Introduction
Plastic products are a newly discovered type of
environmental pollutant. They are lightweight, waterproof, and can
be reused. The technology for producing plastics is mature and the
cost is very low. Their use is ubiquitous around the world, with an
increasing trend. However, they do not degrade well under normal
environmental conditions and thus serve as pollutants in numerous
ecological environments (1,2).
Only 21-26% of plastic waste is properly recycled and incinerated.
The remaining plastic waste is incinerated in open air or discarded
into the environment, leading to water, air, and soil being
polluted by plastic waste (1-3).
Reactions between plastic waste and environmental
particulates/matter result in breakdown of plastic into smaller
pieces of plastic, termed microplastics (MPs) (4-6).
Nanoplastics (NPs) with a diameter of 1-100 nm are formed by
degradation of MPs (diameter, 5 mm) (7) by weathering, solar radiation and
biodegradation (8). MPs have been
reported in foods such as mussels (9), commercial fish (10), sugar (11) and bottled water (12); thus the primary means of entry of
MPs into the human body is likely ingestion.
MPs/NPs reach organs and tissues in the body after
exposure. The liver is one of the first organs to encounter
environmentally harmful substances (such as heavy metals,
biotoxins, chemical residues or particulate matter) absorbed
through the intestine and is subjected to multiple biochemical
stresses such as inflammation and virus (13). The liver also harbors the most
abundant macrophage pool, consisting of resident (Kupffer cells)
and infiltrating bone marrow-derived macrophages (14). Macrophages are the second largest
proportion of cells in the liver and they play key roles in the
development of liver injury (15). Recent research has shown that MPs
cause macrophage recruitment in the liver, and the macrophage
infiltration rate in livers exposed to 1-10 µm MPs is
greater than the large-diameter MPs, these data suggest that
macrophage-driven mechanisms of action potentially play a key role
in PS-MP-induced liver fibrosis (16). Zebrafish exposed to high
concentrations of NPs exhibit notable changes in locomotor
activity, aggression, school formation and predatory avoidance
behaviors, as well as circadian dyskinesia following prolonged
exposure. These changes highlight the potential neurotoxic effects
of NP exposure (17). Expression
of neuronal activity-dependent genes (cFos and Egr1) and synaptic
proteins such as activity-regulated cytoskeleton-associated
protein/activity-regulated gene 3.1, in mice exposed to MPs is
altered, and neuroinflammation in the hippocampus is increased,
followed by behavioral changes via a vagus-dependent pathway
(18). The aforementioned studies
indicated that the nervous system is a potential target organ for
MP exposure. MPs pass through the blood-brain barrier (BBB),
activate microglia and alter neurotransmitter release; however, the
mechanism of microglial cytotoxicity caused by MPs requires further
study.
Inflammation is a defensive response to remove
infections and other toxic irritants, but inflammation can also
cause significant damage and is hypothesized to be the cause of
numerous types of disease such as atherosclerosis (19). Intracellular innate immune
receptor NLRP3, adapter apoptosis-associated speck-like protein
(ASC) and protease caspase-1 form a multi-protein complex called
the NLRP3 inflammasome, which identifies pathogen- and
damage-associated molecular patterns and collects and activates the
inflammatory protease caspase-1 (20). Following long-term exposure to
MPs/NPs, intracellular reactive oxygen species (ROS) production by
mitochondria is increased, oxidative stress reactions occur and the
NLRP3/caspase-1 signal pathway is activated, resulting in granulosa
cell apoptosis and ovarian reserve reduction (21,22).
ROS serve as intracellular signaling molecules,
mediating inflammation, apoptosis and pyroptosis in multiple types
of cell (23). Excess iron
promotes subsequent lipid peroxidation through the production of
ROS via the iron-dependent Fenton reaction. The molecular mechanism
of ferroptosis involves regulating the balance between oxidative
damage and antioxidant defense (24). In addition, ROS are key signaling
molecules that serve an important role in the progression of
inflammatory disorders. Mitochondrial ROS signaling is a primary
regulator of NLRP3 inflammasome activation (25). Thus, ROS serve key roles in the
occurrence of ferroptosis and inflammatory reactions.
Ferroptosis is a special type of non-apoptotic
regulatory cell death mechanism that is characterized by oxidative
modification of phospholipid membranes via iron-dependent
mechanisms, resulting in increased lipid peroxidation downstream of
metabolic dysfunction (26,27). Biochemically, intracellular
glutathione (GSH) is depleted, activity of GSH peroxidase 4 (GPX4)
is reduced and the lipid peroxide cannot be metabolized by the
reduction reaction catalyzed by GPX4. Fe2+ oxidizes
lipids via the Fenton reaction, producing a large number of ROS and
promoting ferroptosis (28,29).
JNK was discovered nearly 35 years ago and was
originally discovered as a member of the pp54
microtubule-associated protein-2 kinase family (30,31). Combined exposure of alumina
nanoparticles and chronic restraint stress compound hippocampal
neuronal ferroptosis and activate the interferon-γ/apoptosis
signal-regulating kinase 1/JNK signaling pathway, leading to
learning and memory dysfunction in rats (32). It is not clear whether NPs, which
are also nano particles, activate JNK and induce ferroptosis of
microglia cells and the mechanism by which JNK is involved in
ferroptosis remains unclear. Heme oxygenase (HO-1) is a key
metabolic enzyme and is considered the central effector of
mammalian stress responses (33,34). It has been hypothesized that HO-1
plays a regulatory role in apoptosis and autophagy, as well as in
pyroptosis, necroptosis and ferroptosis (35). Notably, HO-1 is a key mediator of
ferroptosis induced by erastin in cancer cells; Zinc protoporphyrin
IX, an HO-1 inhibitor, prevents ferroptosis induced by erastin
(36).
To understand the effects of NPs on human health and
provide a basis for toxicological experiments, the present study
investigated the molecular mechanisms of NP-induced microglial cell
toxicity. An experimental in vitro model was established in
which BV2 cells were exposed to NPs to induce an inflammatory
reaction. N-acetylcysteine (NAC) was also used to evaluate the
pivotal role of ROS in the inflammatory response and ferroptosis.
Finally, we explored the toxicity and its mechanism of microglia
induced by NP exposure.
Materials and methods
Characterization of polystyrene
(PS-)NPs
Dragon Green-labeled NPs, which are the primary
components of PS, were purchased from Bangs Laboratories, Inc.
(cat. no. FSDG001). The mean diameter of PS-NPs was 44 nm and the
solid content was 1%. The number of microspheres was
~2.456×1014/ml deionized water. The size and shape of
PS-NPs were detected by transmission electron microscopy (TEM; FEI
Talos F200x). The sample was dispersed into an ethanol solution for
ultrasound and then a few drops of the dispersed liquid were added
onto a copper mesh. After drying, the sample was subjected to an
accelerated voltage of 200 kV using FEI Talos F200X G2, EDS
super-X, and the morphology was captured (high-resolution).
Cell culture and treatment
The mouse microglial cell line BV2 was obtained from
Kunming Cell Bank of the Chinese Academy of Sciences (Kunming,
China). BV2 cells were resuspended and cultured in DMEM (cat. no.
C11995500BT) supplemented with 10% FBS (cat. no. 10099141; both
Gibco; Thermo Fisher Scientific, Inc.) and 1%
Antibiotic-Antimycotic (cat. no. 15240062; Gibco; Thermo Fisher
Scientific, Inc.) and maintained in a humidified incubator supplied
with 5% CO2 at 37°C. Cells were exposed to 0, 25, 50 or
100 µg/ml NPs for 12 h or 24 h at 37°C. For inhibitor
pretreatment groups, the ROS inhibitor NAC (10 µM; cat. no.
S1623; Selleck) and JNK inhibitor SP600125 (2 µM; cat. no.
S1460; Selleck) were added to cells for 2 and 1 h at 37°C,
respectively, and then BV2 cells were exposed to 50 µg/ml
NPs for 12 h or 24 h at 37°C. The nucleus of BV2 cells stained with
DAPI (10 µg/ml; cat. no. C0065; Beijing Solarbio) for 5 h at
room temperature.
Cell viability assay
Cell viability was determined using Cell Counting
Kit-8 (CCK-8; cat. no. CK04; Dojindo Molecular Technologies, Inc.).
NPs (10 mg/ml) were diluted to 0, 25, 50 and 100 µg/ml with
serum-free DMEM and added to cells for 12 or 24 h at 37°C.
Subsequently, 10 µl CCK-8 solution was added for 2 h and the
absorbance was measured at 450 nm using a microplate reader
(SpectraMax 190; Molecular Devices, Inc.).
Determination of intracellular ROS
concentration
BV2 cells were seeded with 3×105 cells
per well in six-well plates and exposed to NPs, as aforementioned,
during the logarithmic growth phase. After 12 h at 37°C, cells were
incubated in DMEM containing 5 µM BODIPY™ 581/591 C11 probe
for 30 min at 37°C (cat. no. D3861, Thermo Fisher Scientific, Inc.)
Cells were digested with Triple-E (Gibco; Thermo Fisher Scientific,
Inc.) for 2 min at 37°C. The cell precipitate was collected by
centrifugation at 4°C, 800 × g for 4 min and washed twice with PBS
(Biological Industries; Sartorius AG). The cells were collected
after being digested and blow down with a pipette, precipitated by
centrifugation at 4°C, 800 × g for 4 min and resuspended in 200
µl PBS. FITC in each group was measured using a flow
cytometer (DxFLEX; Beckman Coulter, Inc.) with an excitation
wavelength of 488 nm and emission wavelength of 510 nm. Annexin-V
labeled with fluorescein FITC was used as a probe. And the software
CytExpert for DxFLEX (version 2.0.0.283; Beckman Coulter, Inc.)
used for analysis.
Determination of oxidative damage
BV2 cells in the logarithmic growth phase were used.
Following exposure of cells to 50 µg/ml NPs for 12 h at
37°C, cells were collected and the protein concentration was
determined using a BCA protein assay kit. Malondialdehyde (MDA)
levels (cat. no. S0131S; Beyotime Institute of Biotechnology) and
activity of superoxide dismutase (SOD; cat. no. A001-3-2; Nanjing
Jiancheng Bioengineering Institute) and GSH were detected using
kits (cat. no. A006-2-1; Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocols.
Inflammatory cytokine analysis
ELISA was performed to analyze inflammatory
cytokines. Cell culture medium was collected and centrifuged for
~10 min (300 × g, 4°C) and the supernatant was carefully collected.
