Scholar
|
|
116
|
Salinas-Santander M, Trevino V, De la
Rosa-Moreno E, Verduzco-Garza B, Sánchez-Domínguez CN,
Cantú-Salinas C, Ocampo-Garza J, Lagos-Rodríguez A, Ocampo-Candiani
J and Ortiz-López R: CAPN3, DCT, MLANA and TYRP1 are overexpressed
in skin of vitiligo vulgaris Mexican patients. Exp Ther Med.
15:2804–2811. 2018.
|
|
117
|
Tang Y, Liu X, Zoltoski RK, Novak LA,
Herrera RA, Richard I, Kuszak JR and Kumar NM: Age-related
cataracts in alpha3Cx46-knockout mice are dependent on a calpain 3
isoform. Invest Ophthalmol Vis Sci. 48:2685–2694. 2007. View Article : Google Scholar
|
|
118
|
Bartoli M, Roudaut C, Martin S,
Fougerousse F, Suel L, Poupiot J, Gicquel E, Noulet F, Danos O and
Richard I: Safety and efficacy of AAV-mediated calpain 3 gene
transfer in a mouse model of limb-girdle muscular dystrophy type
2A. Mol Ther. 13:250–259. 2006. View Article : Google Scholar
|
|
119
|
Selvaraj S, Dhoke NR, Kiley J,
Mateos-Aierdi AJ, Tungtur S, Mondragon-Gonzalez R, Killeen G,
Oliveira VKP, López de Munain A and Perlingeiro RCR: Gene
correction of LGMD2A patient-specific iPSCs for the development of
targeted autologous cell therapy. Mol Ther. 27:2147–2157. 2019.
View Article : Google Scholar
|














