Introduction
Esophageal squamous cell carcinoma (ESCC) accounts
for ~90% of all esophageal cancer cases and is typically in an
advanced stage at the time of diagnosis (1,2).
Cisplatin is the first-line chemotherapeutic agent of choice for
ESCC (3). However, the development
of resistance to cisplatin is a key issue hindering favorable
outcomes (4). For newly treated or
locally advanced ESCC patients, the initial response to
cisplatin-based therapy can be as high as 50% (5). However, most patients will develop
acquired cisplatin resistance, which leads to ESCC recurrence
(6). Cisplatin resistance and
recurrence are the primary factors leading to the failure of
cisplatin therapy in OSCC patients (7). Thus, uncovering the underlying
mechanisms of cisplatin resistance and developing agents that boost
sensitivity to cisplatin may provide novel therapeutic
strategies.
Aberrant gene expression may be a major intrinsic
factor that results in cisplatin resistance (8). Recent studies have found several
molecules and signaling pathways that participate in cisplatin
resistance, such as TGF-β (9),
AKR1C1 (8), and the Wnt pathway
(10). However, none of these have
been applied in clinical practice as indicators for predicting the
response to cisplatin and/or as targets in ESCC cells to improve
their sensitivity to cisplatin.
Aberrations in mitochondrial Ca2+
homeostasis are related to various pathological processes,
especially carcinogenesis (11),
drug resistance (12), and cancer
metastases (13). The
mitochondrial calcium uniporter (MCU) complex channel [including
nuclear-encoded channel-forming elements (such as MCU) and
regulators (MICUs)] is a major mediator of Ca2+
accumulation in the mitochondrial matrix (14). Changes in the expression or
function of MCU complex members are associated with various tumors
and tumor-related phenotypes (15). For example, MCU-mediated
mitochondrial calcium uptake facilitates tumor growth in colorectal
cancer (16). MCU facilitates
breast cancer growth and metastasis via HIF-1α (17). MCU-dependent mitochondrial
Ca2+ promotes reactive oxygen species production and
cancer metastases by suppressing the NAD+/SIRT3/SOD2 axis in
hepatocellular carcinoma (13).
However, the role of MCU in the mechanism of cisplatin resistance
in ESCC remains unclear. Hence, it was hypothesized that targeting
MCU may suppress tumor growth and improve cisplatin resistance in
ESCC.
Materials and methods
Cell culture
In the present study, three esophageal cancer cell
lines, KYSE-150, KYSE-410, and TE-1 (ATCC) were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin, and 100 U/ml streptomycin. Cells were
maintained in a humidified incubator at 37°C supplied with 5%
CO2 air.
Transfection
Based on the MCU sequence, two short hairpin RNAs
(shRNAs) targeting MCU (sh-MCU#1 and sh-MCU#2) and one negative
control shRNA (sh-NC) were designed; the sequences were: sh-MCU#1
(cat. no. PLVE2985), CCG GGA TGA TGT TAC AGT GGT TTA TCT CGA GAT
AAA CCA CTG TAA CAT CAT CTT TTT TG; sh-MCU#2 (cat. no. PLVE2986),
CCG GGA CAT TGG TCC AGC AAC TAT ACT CGA GTA TAG TTG CTG GAC CAA TGT
CTT TTT TG; and sh-NC (cat. no. PLVT7) CCG GGT TCT CCG AAC GTG TCA
CGT ACT CGA GTA CGT GAC ACG TTC GGA GAA CTT TTT TG. The lentiviral
vector for MCU overexpression (OE-MCU, Plasmid no.