The levels of 1L-1β and IL-18 in supernatant of each sample were
measured using corresponding mouse ELISA kits [cat. nos.
EK201B/3-96 and EK218-96, respectively; both Hangzhou Lianke
Biotechnology Co., Ltd.), according to the manufacturer's
protocols.
Western blot analysis
RIPA lysis buffer (cat. no. P0013B) containing PMSF
(cat. No. ST505; both Beyotime Institute of Biotechnology) was used
to lyse BV2 cells treated with 0, 25, 50 and 100 µg/ml NPs
for 12 h at 37°C, on ice for 30 min and centrifuged at 15,000 × g
for 15 min at 4°C. The supernatant was obtained and the total
protein concentration was determined using a BCA kit (Beyotime
Institute of Biotechnology) and adjusted to the same quantity
(44-46 µg) with PBS. The protein (44-46 µg/lane; 20
µl) was loaded on a 10% SDS gel, resolved using SDS-PAGE and
transferred to PVDF membranes (MilliporeSigma). The membranes were
blocked with blocking buffer (containing 5% milk powder) for 1 h at
room temperature, washed three times with PBST (containing 0.1%
Tween-20) and incubated with primary antibodies at 4°C for 12 h.
The following day, the membrane was incubated with
fluorescent-labeled goat anti-rabbit or goat anti-mouse antibody
for 1 h at 37°C. Blots were visualized with Odyssey® CLx Imaging
System (cat. no. 9140-00, LI-COR Biosciences). The gray values of
the protein bands were analyzed using ImageLab™ Software (version
no. 1.53 e). The primary antibodies were as follows: β-actin
(1:1,000, cat. no. 4970S, Cell Signaling Technology, Inc.), NLRP3
(1:1,000, cat. no. ab263899, Abcam), cyclooxygenase (COX)-2
(1:1,000, cat. no. 12282, Cell Signaling Technology, Inc.), pro
caspase-1 + p10 + p12 (1:1,000, cat. no. ab179515, Abcam), ferritin
heavy chain 1 (FTH1) (1:1,000, cat. no. 3998, Cell Signaling
Technology, Inc.), HO-1 (1:1,000, cat. no. 43966, Cell Signaling
Technology, Inc.), ASC/Target of methylation-induced silencing
(TMS1) (1:1,000, cat. no. 67824, Cell Signaling Technology, Inc.),
GPX4 (1:1,000, cat. no. ab125066, Abcam), Acyl-CoA synthetase
long-chain family member 4 (FACL4) (1:1,000, cat. no. ab155282;
Abcam), Solute carrier family 7 member 11 (XCT) (1:1,000, cat. no.
ab175186; Abcam), transferrin receptor (TFRC; 1:5,000, cat. no.
ab269513, Abcam), JNK1 + JNK2 + JNK3 (1:1,000, cat. no. ab179461,
Abcam) and JNK1 + JNK2 + JNK3 [phosphorylated (p)-T183 + T183 +
T221; 1:5,000, cat. no. ab124956, Abcam]. The secondary anti-bodies
were: IRDye® 680RD Goat anti-rabbit IgG (1:10,000, cat.
no. 926-68071, LI-COR Biosciences) and IRDye® 800CW goat
anti-mouse IgG (1:10,000, cat. no. 926-32210, LI-COR Biosciences).
β-actin was used as an internal loading control for
normalization.
Cellular iron level detection
BV2 cells were seeded in black 96-well plates at a
density of 5×104 cells/well and treated with NPs for 12
h, as aforementioned. The cells were incubated in 1X Hoechst
(Beyotime Institute of Biotechnology) for 10 min at 37°C, then with
1 µM FerroOrange (Dojindo Molecular Technologies, Inc.) for
30 min at 37°C and the iron concentration in the cytoplasm was
observed using a fluorescence microscope (×200).
Statistical analysis
Data were analyzed using GraphPad Prism version
9.0.0 (GraphPad Software, Inc.; Dotmatics, Inc.). Data were
analyzed using ANOVA (one-way) followed by Dunnett's post hoc test.
Data are presented as the mean ± SD of three independent repeats.
P<0.05 was considered to indicate a statistically significant
difference.
Results
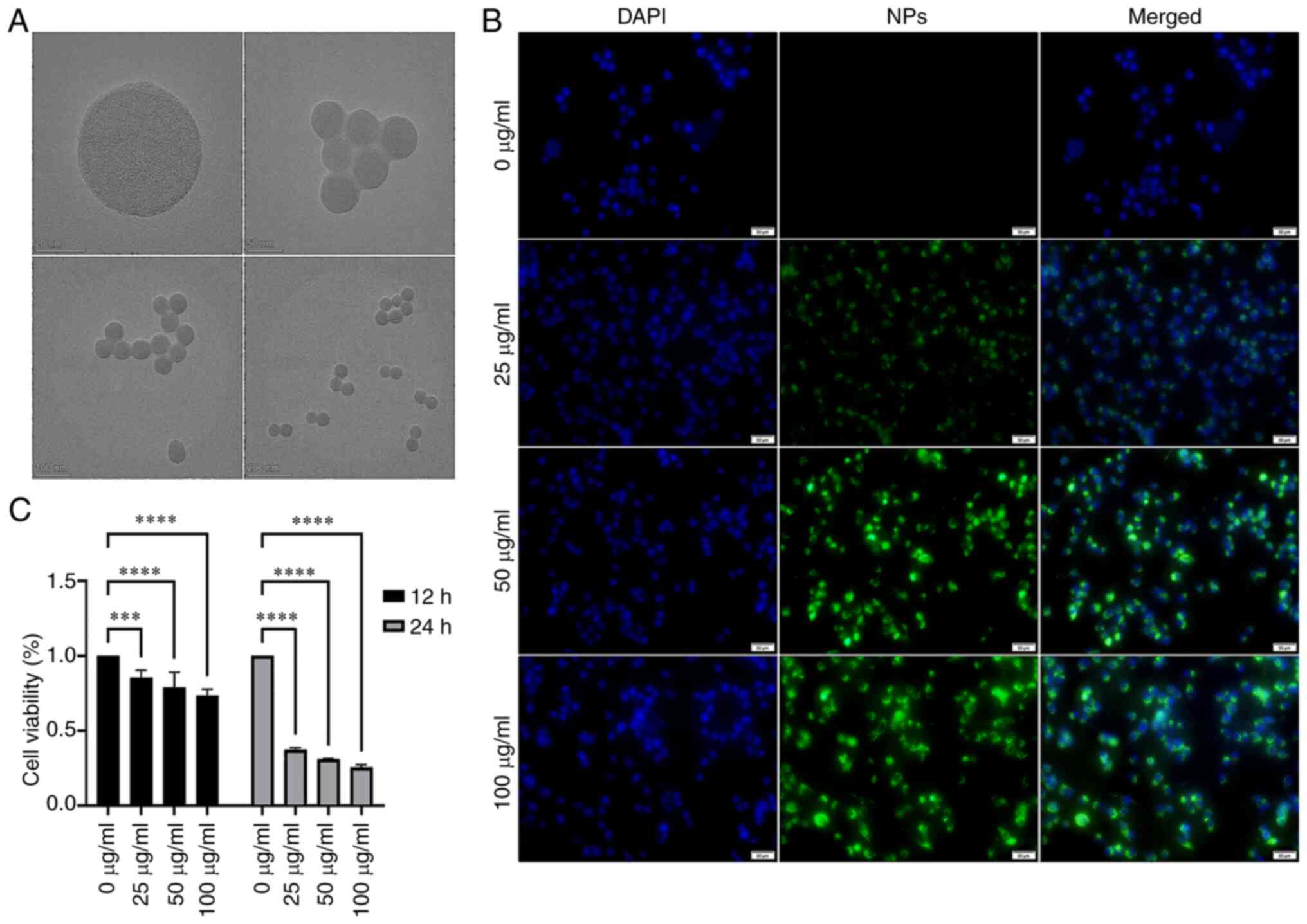
Cytotoxicity of NPs to BV2 cells
The mean diameter of NPs was ~50 nm when single or
multiple fluorescent microspheres were observed by TEM (Fig. 1A). BV2 cells exhibited green
fluorescence following phagocytosis, and the intensity of
fluorescence increased in a dose-dependent manner (Fig. 1B). This suggested that NPs were
phagocytosed by microglia. The viability of BV2 cells after treated
with NPs 12 h decreased obviously. The viability of BV2 cells
exposed to 25, 50, 100 µg/ml NPs were lower than 0
µg/ml. After exposed to NPs 24 h, the viability of BV2 cells
exposed to 25, 50, 100 µg/ml NPs substantially reduced and
this effect was dose-and time-dependent (Fig. 1C).
NPs induce oxidative damage in BV2
cells
To investigate whether NPs cause oxidative damage to
BV2 cells, FITC following exposure to NPs for 12 h was measured.
FITC in BV2 cells exposed to 100 µg/ml NPs increased
significantly (Fig. 2A). MDA
content increased 1.5x at 100 µg/ml compared with the
untreated control group and was also notably higher than that of
cells treated with lower concentrations of NPs (Fig. 2C). The activity of GSH (Fig. 2D) and SOD (Fig. 2B) decreased as the NP
concentration increased. The activity of GSH and SOD decreased 1.6x
and 1.2x, respectively, at 100 µg/ml NP compared with the
control group. To confirm the impact of lipid oxidation on cell
death, ROS inhibitor NAC was used. Compared with the 50
µg/ml NP group, the content of FITC (Fig. 2E) and MDA (Fig. 2G) in cells co-treated with NPs and
NAC was significantly decreased. However, compared with the 50
µg/ml NP group, the activity of GSH (Fig. 2H) and SOD (Fig. 2F) in cells co-treated with NPs and
NAC increased significantly.
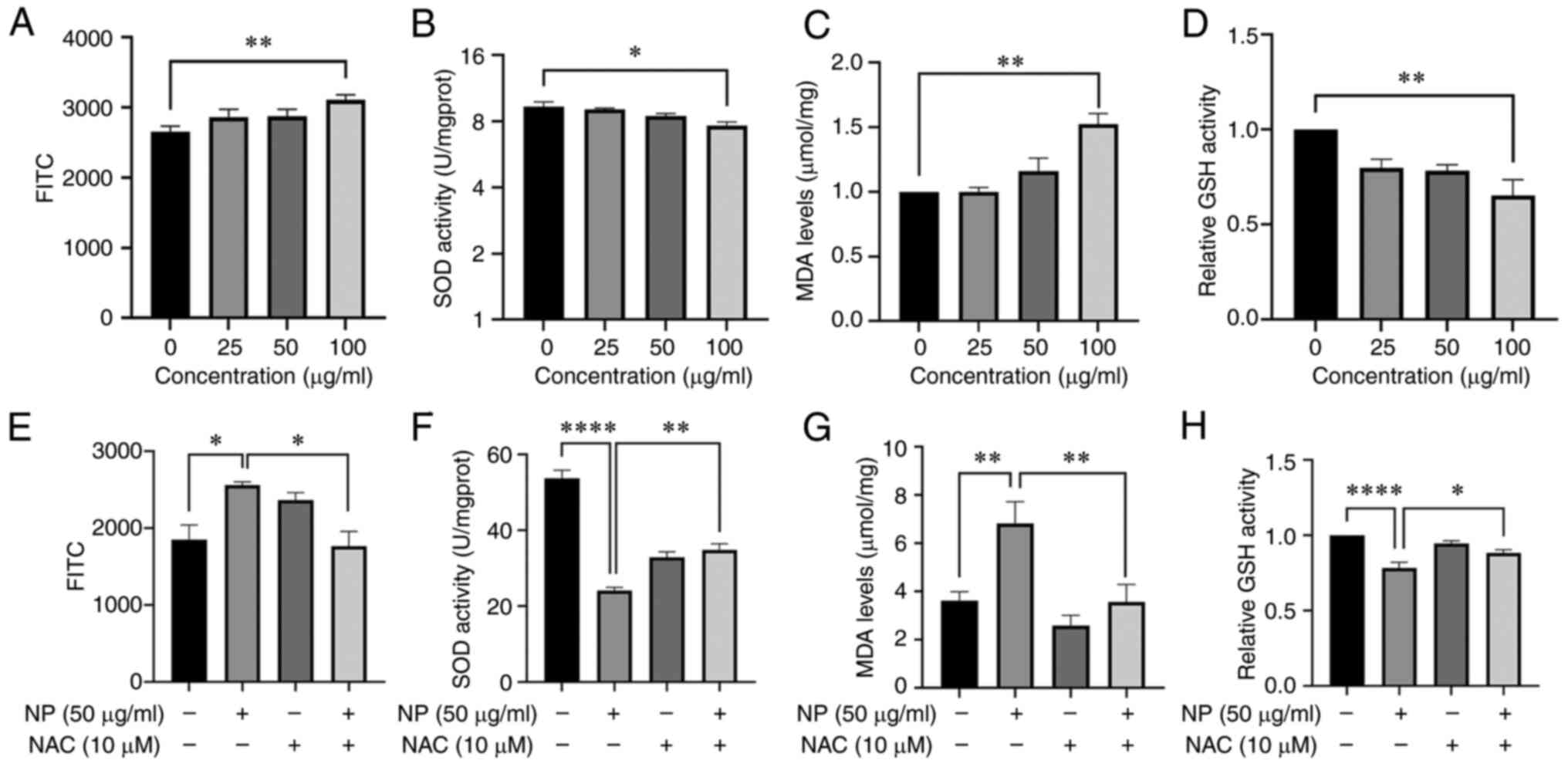 | Figure 2NP induces oxidative stress in BV2
cells. Following treatment with NPs, (A) FITC and (C) MDA levels
increased in a dose-dependent manner, whereas (B) SOD and (D) GSH
activity decreased. (E) FITC, (F) SOD, (G) MDA and (H) GSH in BV2
cells pretreated with 10 µM NAC followed by treatment with
50 µg/ml NP for 12 h. ROS, reactive oxygen species; MDA,
malondialdehyde; SOD, superoxide dismutase; GSH, glutathione; NAC,
N-acetylcysteine; NP, nanoplastic. *P<0.05,
**P<0.01, ****P<0.0001. |
NPs induce ferroptosis in BV2 cells
To verify whether NPs induced ferroptosis in BV2
cells, expression of ferroptosis-related proteins, including lipid
peroxidation-associated indicators GPX4 and ACSL4 and XCT, was
assessed. The protein levels of GPX4 and XCT in BV2 cells were
downregulated by NPs (Fig. 3C and
H) and protein expression of ACSL4 was upregulated (Fig. 3F). These results suggested that
NPs stimulated lipid peroxidation in BV2 cells. The expression of
TFRC and FTH1, which are responsible for ferritin transport
(14), was also significantly
altered; NPs downregulated the expression of FTH1 protein, while
the levels of TFRC were increased (Fig. 3B and D). Using fluorescence
microscopy, the mean fluorescence intensity of intracellular ions
was shown to increase in a dose-dependent manner (Fig. 3E and I). Expression of COX-2
(Fig. 3G) also increased in a
concentration-dependent manner.
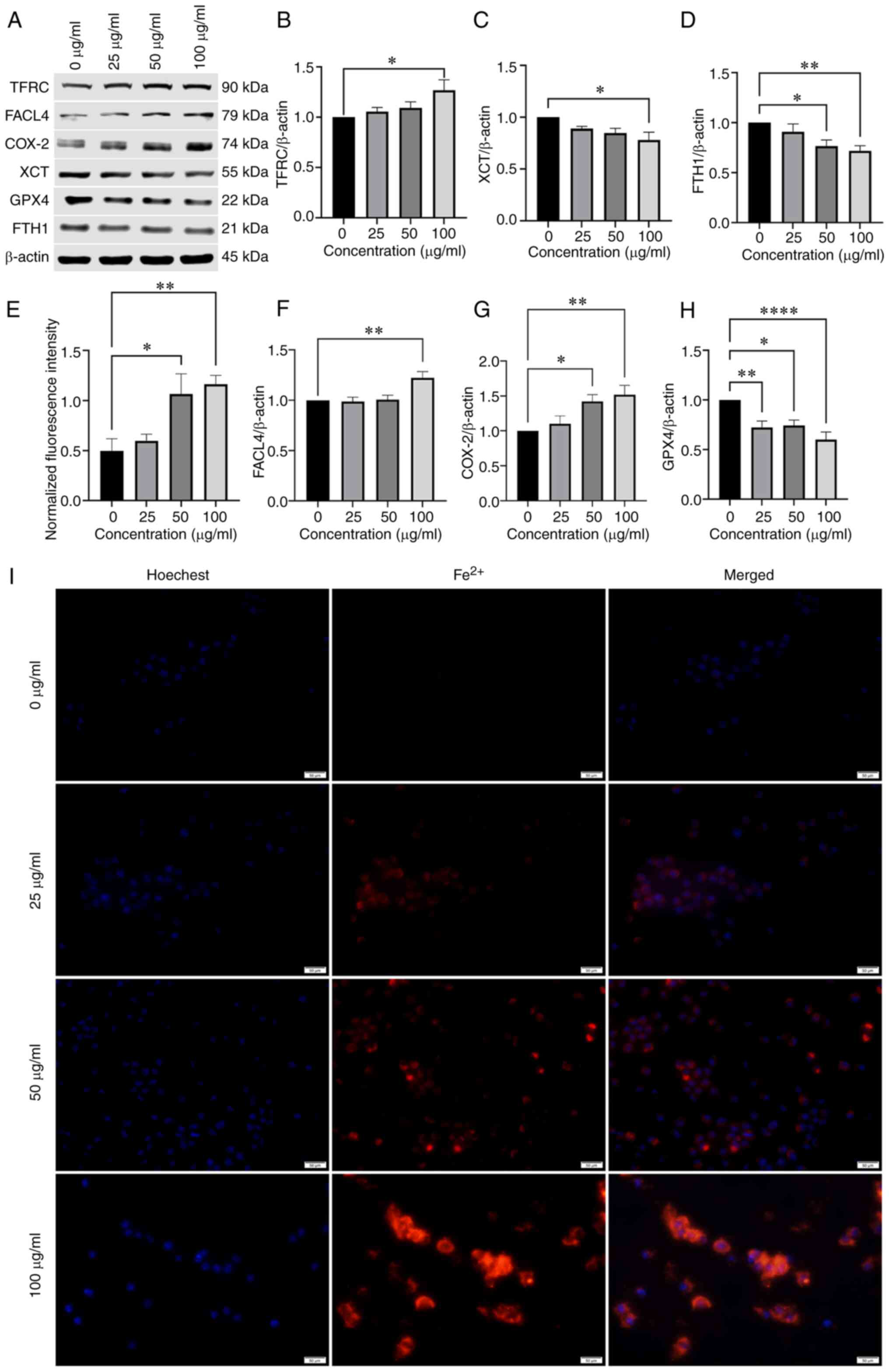 | Figure 3NP induces ferroptosis in BV2 cells.
(A) Western blot analysis of ferroptosis-associated protein
expression in BV2 cells treated with 0, 25, 50, or 100 µg/ml
NPs for 12 h. (B) TFRC, (C) XCT, (D) FTH1, (E) Fluorescence
intensity of Fe2+, (F) FACL4, (G) COX-2, (H) GPX4, (I)
Effect of NPs on Fe2+ content in BV2 cells using
fluorescence microscope. NP, nanoplastic; TFRC, transferrin
receptor; FACL4, acyl-CoA synthetase long-chain family member 4;
COX-2, cyclooxygenase-2; XCT, solute carrier family 7 member 11;
GPX4, glutathione peroxidase; FTH1, ferritin heavy chain 1.