200707HE6725-5R12) and negative control vector (OE-NC, pcDNA3.1)
were purchased from Sangon Biotech, co., Ltd. Cells were treated
with the overexpression vectors to construct stable MCU
overexpression cells. The lentiviral plasmid (pMAGic 7.1) was
purchased from Sangon Biotech, co., Ltd. Production of 3rd
generation lentiviruses was performed using a ratio of lentiviral
plasmid, packaging vector, and envelope of 3:2:1. A total of 42
µg lentiviral plasmid was used for transfection of 293T
cells (The Chinese Academy of Sciences). Lentiviral particles were
collected using an EZ-10 Spin Column Plasmid Mini-Prep Kit (Sangon
Biotech, co., Ltd.). Three esophageal cancer cell lines KYSE-150,
KYSE-410, and TE-1 were treated with the lentiviral particles with
an MOI of 10, and incubated for 12 h. Subsequently, the medium was
removed and replaced with supplemented media, and cells were
cultured for a further 72 h. Puromycin (2 µg/ml) was used
for the selection of stably transfected cell lines, and 1
µg/ml was used for the maintenance of stably transfected
cells. Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and 200 pmol shRNA were used to transfect cells
in a 60 mm dish. After mixing the shRNA, Lipofectamine®
2000, and serum-free medium, the mixture was incubated at room
temperature for 20 min, added to cells, then the cells were
cultured for 24 h for subsequent experimentation.
Western blotting
Cells or tissues were lysed for 20 min on ice with
RIPA lysis buffer (Nanjing KeyGen Biotech Co., Ltd.) containing
Sodium Deoxycholate, 1% Triton X-100, 0.1% SDS, PMSF, and protease
and phosphatase inhibitors. Protein concentration was calculated
using a BCA protein assay kit. A total of 30 µg protein was
loaded per lane of 10% SDS-gel, resolved using SDS-PAGE, and
transferred to a PVDF membrane (MilliporeSigma)/Membranes were
subsequently blocked using 5% skimmed milk for 1 h at room
temperature, and incubated with primary antibodies at 4°C
overnight. The primary antibodies used were: Anti-MCU (1:1,000;
cat. no. sc-515930; Santa Cruz Biotechnology, Inc.), anti-MICU2
(1:1,000; cat. no. ab101465; Abcam), anti-MICU1 (1:1,000; cat. no.
ab190114; Abcam), anti-PD-L1 (1:1,000; cat. no. 66248-1-Ig;
ProteinTech Group, Inc.), anti-CyclinD1 (1:5,000; cat. no.
60186-1-Ig; ProteinTech Group, Inc.), anti-Ki-67 (1:1,000; cat. no.
ab16667), anti-Vimentin (1:2,000; cat. no. ab92547; Abcam),
anti-β-catenin (1:1,000; cat. no. 51067-2-AP; ProteinTech Group,
Inc.), and anti-β-actin (1:2,000; cat. no. 20536-1-AP; ProteinTech
Group, Inc.). Subsequently, the membranes were incubated with
HRP-conjugated secondary antibody (1:5,000; cat. no. Zb-2301;
OriGene Technologies, Inc.) at room temperature for 1 h. The bands
were developed using an ECL kit (Thermo Fisher Scientific, Inc.)
and analyzed using ImageJ (National Institutes of Health). β-actin
served as the loading control.
Colony formation assay
The cells were plated in six-well plates
(1×103 cells/well) in 2 ml supplemented media, and
incubated for 14 days. Subsequently, the cells were fixed using
methanol for 30 min at room temperature, followed by staining with
0.1% crystal violet at room temperature for 20 min. A total of 50
stable cells constituted a colony under an optical inverted
microscope (magnification, ×100). Each well was divided into three
fields of view to count the number of colony.
Transwell migration assay
Cell migration was assessed using Transwell assays
(Corning, Inc.). The cells were diluted to 1×105/ml
using serum-free RPMI-1640 medium. Then, 200 µl cell
suspension was added to the upper chamber of the Transwell insert,
and 600 µl medium supplemented with 20% FBS was added to the
lower chamber. The cultures were incubated in a 37°C incubator for
48 h. After removing media from the upper chamber, the cells were
washed using 600 µl PBS three times, and the cells that had
migrated were stained using crystal violet at room temperature for
20 min. The number of cells that had migrated was counted using an
inverted light microscope (magnification, ×200) and imaged.
Wound healing assay
Cells were plated in a six-well plate
(5×105 cells/well). Once a confluent monolayer had
formed, a linear scratch was made in the monolayer of cells using a
200 µl pipette tip. Subsequently, the cells were washed and
cultured for 48 h in supplemented serum-free RPMI-1640 media
(18). Images were taken at 0 and
48 h using a light microscope (magnification, ×40). The ratio of
wound closure was used to quantify migration.