*P<0.05, **P<0.01,
****P<0.0001. |
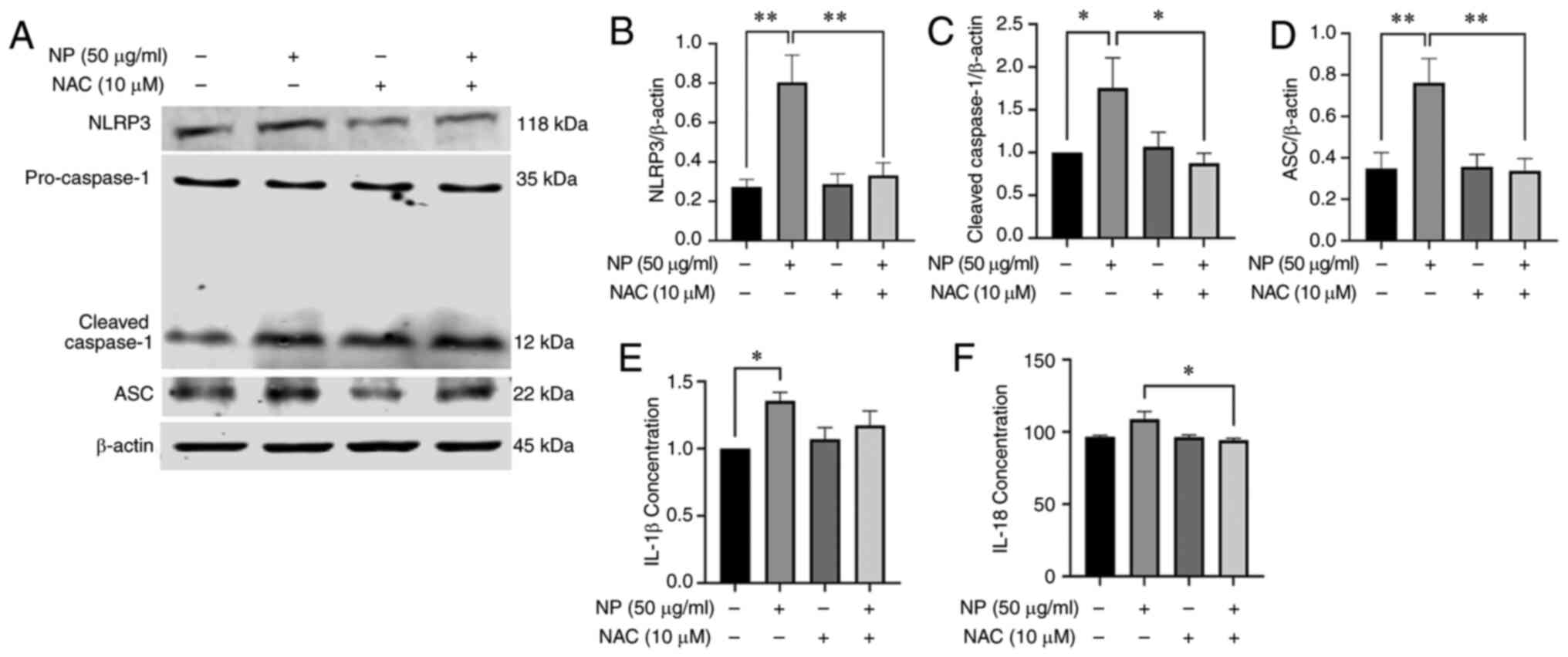
NPs induce release of inflammatory
factors and activate JNK, HO-1 in BV2 cells
The expression of inflammatory response-associated
proteins in BV2 cells was determined using western blotting. The
expression of NLRP3 and ASC in BV2 cells treated with 100
µg/ml NPs was significantly higher than in the control group
(Fig. 4B and D). The ratio of
cleaved-caspase-1 to pro-caspase-1 protein increased as the dose of
NP increased, with 100 µg/ml NP increasing the ratio
significantly compared with the control group (Fig. 4C). The expression levels of IL-1β
increased evidently in cells treated with NPs compared with the
control in a dose-dependent manner (Fig. 4E). To explore the impact of JNK
and HO-1 in NP-induced cytotoxicity, expression of p-JNK, JNK and
HO-1 was detected using western blotting. Phosphorylation of JNK
increased following treatment of NPs and the increase in p-JNK and
HO-1 levels was dose-dependent (Fig.
4H and I). These results suggested that NPS induced activation
of JNK and HO-1.
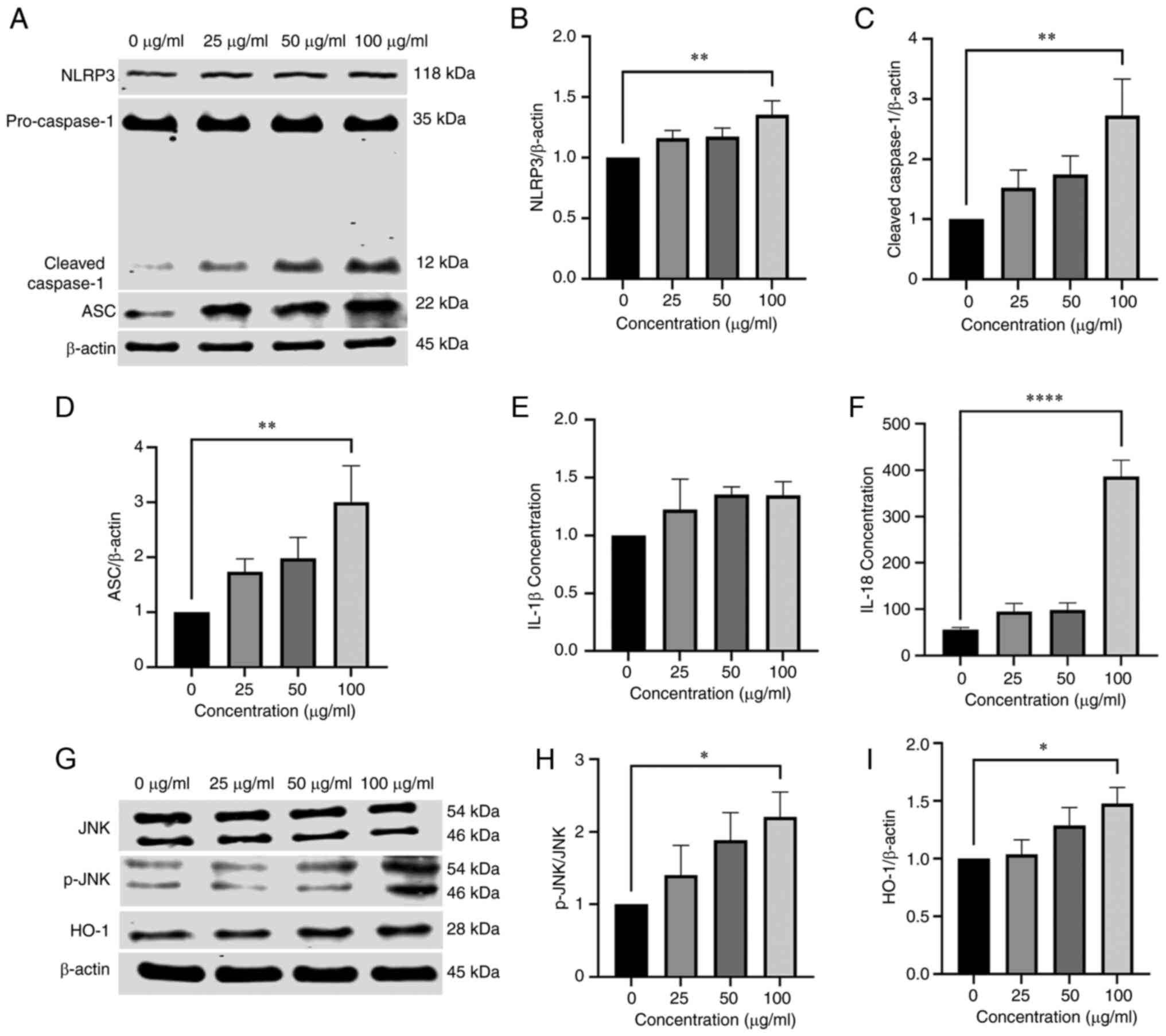 | Figure 4NPs cause inflammation and activate
JNK and HO-1 in BV2 cells. (A) Detection of (B) NLRP3, (C)
cleaved-caspase-1 and (D) ASC expression in BV2 cells by western
blotting. Detection of IL-18 and (E) IL-1β and (F) IL-18 levels
using ELISA. (G) Western blot analysis of the expression of (H)
JNK, p-JNK and (I) HO-1 in BV2 cells treated with 0, 25, 50 or 100
µg/ml NP for 12 h. NP, nanoplastic; HO-1, heme oxygenase 1;
ASC, apoptosis-associated speck-like protein; p-, phosphorylated.
*P<0.05, **P<0.01,
****P<0.0001. |
Increased ROS levels induce
ferroptosis
ROS and lipid peroxidation induce cell death, such
as by ferroptosis (14). NAC was
used to determine whether ROS was involved in the regulation of
ferroptosis. Cells were treated with 50 µg/ml NP and NAC to
determine whether NPs induced ferroptosis was mediated by ROS. As
aforementioned, NAC significantly decreased the expression of ROS,
SOD, MDA and GSH and also decreased NP-induced oxidative damage.
NAC inhibited the decrease in GPX4 expression (Fig. 5G). Compared with cells treated
with NPs alone, the expression of FACL4 and TFRC decreased and the
protein expression levels of XCT were increased following NAC
pretreatment (Fig. 5B, C and F).
Finally, compared with 0 µg/ml NPs group, the fluorescence
intensity of the 50 µg/ml NPs group significantly grew
larger. The fluorescence intensity in cells co-treated with NPs and
NAC was significantly inhibited (Fig.
5E and I).
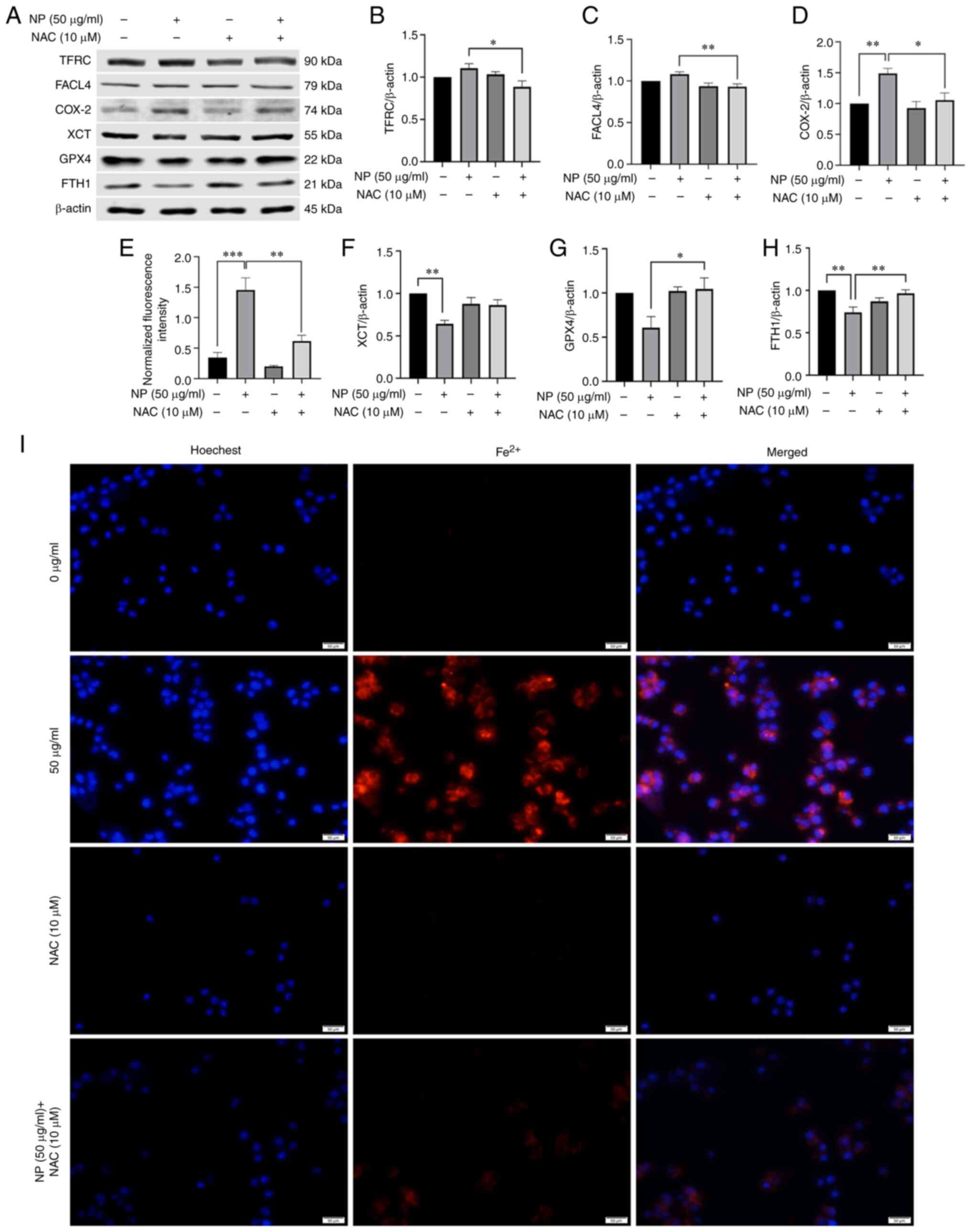 | Figure 5NAC inhibits NP-induced ferroptosis
in BV2 cells. (A) BV2 cells were pretreated with SP600125 (2
µM) for 1 h and exposed to NPs for 12 h. The expression
levels of ferroptosis-associated proteins were detected by western
blotting. (B) TFRC, (C) FACL4, (D) COX-2, (E) Fluorescence
intensity of Fe2+, (F) XCT, (G) GPX4 and (H) FTH1
expression in BV2 cells pretreated with 10 µM NAC followed
by treatment with 50 µg/ml NP for 12 h. (I) Mean
fluorescence intensity of Fe2+ in BV2 cells determined
using fluorescence microscopy. NP, nanoplastic; NAC,
N-acetylcysteine; TFRC, transferrin receptor; FACL4, acyl-CoA
synthetase long-chain family member 4; COX-2, cyclooxygenase-2;
XCT, solute carrier family 7 member 11; GPX4, glutathione
peroxidase; FTH1, ferritin heavy chain 1. *P<0.05,
**P<0.01, ***P<0.001. |
Inflammatory response is induced by NPs
in a ROS-mediated-manner
To determine the effect of ROS on inflammatory
reactions, NAC was used to inhibit ROS and levels of inflammatory
cytokines, intracellular inflammation-associated proteins and
inflammatory factors were assessed following NP treatment. Western
blotting showed that NAC treatment resulted in downregulation of
NLRP3, ASC and cleaved caspase-1 protein expression in BV2 cells
(Fig. 6A-D). Following
pretreatment with NAC, the expression of IL-18 and IL-1β also
decreased significantly compared with cells treated with NPs alone
(Fig. 6E and F). These results
showed the key role of ROS in NP-induced inflammatory
responses.
Ferroptosis is induced by the
JNK/HO-1/FTH1 signaling pathway in a ROS mediated-manner
The aforementioned results indicated the important
role of ROS in the inflammatory response and ferroptosis. NAC
treatment inhibited JNK phosphorylation and downregulated HO-1
expression (Fig. 7B and C). To
verify the role of JNK in ferroptosis, cells treated with NP were
treated with JNK inhibitor SP600125. SP600125 inhibited
downregulation of GPX4 and XCT (Fig.
7I and J), downregulated expression of TFRC and FACL4 protein
(Fig. 7E and F) and upregulated
the expression of the FTH1 protein (Fig. 7G). Compared with 0 µg/ml NP
group, the fluorescence intensity of the 50 µg/ml NPs group
significantly grew larger. The mean fluorescence intensity of
intracellular iron ions was also significantly decreased following
pretreatment of BV2 cells with SP600125 compared with 50
µg/ml NPs group (Fig. 7K and
L).
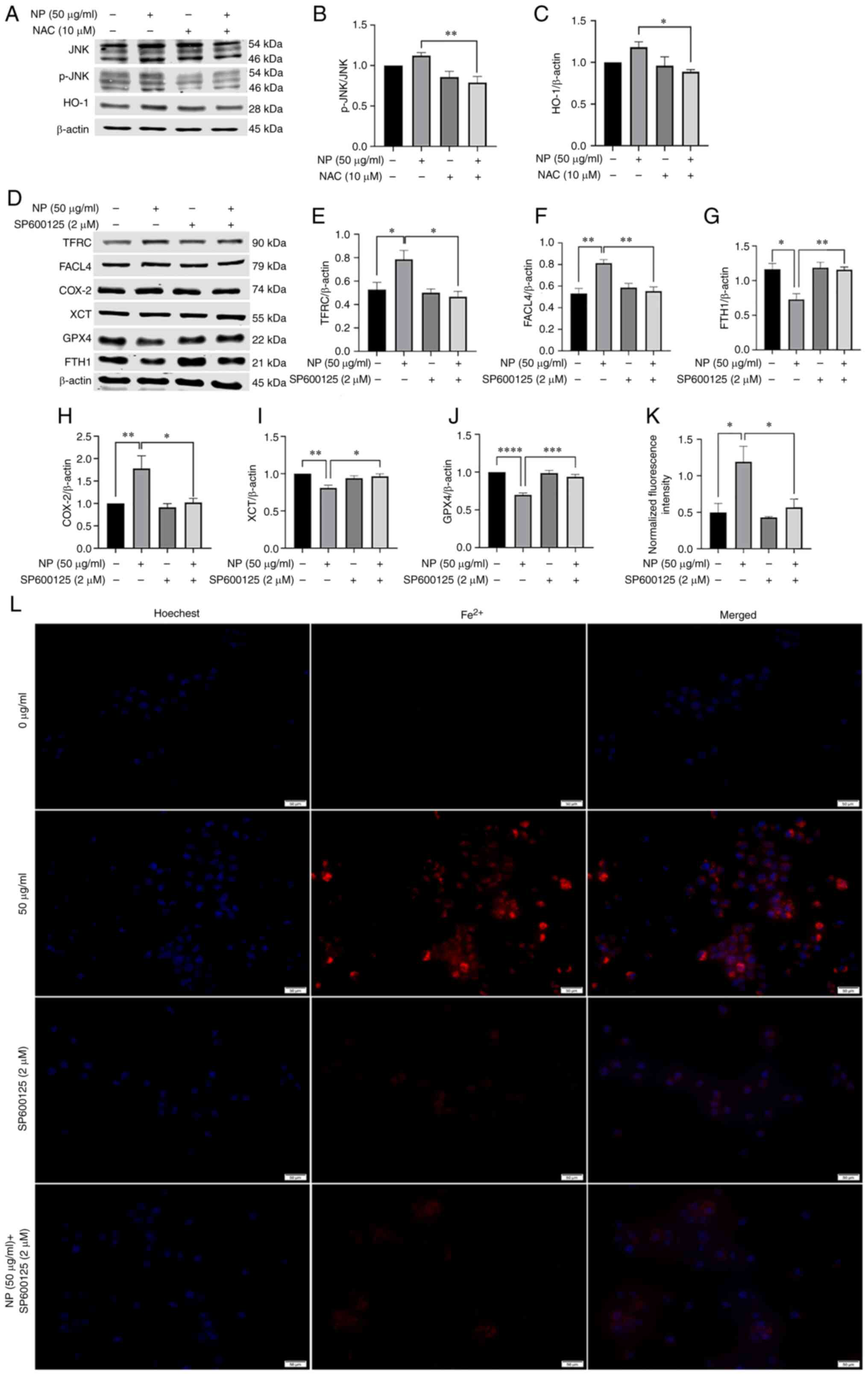 | Figure 7NPs regulate ROS to activate the
JNK/HO-1/FTH1 signaling pathway to induce ferroptosis in BV2 cells.
(A) NAC treatment downregulated expression of (B) JNK and (C) HO-1.
(D) BV2 cells were pretreated with SP600125 (2 µM) for 1 h
and exposed to NPs for 12 h. The expression levels of
ferroptosis-associated proteins were detected by western blotting.
NAC treatment downregulated expression of (E) TFRC, (F) FACL4 and
(H) COX-2, upregulated expression of (G) FTH1, (I) XCT, (J) GPX4.
(K) Fluorescence intensity of Fe2+. (L) SP600125
treatment downregulated fluorescence intensity of Fe2+.
NP, nanoplastic; ROS, reactive oxygen species; HO-1, heme oxygenase
1; FTH1, ferritin heavy chain 1; p-, phosphorylated; TFRC,
transferrin receptor; FACL4, acyl-CoA synthetase long-chain family
member 4; COX-2, cyclooxygenase-2; XCT, solute carrier family 7
member 11; GPX4, glutathione peroxidase. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. |
Discussion
NPs are widely present in the environment, have
disadvantageous effects on organisms and have received increasing
attention (16-18,37). The number of NPs present in the
environment is unknown as there are no effective methods to measure
this. Due to the small size of NPs, they are extensively spread in
aqueous environments and easily ingested by organisms (37). The aim of the present study was to
investigate the possible effects of NPs on microglia.