Measurement of the mitochondrial membrane
potential (MMP)
The MMP (Δψm) of the cells was measured using a JC-1
kit (Beyotime Institute of Biotechnology). The cells were rinsed
with PBS, followed by incubation with 10 µM JC-1 at 37°C for
30 min. Subsequently, the supernatant was aspirated. The cells were
washed twice with JC-1, and 2 ml media was added. Images were
acquired using a fluorescent microscope (Olympus Corporation).
Immunofluorescence staining. Cells or tissues
were fixed using 4% paraformaldehyde for 20 min at room
temperature, and subsequently, 0.2% Triton X-100 was added for 3
min to permeabilize cells. Non-specific binding was blocked using
goat serum for 15 min at 37°C, followed by incubation with the
primary antibody against anti-MCU (1:20), anti-MICU2 (1:100),
anti-MICU1 (1:100), anti-PD-L1 (1:50), or anti-CD34 (1:100; cat.
no. 14486-1-AP; ProteinTech Group, Inc.) overnight at 4°C. The
following day, samples were washed with PBS three times, and the
sections were incubated with Alexa Fluor® 488 Conjugate
(1:100; cat. no. ZF-0512; OriGene Technologies, Inc.) or Alexa
Fluor® 594 Conjugate (1:100; cat. no. ZF-0513; OriGene
Technologies, Inc.) secondary antibodies at 37°C for 2 h. Nuclei
were counterstained, with DAPI (MilliporeSigma, USA) at room
temperature until the samples were dry. Images were acquired using
a fluorescent microscope (magnification, ×400) (Olympus
Corporation).
Generation of cisplatin-resistant
cells
Cisplatin-resistant KYSE-150 (KYSE-150-CDDP),
KYSE-410 (KYSE-410-CDDP), and TE-1 (TE-1-CDDP) cells were generated
through stepwise exposure to increasing doses of cisplatin
(MilliporeSigma) (19-21). Cells were first exposed to
cisplatin at concentration of 5 µM for 6 months.
Subsequently, the cells were maintained in cisplatin-free RPMI-1640
medium for 3 days. When confluence reached the initial confluence
prior to treatment, the cells were treated with a higher
concentration of cisplatin (10, 20, 40, or 60 µM). The
concentration was gradually elevated every few passages until a
concentration of 60 µM was reached (within 2 months). For
subsequent maintenance of cisplatin resistance in cells, 60
µM cisplatin was used in general culture.
Mouse xenograft models
BALB/c nude male mice (weighing 16-20 g, 5 weeks
old; Beijing Vital River Laboratory Animal Technology Co., Ltd.)
were kept at 24±2°C, in a humid environment (60±10%), with a 12/12
light-dark cycle, and free access to food and water. The mice were
randomly separated into four groups (n=5 per group). The mice were
subcutaneously injected with sh-NC-transfected KYSE-150 cells
(Control), sh-NC-transfected KYSE-150-CDDP cells (CDDP),
sh-MCU-transfected KYSE-150 cells (sh-MCU), or sh-MCU-transfected
KYSE-150-CDDP cells (CDDP+sh-MCU) (2×106 in 0.1 ml) in
the back. The tumor volumes were measured weekly using the
following formula: volume=0.5× length × width2. After 21
days, mice were euthanized by cervical dislocation following
anesthesia by injection of sodium pentobarbital (50 mg/kg)
intraperitoneally, and the tumors were isolated, weighed, and
prepared for subsequent analysis. The protocol used strictly
adhered to the Guidelines for the Care and Use of Laboratory
Animals, and was approved by the Ethics Committee of Ningxia
Medical University (approval no. 2020-880).
Immunohistochemistry (IHC) staining
Tissues from the subcutaneous tumors of nude mice
were collected and fixed in 10% neutral formalin solution at room
temperature for 24 h. The xenograft tumors were collected and
embedded in paraffin, sectioned into 4 µm slices, dewaxed,
repaired under high pressure for 3 min in EDTA (ZLI-9072;
ZSGB-BIO), incubated with 3% hydrogen peroxide at 37°C for 15 min
as a quenching step, then blocked with 10% goat serum at 37°C for
30 min. Then, the sections were incubated with primary antibodies
at 4°C overnight. The primary antibodies used were: Anti-MCU (1:20;
sc-515930; SANTA CRUZ, USA), anti-MICU2 (1:100), anti-MICU1
(1:100), anti-PD-L1 (1:100), anti-Ki-67 (1:100), anti-Vimentin
(1:150; cat. no. 10366-1-AP; ProteinTech Group, Inc.), or
anti-E-cadherin (1:100; cat. no. 20874-1-AP; ProteinTech Group,
Inc.). The sections were stained with HRP-conjugated secondary
antibodies (cat. no. PV-9000; OriGene Technologies, Inc.) at room
temperature for 1 h followed by DAB chromogenic solution, and
counterstaining with hematoxylin at room temperature for 3 min.
Samples were finally sealed, and imaged using a microscope
(magnification, ×400) (BX61, Olympus Corporation), and analyzed
using IPP version 6.0 (Media Cybernetics).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from KYSE-150, KYSE-410, and
TE-1 cells using TRIzol®. RevertAid First Strand cDNA
Synthesis Kit (Thermo Fisher Scientific, Inc.) was used to
synthesize cDNA [using 5 µg RNA, 1 µl Oligo(dT)18
primer (100 µM), 1 µl Random Hexamer primers (100
µM), 4 µl 5x Reaction Buffer, 1 µl RiboLock
RNase Inhibitor (20 U/µl), 2 µl dNTP Mix (10 mM), and
1 µl RevertAid M-MuLV RT (200 U/µl), made to a final
volume of 20 µl using nucleotide-free water]. PowerUp™ SYBR™
Green MasterMix (Applied Biosystems, Thermo Fisher Scientific,
Inc.) was used for qPCR. The thermocycling conditions were: 20 sec
initial denaturation at 95°C; followed by 35 cycles of 1 sec at
95°C for denaturation, and 20 sec at 60°C for annealing and
extension. The primers were designed by Sangon Biotech, Co., Ltd.,
and the sequences were: MCU forward, 5′-TCC AGA AGC CAG AGA CAG
AC-3′ and reverse, 5′-TGT CGG AGA GGC AGA TGT AC-3′. GAPDH forward,
5′-CAA GGT CAT CCA TGA CAA CTT TG-3′ and reverse, 5′-GTC CAC CAC
CCT GTT GCT GTA G-3′. GAPDH was used as the housekeeping gene. Gene
expression was normalized using the 2−ΔΔCq method
(22).
Statistical analysis
All statistical analysis was performed using SPSS
version 22.0 (IBM Corp.). Each assay was performed in triplicate.
Data are presented as the mean ± SD. Differences between ≥3 groups
were compared using a one-way ANOVA followed by Bonferroni
corrections. P<0.05 was considered to indicate a statistically
significant difference.
Results
MCU increases the proliferative capacity
of ESCC cells
To investigate the biological role of MCU on ESCC
progression, MCU was stably overexpressed or knocked down in three
ESCC cell lines (KYSE-150, KYSE-410, and TE-1 cells; Fig. 1A and B). The transfection efficacy
was confirmed using western blotting and RT-qPCR. The results
showed that transfection of the OE-MCU vector resulted in
overexpression of MCU at the protein and mRNA level in KYSE-150,
KYSE-410, and TE-1 cells (Figs.
1C-F and S1D-F). Moreover,
both sh-MCU#1 and sh-MCU#2 significantly reduced MCU protein and
mRNA expression levels compared with the sh-NC transfected cells in
all three ESCC cell lines (Figs.
1G-J and S1A-C). These
results indicated that MCU was separately successfully
overexpressed as well as knocked down in the ESCC cells. the
proliferative capacity of ESCC cells was determined using a colony
formation assay; MCU overexpression significantly increased the
proliferation of KYSE-150, KYSE-410, and TE-1 cells (Fig. 1K-P). Conversely, MCU knockdown
substantially decreased the proliferation of ESCC cells. These
results suggested that MCU regulated the proliferation of ESCC
cells.