Microglia are key cells of the immune system present
in the central nervous system (38). Microglia comprise the innate
immune system of the central nervous system and are key cellular
mediators of neuroinflammatory processes. They serve a key role in
central nervous system disease, including infection and acute and
chronic neuroinflammatory responses (38). Chronic microglial activation is a
key component of neurodegenerative disease and this chronic
neuroinflammatory component likely contributes to neuronal
dysfunction, injury and loss, leading to disease progression
(38). BV2 cells are immortalized
cell lines of mouse microglia and were selected as the experimental
model. Due to their small particle size, NPs can enter tissues and
organs and cross the BBB. The breakdown of the BBB may result in an
inflammatory response and neurodegeneration (40). Studies have shown that PS-NPs
increase the permeability of the BBB, decrease dendritic spine
density and induce an inflammatory response in the hippocampus and
PS-NPs in microglia induce microglial activation and neuron damage
in the mouse brain (39,40). The aforementioned results suggest
that PS-NPs can pass through the BBB and induce neurotoxicity in
mammals, potentially by inducing activation of microglia.
Microglia, the resident macrophages of the central nervous system,
rapidly activate in nearly all types of neurological disease. These
activated microglia become highly motile, secrete inflammatory
cytokines, migrate to the lesion area and phagocytose cell debris
or damaged neurons. Even a minimal stimulus activates microglia and
causes inflammation-induced neuronal damage (41). Kwon et al (42) found that PS-MPs in the brain
co-localize with Ionized calcium binding adapter molecule 1, a
marker of microglia, in all three regions (hippocampus, cortex,
cerebellum). Shan et al (39) found that PS-NPs in microglia
result in the activation of quiescent microglia, evidenced by
transformation from a ramified to an amoeboid phenotype,
enlargement of cell nuclei, stubby branches and reduced end-point
voxels under Sholl analysis. The aforementioned results indicate
that MPs can enter microglia and microglia can be activated. In the
present study, the fluorescent signal of NPs in BV2 cells became
stronger as the concentration of NPs increased.
ROS-mediated lipid peroxidation is key to
ferroptosis. Cells have evolved a counterbalancing system to
neutralize excess ROS, an antioxidant system consisting of
enzymatic antioxidants such as SOD, catalase, GPX4 and thioredoxin
(43). In the present study, when
exposed to 100 µg/ml NPs, the levels of ROS increased
significantly compared with the control group. The levels of GSH
and SOD decreased, whereas MDA levels increased. From these
results, it was speculated that NPs induced oxidative damage in BV2
cells.
Inflammation is a common immune response that leads
to numerous types of dangerous and complex disease, such as
atherosclerosis. Inflammasomes, which are multiprotein complexes,
comprise Nod-like receptor (NLR), ASC and pro-caspase-1. Following
assembly of inflammasomes, caspase-1 is activated and hydrolyzed
into two products, forming a dimer to become mature cleaved
caspase-1 (44). Activated
caspase-1 cleaves pre-IL-1β and pre-IL-18 into mature IL-1β and
IL-18. In the present study, expression of NLRP3, ASC and pro
caspase-1 increased significantly following NP exposure and the
expression of IL-18 and IL-1β also increased. ROS were shown to be
the key mechanism triggering the formation and activation of the
NLRP3 in the present study. Monocytes (MOs) serve as precursors of
macrophage and effector cells, survey peripheral tissue and
maintain endothelial integrity. Following inflammatory stimulus,
MOs circulating in the bloodstream migrate to tissue and
differentiate into either macrophages or MO-derived dendritic cells
(MODCs) (45). MOs and MODCs
release pro-inflammatory or anti-inflammatory cytokines that
activate or suppress inflammation, respectively. In a recent study
(46), PVC particles were shown
to increase secretion of both pro-(IL-6 and TNF) and
anti-inflammatory cytokines (IL-10) by MOs. The increase of both
proand anti-inflammatory cytokine levels indicates the presence of
a counterbalancing mechanism. NP exposure induces secretion of pro-
and anti-inflammatory cytokines in primary human MOs and DCs
(46).
Lysosome rupture is a mechanism of NLRP3
inflammasome activation. During lysosome rupture, cathepsin B is
released from the lysosome. A previous study (19) showed that cathepsin B binds to the
leucine-rich repeat domain of NLRP3 and activates NLRP3. Cathepsin
B is also involved in the activation of MAPKs, one of which is JNK.
In addition, JNK inhibition decreases levels of IL-1β and
activation of caspase-1; JNK1 and JNK2 participate in IL-1β
cleavage (19). Ca2+
also activates JNK, a necessary kinase for ASC speck formation, by
activating tat-associated kinase/JNK pathway (47). MAPKs control cellular activity,
including gene expression, mitosis, differentiation, cell survival
and apoptosis (48). In
particular, MAPKs (ERK, p38 and JNK) have key roles in regulating
inflammatory and immune responses (49). A previous study (50) indicated that ROS was upstream of
MAPKs/NF-κB/NLRP3 and regulated expression of components involved
in this pathway. Furthermore, to explore the sequence of MAPKs,
nuclear NF-κB and NLRP3 in the inflammatory response induced by
high glucose (HG), specific inhibitors of p38 (SB203580), ERK
(PD98059) and JNK (SP600125) were used to pretreat osteoclasts
(OCs) before induction with HG. SP600125, PD98059 and SB203580
significantly inhibited production of nuclear NF-κB, p-IκB,
inhibitor of κB kinase and NLRP3 components (ASC, caspase-1, IL-18,
IL-1β and NLRP3) and secretion of IL-1β and IL-18. These results
showed that p-JNK, p-ERK1/2 and p-p38 mediated the activation of
nuclear NF-κB-associated proteins and the NLRP3 inflammasome by HG
in OCs. Together, these data demonstrated that the ROS/MAPK/NF-kB
pathway regulates the HG-induced NLRP3 inflammasome response in
vitro (50). Adiponectin
suppresses palmitate-mediated NLRP3 inflammasome activation in
hepatocytes via AMPK/JNK/ERK1/NF-κB/ROS signaling pathways
(51). Past studies have shown
that the JNK/c-Jun pathway serves crucial roles in the microglial
response and kaempferol attenuates retinal ganglion cell death by
suppressing the NLRP3 inflammasome through JNK pathways in acute
glaucoma (52,53). A previous study (54) indicated that indirect traumatic
optic neuropathy induces significant retinal ganglion cell (RGC)
death and axonal degeneration and activates JNK/c-Jun signaling,
which could further induce the microglial response and NLRP3
inflammasome activation. Moreover, JNK disruption suppresses NLRP3
inflammasome activation in microglia and prevents RGC death and
axonal degeneration (54). A
previous study found that spleen tyrosine kinase (Syk) and JNK are
rapidly phosphorylated during Staphylococcus aureus
infection. Moreover, a Syk/JNK inhibitor and Syk/JNK small
interfering RNA not only decrease NLRP3 inflammasome-associated
molecule expression at the protein and mRNA levels, ASC speck
formation and IL-1β and IL-18 release but also rescue decreased
NIMA-related kinase 7 (NEK7) expression following suppression of
the NEK7/NLRP3 pathway in macrophages (55). Syk/JNK phosphorylation levels and
NLRP3 inflammasome-associated molecule expression are decreased by
blockade of K+ efflux (55). The aforementioned studies indicate
that NPs induce JNK to activate NLRP3 inflammatory vesicles and
induce inflammatory response. The present study was not designed to
prove the chronological association between activation of JNK and
inflammatory response.
In the present study, NLRP3 was activated and
protein expression levels of ASC, cleaved caspase-1, IL-18 and
IL-1β increased significantly following exposure to NPs. NLRP3,
ASC, cleaved caspase-1, IL-18, and IL-1β were all significantly
downregulated by ROS inhibitor NAC. Thus, it was suggested that NPs
induced microglia toxicity by increasing ROS levels, in turn
inducing inflammatory responses.
System XC− is an amino acid antiporter
that is abundant in the phospholipid bilayer. It transports
extracellular cystine and intracellular glutamate at a ratio of 1:1
and reduces cystine to cysteine in the cell. Inhibition of the
activity of system XC− decreases GSH synthesis by
inhibiting cysteine uptake, leading to decreased GPX activity and
decreased cellular antioxidant capacity, accumulation of lipid ROS
and finally oxidative damage and ferroptosis (56). As a member of the GPX family, GPX4
primarily inhibits lipid peroxidation and serves an important role
in the occurrence of ferroptosis. GSH serves as a cofactor in
reduction of peroxide to the corresponding alcohol by GPX4. Thus,
intracellular GSH is key for GPX4 activity (57,58). The results of the present study
showed that exposure to NPs inhibited activity of system
XC−; levels of GSH were significantly decreased and
activity of GPX4 was also inhibited. The aforementioned results
demonstrated that NPs may cause lipid peroxidation, leading to
ferroptosis.
Iron is a key trace element that participates in
normal hematopoietic and immune function of the human body.
Excessive iron cause lesions and damage to numerous types of tissue
and organ. The key to the induction of ferroptosis is the
accumulation of a large quantity of free iron in cells. Cells take
up iron primarily via TF and TFRC 1, which are the primary iron
uptake proteins. Excess iron must be removed via FTH1 and FTL,
thereby avoiding the formation of ROS (14). The present study demonstrated that
intracellular ferritin levels increased following NP exposure and
the expression of TFRC protein increased in a dose-dependent
manner. The expression of FTH1 decreased in a dose-dependent manner
due to accumulation of excess ferritin in the cells. Lipid
metabolism is a key factor in ferroptosis. Polyunsaturated fatty
acids are susceptible to lipid peroxidation and are key elements of
ferroptosis (15). Acyl CoA
synthase long-chain family member 4 (ACSL4) and
lysophosphatidyltransferase 3 (LPCAT3) activate polyunsaturated
fatty acids. ACSL4 catalyzes free arachidonic acid (AA)/adrenic
acid (AdA) binding to CoA to form AA/AdA-CoA derivatives. LPCAT3
catalyzes the biosynthesis of AA/AdA-CoA and membrane
phosphatidylethanolamine (PE) to form AA/AdA-PE, which is an
intermediate process to activate ferroptotic signals (59). Therefore, decreasing ACSL4 and
LPCAT3 levels inhibits ferroptosis (60). In the present study, expression
levels of FACL4 were significantly increased following exposure to
NPs. Following pretreatment of BV2 cells with NAC, compared with
the 50 µg/ml NP group, system XC− and GSH
activity were increased and the downregulation of GPX4 was
reversed. Ferritin accumulation and transferrin expression
decreased, FTH1 levels increased, lipid peroxidation was inhibited
and FACL4 expression was downregulated. The aforementioned results
demonstrated that lipid peroxidation and accumulation of ROS may be
involved in multiple pathways associated with ferroptosis. NPs may
induce lipid peroxidation and ROS accumulation, thus causing
microglial ferroptosis. The primary function of system
XC− is to transport cystine and glutamate in and out of
cells. Cysteine participates in GSH synthesis via system
XC−. GPX4 converts GSH to GSSG and inhibits ROS
accumulation. Fe3+ binding to transferrin on the cell
membrane is converted to Fe2+ and induces ferroptosis
through lipid peroxidation via the Fenton reaction. NLRP3 is
activated and interacts with ASC, resulting in cleavage of
caspase-1 to mature cleaved caspase-1. Activated caspase-1 converts
pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18 (Fig. 8).
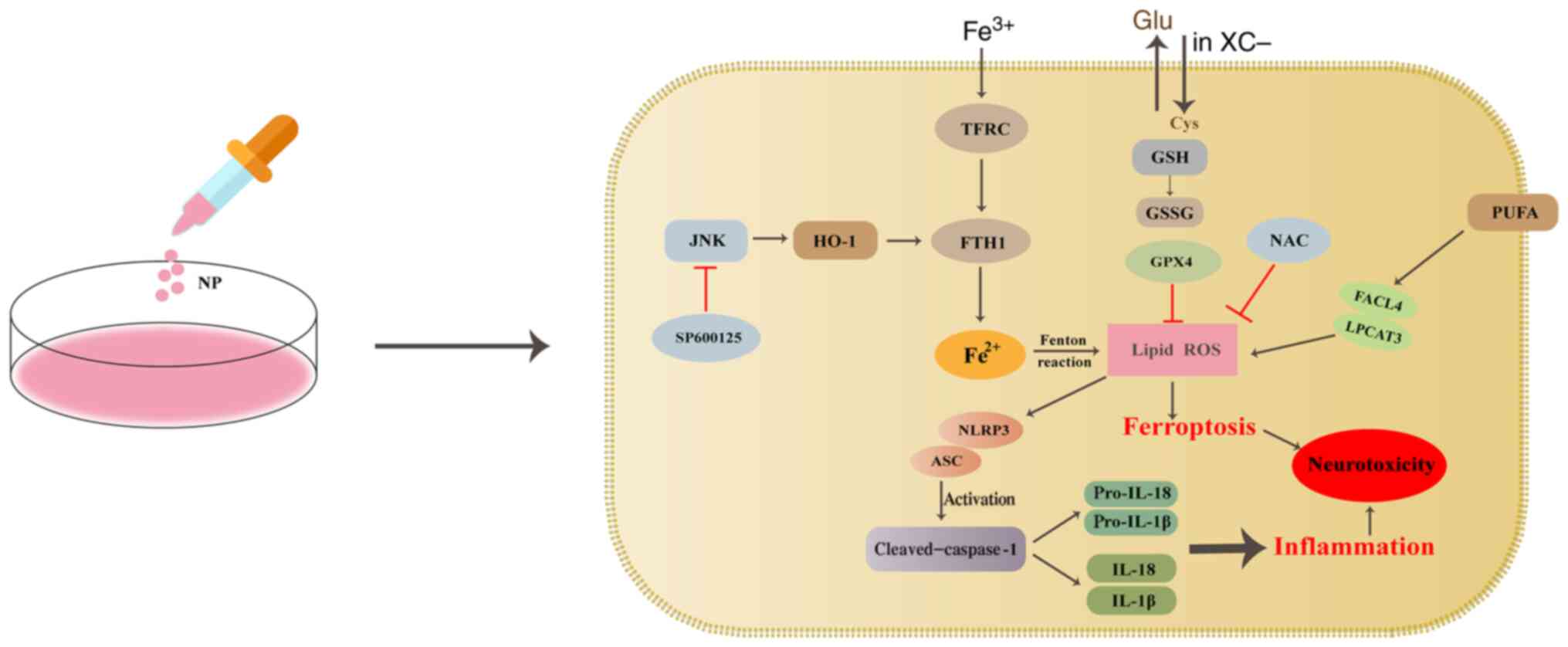 | Figure 8Schematic illustration of NP-induced
inflammatory reactions and ferroptosis in BV2 cells. The primary
function of system XC− is to transport cystine and
glutamate in and out of cells. Cysteine participates in GSH
synthesis via system XC−. GPX4 converts GSH to GSSG and
inhibits ROS accumulation. Fe3+ binding to transferrin
on the cell membrane is converted to Fe2+ and induces
ferroptosis through lipid peroxidation via the Fenton reaction.
NLRP3 is activated and interacts with ASC, resulting in cleavage of
caspase-1 to mature cleaved caspase-1. Activated caspase-1 converts
pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18. GSH,
glutathione; GPX4, glutathione peroxidase 4; ROS, reactive oxygen
species; NP, nanoplastic; GSSG, glutathione disulfide; ASC,
apoptosis-associated speck-like protein; HO-1, heme oxygenase 1;
FTH1, ferritin heavy chain 1; TFRC, transferrin receptor; Glu,
glutamate; Cys, cysteine; LPCAT3, lysophosphatidyltransferase 3;
PUFA, polyunsaturated fatty acid. |
JNK is activated by cytokines and environmental
stressors such as ultraviolet irradiation and oxidative stress and
belongs to a subset of MAP agonists (61). In the present study, NPs activated
JNK. It was hypothesized that ROS generated following exposure to
NPs activated the JNK pathway. To confirm this, cells were treated
with ROS inhibitor NAC; expression of JNK in the NP + NAC group was
notably lower than in the 50 µg/ml NPs group. Therefore, the
increase in ROS was caused by activation of JNK. Previous studies
have suggested that the regulation of HO-1 is mediated via MAPKs,
including the ERK, JNK and p38 MAPK pathways (62,63). In the present study, following
addition of the JNK inhibitor SP600125, the expression of HO-1
decreased, expression of FTH1 was increased and iron accumulation
was decreased. Therefore, it was hypothesized that NPs induced
ferroptosis in BV2 cells via the JNK/HO-1/FTH1 pathway.
There are limitations to this study. A limitation is
that the present study was an in vitro study of NP-induced
phenotype and mechanism. And the present study was not designed to
prove that the phagocytosis of microglia occurred in a relatively
short time. Future experiments should explore whether phagocytosis
of pathogens and/or cell debris by microglia is affected by
exposure to NPs.
The present study showed that NPs entered BV2 cells
and induced oxidative stress and inflammatory responses. NPs also
led to ferroptosis of BV2 cells via increased lipid peroxidation
and ROS accumulation. ROS served a pro-ferroptotic role in
NP-induced inflammatory responses and ferroptosis. NPs potentially
induced ferroptosis by regulating the JNK/HO-1/FTH1 signaling
pathway. The present study high-lighted a novel avenue for the
study of the pathogenesis and treatment of NP-induced central
nervous system disease.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JYS, YHW, YLD, WXZ, GQC and ZJD conceived and
designed the study. ZDL, GQC, JYS, YHW, YLD, WXZ and JB performed
the experiments. JYS wrote the manuscript. GQC and ZJD provided
funding and project administration, acquired and analyzed the data
and critically reviewed and edited the manuscript. All authors have
read and approved the final manuscript. All authors confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
NP
|
nanoplastic
|
|
ASC
|
apoptosis-associated speck-like
protein
|
|
GSH
|
glutathione
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
|
GPX4
|
glutathione peroxidase 4
|
|
HO-1
|
heme oxygenase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
FTH1
|
ferritin heavy chain 1
|
|
TFRC
|
transferrin receptor
|
|
COX-2
|
cyclooxygenase-2
|
|
XCT
|
solute carrier family 7 member 11
|
|
NAC
|
N-acetylcysteine
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81602893), the Natural Science
Foundation of Shandong Province (grant nos. ZR2015YL049,
ZR2021MH218 and ZR2022MH184), the Key Technology Research and
Development Plan of Shandong Province (grant no. 2018GSF118018),
Jinan Science and Technology Project (grant nos. 201907022 and
202019183) and the Innovation Project of Shandong Academy of
Medical Science and Academic Promotion Programme of Shandong First
Medical University (grant no. 2019QL001).
References
|
1
|
Geyer R, Jambeck JR and Law KL:
Production, use, and fate of all plastics ever made. Sci Adv.
3:e17007822017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rhodes CJ: Plastic pollution and potential
solutions. Sci Prog. 101:207–260. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu J, Lezama N, Gasper J, Kawata J,
Morley S, Helmer D and Ciminera P: Burn pit emissions exposure and
respiratory and cardiovascular conditions among airborne hazards
and open burn pit registry participants. J Occup Environ Med.
58:e249–e255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kubowicz S and Booth AM: Biodegradability
of plastics: Challenges and misconceptions. Environ Sci Technol.
51:12058–12060. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Avio CG, Gorbi S and Regoli F: Plastics
and microplastics in the oceans: From emerging pollutants to
emerged threat. Mar Environ Res. 128:2–11. 2017. View Article : Google Scholar
|
|
6
|
Waring RH, Harris RM and Mitchell SC:
Plastic contamination of the food chain: A threat to human health?
Maturitas. 115:64–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jambeck JR, Geyer R, Wilcox C, Siegler TR,
Perryman M, Andrady A, Narayan R and Law KL: Marine pollution.
Plastic waste inputs from land into the ocean. Science.
347:768–771. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thompson RC, Olsen Y, Mitchell RP, Davis
A, Rowland SJ, John AW, McGonigle D and Russell AE: Lost at sea:
Where is all the plastic? Science. 304:8382004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, Qu X, Su L, Zhang W, Yang D,
Kolandhasamy P, Li D and Shi H: Microplastics in mussels along the
coastal waters of China. Environ Pollut. 214:177–184. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Neves D, Sobral P, Ferreira JL and Pereira
T: Ingestion of microplastics by commercial fish off the Portuguese
coast. Mar Pollut Bull. 101:119–126. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liebezeit G and Liebezeit E: Non-pollen
particulates in honey and sugar. Food Addit Contam Part A Chem Anal
Control Expo Risk Assess. 30:2136–2140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ossmann BE, Sarau G, Holtmannspotter H,
Pischetsrieder M, Christiansen SH and Dicke W: Small-sized
microplastics and pigmented particles in bottled mineral water.