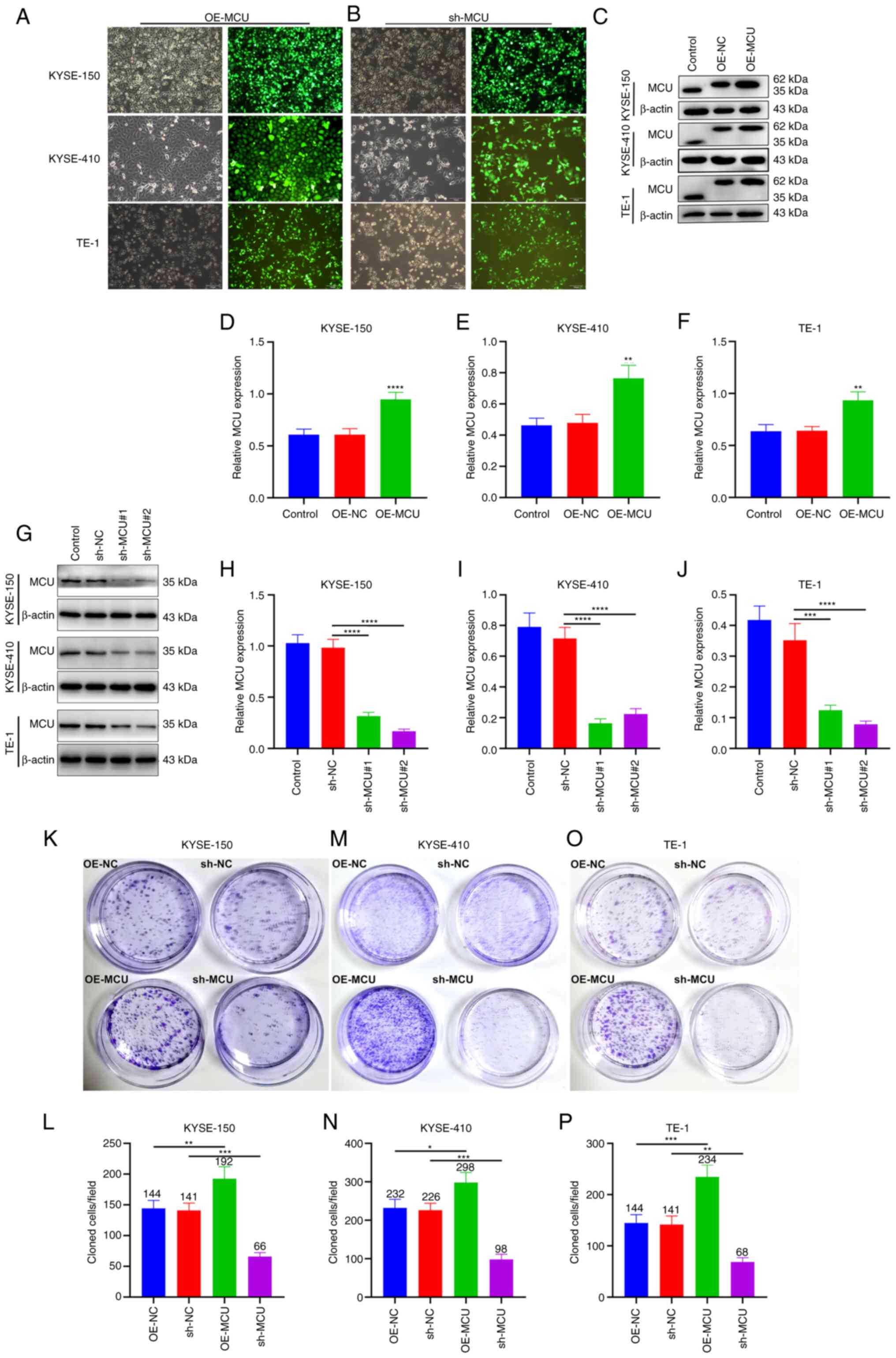 | Figure 1MCU accelerates the proliferative
capacity of ESCC cells. (A and B) Construction of
MCU-overexpressing or MCU-knockdown KYSE-150, KYSE-410, and TE-1
cells. GFP-PURO-MCU transfection was observed using a fluorescent
microscope (magnification, ×100). Scale bar, 100 µm. (C-F)
MCU expression was examined in MCU-overexpressing KYSE-150,
KYSE-410, and TE-1 cells by western blotting.