Water Res. 141:307–316. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marra F and Tacke F: Roles for chemokines
in liver disease. Gastroenterology. 147:577–594.e1. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
MacKenzie EL, Iwasaki K and Tsuji Y:
Intracellular iron transport and storage: From molecular mechanisms
to health implications. Antioxid Redox Signal. 10:997–1030. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar :
|
|
16
|
Wang S, Chen L, Shi X, Wang Y and Xu S:
Polystyrene microplastics-induced macrophage extracellular traps
contributes to liver fibrotic injury by activating
ROS/TGF-β/Smad2/3 signaling axis. Environ Pollut. 324:1213882023.
View Article : Google Scholar
|
|
17
|
Sarasamma S, Audira G, Siregar P, Malhotra
N, Lai YH, Liang ST, Chen JR, Chen KH and Hsiao CD: Nanoplastics
cause neurobehavioral impairments, reproductive and oxidative
damages, and biomarker responses in zebrafish: Throwing up alarms
of widespread health risk of exposure. Int J Mol Sci. 21:14102020.
View Article : Google Scholar
|
|
18
|
Lee CW, Hsu LF, Wu IL, Wang YL, Chen WC,
Liu YJ, Yang LT, Tan CL, Luo YH, Wang CC, et al: Exposure to
polystyrene microplastics impairs hippocampus-dependent learning
and memory in mice. J Hazard Mater. 430:1284312022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoseini Z, Sepahvand F, Rashidi B,
Sahebkar A, Masoudifar A and Mirzaei H: NLRP3 inflammasome: Its
regulation and involvement in atherosclerosis. J Cell Physiol.
233:2116–2132. 2018. View Article : Google Scholar
|
|
20
|
Kim YG, Kim SM, Kim KP, Lee SH and Moon
JY: The role of inflammasome-dependent and inflammasome-independent
NLRP3 in the kidney. Cells. 8:13892019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hou J, Lei Z, Cui L, Hou Y, Yang L, An R,
Wang Q, Li S, Zhang H and Zhang L: Polystyrene microplastics lead
to pyroptosis and apoptosis of ovarian granulosa cells via
NLRP3/Caspase-1 signaling pathway in rats. Ecotoxicol Environ Saf.
212:1120122021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang F, Salvati A and Boya P:
Lysosome-dependent cell death and deregulated autophagy induced by
amine-modified polystyrene nanoparticles. Open Biol. 8:1702712018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Latunde-Dada GO: Ferroptosis: Role of
lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta
Gen Subj. 1861:1893–1900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Forrester SJ, Kikuchi DS, Hernandes MS, Xu
Q and Griendling KK: Reactive oxygen species in metabolic and
inflammatory signaling. Circ Res. 122:877–902. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Friedmann Angeli JP, Krysko DV and Conrad
M: Ferroptosis at the crossroads of cancer-acquired drug resistance
and immune evasion. Nat Rev Cancer. 19:405–414. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Conrad M and Pratt DA: The chemical basis
of ferroptosis. Nat Chem Biol. 15:1137–1147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kyriakis JM and Avruch J: pp54
microtubule-associated protein 2 kinase. A novel serine/threonine
protein kinase regulated by phosphorylation and stimulated by
poly-L-lysine. J Biol Chem. 265:17355–17363. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pulverer BJ, Kyriakis JM, Avruch J,
Nikolakaki E and Woodgett JR: Phosphorylation of c-jun mediated by
MAP kinases. Nature. 353:670–674. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang H, Jiao W, Cui H, Sun Q and Fan H:
Combined exposure of alumina nanoparticles and chronic stress
exacerbates hippocampal neuronal ferroptosis via activating
IFN-ү/ASK1/JNK signaling pathway in rats. J Hazard Mater.
411:1251792021. View Article : Google Scholar
|
|
33
|
Abraham NG and Kappas A: Pharmacological
and clinical aspects of heme oxygenase. Pharmacol Rev. 60:79–127.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ryter SW, Alam J and Choi AM: Heme
oxygenase-1/carbon monoxide: From basic science to therapeutic
applications. Physiol Rev. 86:583–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ryter SW: Heme oxgenase-1, a cardinal
modulator of regulated cell death and inflammation. Cells.
10:5152021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kwon MY, Park E, Lee SJ and Chung SW: Heme
oxygenase-1 accelerates erastin-induced ferroptotic cell death.
Oncotarget. 6:24393–24403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shen M, Zhang Y, Zhu Y, Song B, Zeng G, Hu
D, Wen X and Ren X: Recent advances in toxicological research of
nanoplastics in the environment: A review. Environ Pollut. 252(Pt
A): 511–521. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Streit WJ, Mrak RE and Griffin WS:
Microglia and neuroinflammation: A pathological perspective. J
Neuroinflammation. 1:142004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shan S, Zhang Y, Zhao H, Zeng T and Zhao
X: Polystyrene nanoplastics penetrate across the blood-brain
barrier and induce activation of microglia in the brain of mice.
Chemosphere. 298:1342612022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin H, Yang C, Jiang C, Li L, Pan M, Li D,
Han X and Ding J: Evaluation of neurotoxicity in BALB/c mice
following chronic exposure to polystyrene microplastics. Environ
Health Perspect. 130. pp. 1070022022, View Article : Google Scholar
|
|
41
|
Fu R, Shen Q, Xu P, Luo JJ and Tang Y:
Phagocytosis of Microglia in the Central Nervous System Diseases.
Mol Neurobiol. 49:1422–1434. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kwon W, Kim D, Kim HY, Jeong SW, Lee SG,
Kim HC, Lee YJ, Kwon MK, Hwang JS, Han JE, et al: Microglial
phagocytosis of polystyrene microplastics results in immune
alteration and apoptosis in vitro and in vivo. Sci Total Environ.
807(Pt 2): 1508172022. View Article : Google Scholar
|
|
43
|
Nelson BA: A comprehensive program for
pregnant adolescents: Parenting and prevention. Child Welfare.
68:57–60. 1989.PubMed/NCBI
|
|
44
|
Sollberger G, Strittmatter GE,
Garstkiewicz M, Sand J and Beer HD: Caspase-1: The inflammasome and
beyond. Innate Immun. 20:115–125. 2014. View Article : Google Scholar
|
|
45
|
Teh YC, Ding JL, Ng LG and Chong SZ:
Capturing the fantastic voyage of monocytes through time and space.
Front Immunol. 10:8342019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weber A, Schwiebs A, Solhaug H, Stenvik J,
Nilsen AM, Wagner M, Relja B and Radeke HH: Nanoplastics affect the
inflammatory cytokine release by primary human monocytes and
dendritic cells. Environ Int. 163:1071732022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Okada M, Matsuzawa A, Yoshimura A and
Ichijo H: The lysosome rupture-activated TAK1-JNK pathway regulates
NLRP3 inflammasome activation. J Biol Chem. 289:32926–32936. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dickinson RJ and Keyse SM: Diverse
physiological functions for dual-specificity MAP kinase
phosphatases. J Cell Sci. 119(Pt 22): 4607–4615. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ajizian SJ, English BK and Meals EA:
Specific inhibitors of p38 and extracellular signal-regulated
kinase mitogen-activated protein kinase pathways block inducible
nitric oxide synthase and tumor necrosis factor accumulation in
murine macrophages stimulated with lipopolysaccharide and
interferon-gamma. J Infect Dis. 179:939–944. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
An Y, Zhang H, Wang C, Jiao F, Xu H, Wang
X, Luan W, Ma F, Ni L, Tang X, et al: Activation of
ROS/MAPKs/NF-kB/NLRP3 and inhibition of efferocytosis in
osteoclast-mediated diabetic osteoporosis. FASEB J. 33:12515–12527.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dong Z, Zhuang Q, Ye X, Ning M, Wu S, Lu L
and Wan X: Adiponectin Inhibits NLRP3 inflammasome activation in
nonalcoholic steatohepatitis via AMPK-JNK/ErK1/2-NFκB/ROS signaling
pathways. Front Med (Lausanne). 7:5464452020. View Article : Google Scholar
|
|
52
|
Welsbie DS, Ziogas NK, Xu L, Kim BJ, Ge Y,
Patel AK, Ryu J, Lehar M, Alexandris AS, Stewart N, et al: Targeted
disruption of dual leucine zipper kinase and leucine zipper kinase
promotes neuronal survival in a model of diffuse traumatic brain
injury. Mol Neurodegener. 14:442019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lin C, Wu F, Zheng T, Wang X, Chen Y and
Wu X: Kaempferol attenuates retinal ganglion cell death by
suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via JNK and
NF-κB pathways in acute glaucoma. Eye (Lond). 33:777–784. 2019.
View Article : Google Scholar
|
|
54
|
Chu X, Wang C, Wu Z, Fan L, Tao C, Lin J,
Chen S, Lin Y and Ge Y: JNK/c-Jun-driven NLRP3 inflammasome
activation in microglia contributed to retinal ganglion cells
degeneration induced by indirect traumatic optic neuropathy. Exp
Eye Res. 202:1083352021. View Article : Google Scholar
|
|
55
|
Liu R, Liu R, Liu C, Gao A, Wang L, Tang
H, Wu Q, Wang X, Tian D, Qi Z and Shen Y: NEK7-Mediated Activation
of NLRP3 inflammasome is coordinated by potassium Efflux/Syk/JNK
signaling during staphylococcus aureus infection. Front Immunol.
12:7473702021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao JY, Poddar A, Magtanong L, Lumb JH,
Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E, et al:
A genome-wide haploid genetic screen identifies regulators of
glutathione abundance and ferroptosis sensitivity. Cell Rep.
26:1544–1556.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Forcina GC and Dixon SJ: GPX4 at the
crossroads of lipid homeostasis and ferroptosis. Proteomics.
19:e18003112019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yan N and Zhang JJ: The emerging roles of
ferroptosis in vascular cognitive impairment. Front Neurosci.
13:8112019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar
|
|
61
|
Li DD, Wang LL, Deng R, Tang J, Shen Y,
Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anti-cancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2009. View Article : Google Scholar
|
|
62
|
Otterbein LE, Bach FH, Alam J, Soares M,
Tao Lu H, Wysk M, Davis RJ, Flavell RA and Choi AM: Carbon monoxide
has anti-inflammatory effects involving the mitogen-activated
protein kinase pathway. Nat Med. 6:422–428. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yang J, Mo J, Dai J, Ye C, Cen W, Zheng X,
Jiang L and Ye L: Cetuximab promotes RSL3-induced ferroptosis by
suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant
colorectal cancer. Cell Death Dis. 12:10792021. View Article : Google Scholar : PubMed/NCBI
|