**P<0.01, ****P<0.0001 vs. OE-NC group.
(G-J) KYSE-150, KYSE-410, and TE-1 cells were transfected with
shRNAs targeting MCU or sh-NC for 48 h, and MCU expression was
detected by western blot. ***P<0.001,
****P<0.0001 vs. sh-NC group. (K-P) Following stable
overexpression or knockdown of MCU in KYSE-150, KYSE-410, and TE-1
cells, the proliferative capacity was assessed using a colony
formation assay. *P<0.05, **P<0.01,
***P<0.001 vs. OE-NC or sh-NC group. Data are
presented as the mean ± SD. MCU, mitochondrial calcium uniporter;
ESCC, esophageal squamous cell carcinoma; PURO, puromycin; OE,
overexpression; shRNA, short hairpin RNA; NC, negative control. |
MCU facilitates the acquisition of
metastatic phenotypes and the MMP of ESCC cells
The effect of MCU on cell migration and invasion was
investigated using Transwell and wound healing assays. The results
of the Transwell assays showed that stably overexpressed MCU
significantly increased cell migration of KYSE-150, KYSE-410, and
TE-1 cells (Fig. 2A-D).
Conversely, MCU knockdown substantially reduced the migratory
capacity of the ESCC cell lines (Fig.
2A-D). Furthermore, the wound healing assay showed that MCU
overexpression significantly increased wound closure in all three
cell lines (Fig. 2E-H). In
contrast, wound closure was significantly reduced following MCU
knockdown. Using JC-1 staining, the MMP was investigated in the
three cell lines. MCU overexpression markedly increased the MMP
(Fig. 2I-L), whereas MCU knockdown
resulted in a significant decrease in the MMP of cells.
Collectively, these results indicated that MCU regulated the
acquisition of a metastatic phenotype and the MMP of ESCC cells,
suggesting that the effect of MCU on ESCC cell metastasis is
associated with its ability to modulate the MMP.
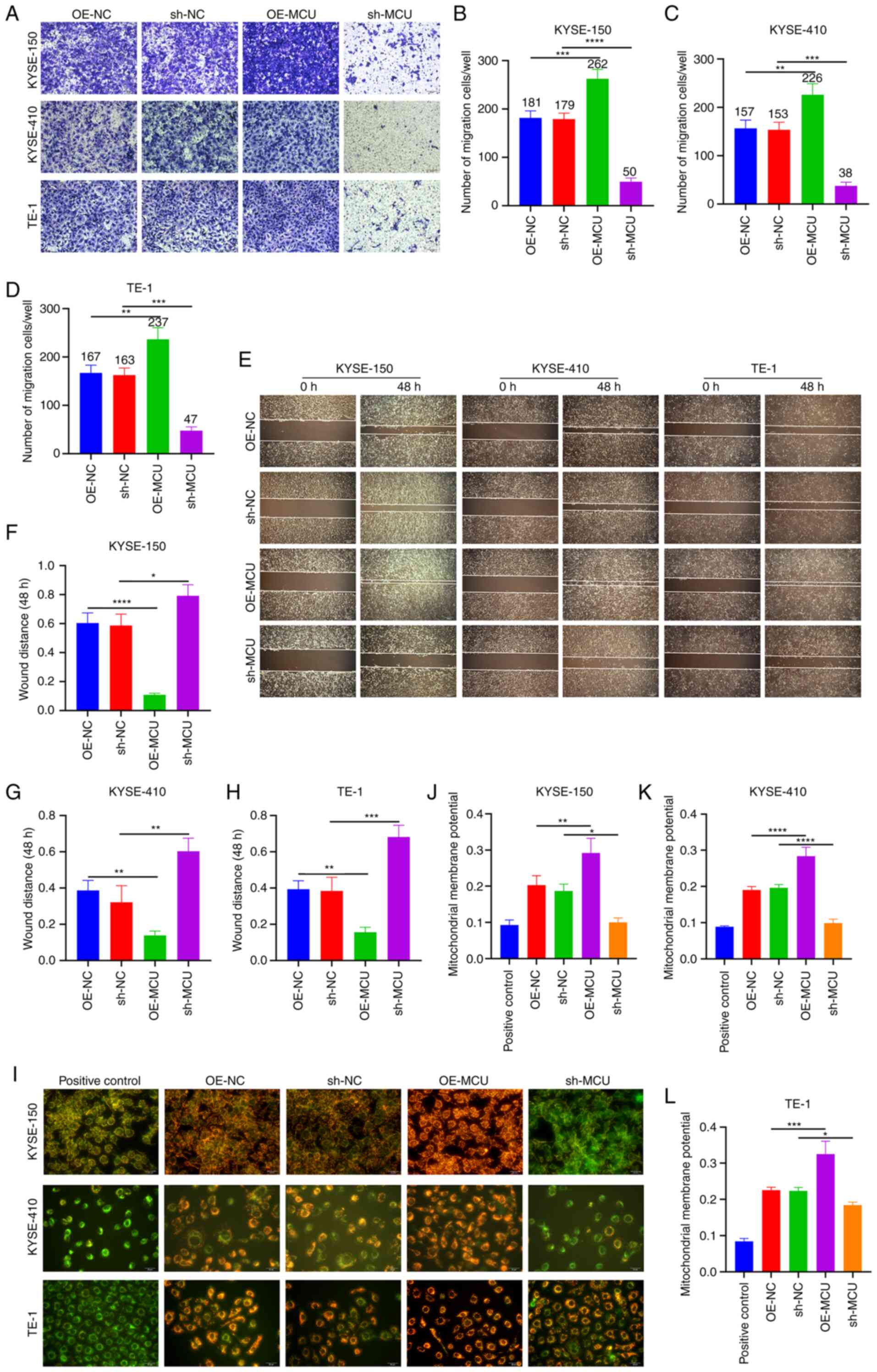 | Figure 2MCU facilitates a metastatic
phenotype and regulates the MMP of ESCC cells. (A-D) MCU was stably
overexpressed or knocked down in KYSE-150, KYSE-, and TE-1 cells.
The migration of cells was assessed using Transwell assays. Scale
bar, 50 µm. (E-H) Wound healing assays were performed using
the established ESCC cells. Scale bar, 200 µm. (I-L) The MMP
was detected in the established ESCC cells. Scale bar, 20
µm. n=3. Data are presented as the mean ± SD.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. MCU,
mitochondrial calcium uniporter; ESCC, esophageal squamous cell
carcinoma; MMP, mitochondrial membrane potential; OE,
overexpression; shRNA, short hairpin RNA; NC, negative control. |
MCU increases MICU2, MICU1, and PD-L1
expression in ESCC cells
The regulatory roles of MCU on MICU2, MICU1, and
PD-L1 expression were determined through western blotting. In
KYSE-150 cells, MCU overexpression markedly increased the
expression of MICU2, MICU1, and PD-L1. In contrast, MICU2, MICU1,
and PD-L1 expression was significantly reduced when MCU expression
was knocked down the three ESCC cell lines (Fig. S2). Immunofluorescence analysis was
used to determine whether MCU affected MICU2, MICU1, and PD-L1
expression. Consistent with the results of western blotting, MCU
overexpression significantly increased MICU2, MICU1, and PD-L1
expression in the three cell lines (Figs. 3 and S3). In contrast, following MCU
knockdown, MICU2, MICU1, and PD-L1 expression was significantly
decreased. Thus, MCU increased MICU1, MICU2, and PD-L1 expression
in ESCC cells, suggesting a possible regulatory association between
MCU and PD-L1.
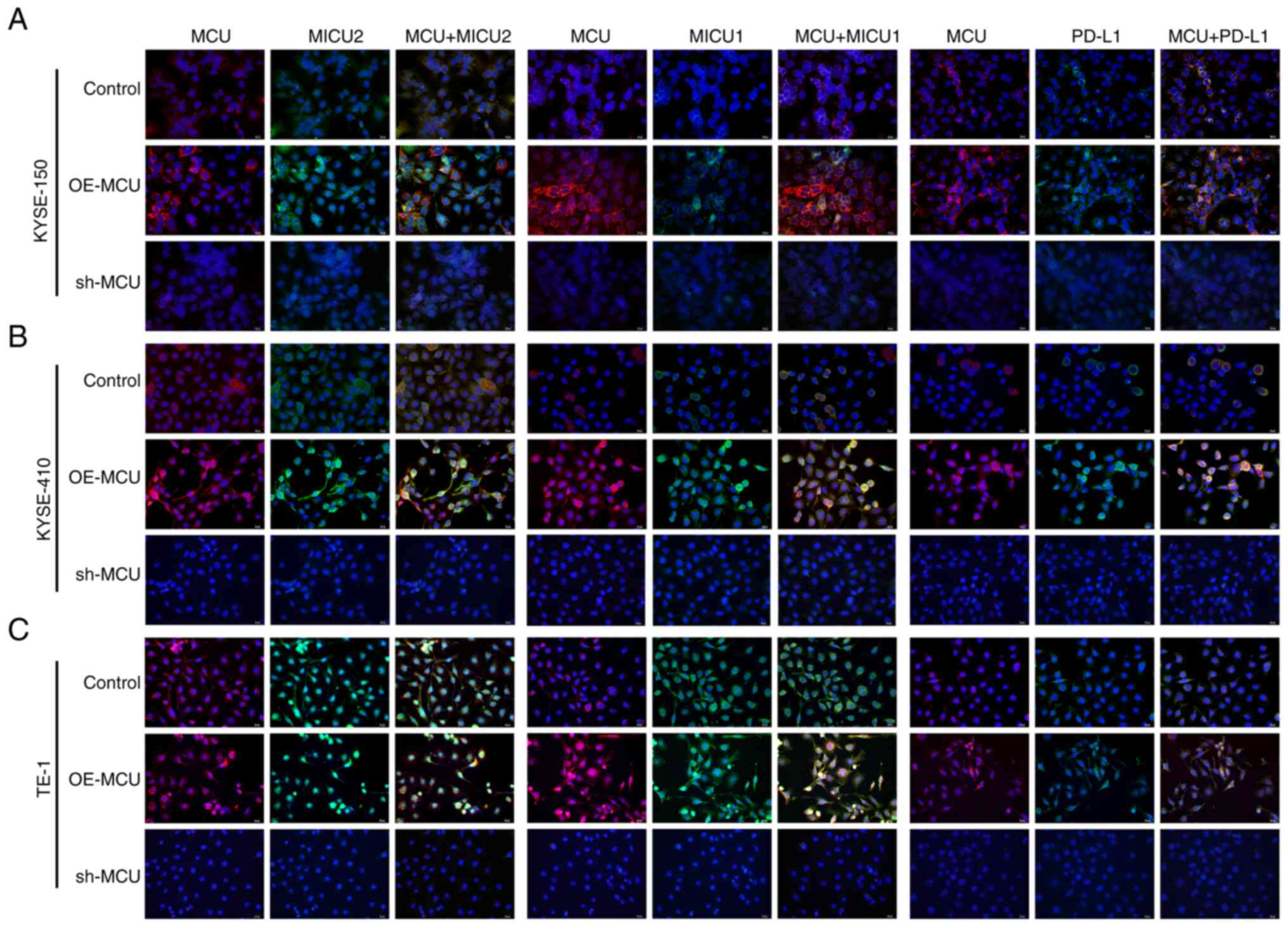 | Figure 3MCU increases MICU1, MICU2, and PD-L1
expression in ESCC cells. (A-C) Immunofluorescence analysis was
used to detect the expression of MCU, MICU1, MICU2, and PD-L1 in
KYSE-150, KYSE-410, and TE-1 with MCU overexpressed or knocked
down. Scale bar, 20 µm. MCU, mitochondrial calcium
uniporter; ESCC, esophageal squamous cell carcinoma. |
MCU overexpression upregulates MICU2,
MICU1, and PD-L1 expression in cisplatin-resistant ESCC cells
Here, cisplatin-resistant KYSE-150 (KYSE-150-CDDP),
KYSE-410 (KYSE-410-CDDP), and TE-1 (TE-1-CDDP) cells were
established through a stepwise increase in exposure to higher
concentrations of cisplatin. In cisplatin-resistant ESCC cells, the
effects of MCU on MICU2, MICU1, and PD-L1 expression were assessed.
Western blotting confirmed that MCU was stably overexpressed and
knocked down in KYSE-150-CDDP cells (Fig. 4A and B). Moreover, MCU
overexpression substantially increased the expression of MICU2,
MICU1, and PD-L1 in KYSE-150-CDDP cells (Fig. 4C-E). Conversely MCU knockdown
substantially decreased the expression of MICU2, MICU1, and PD-L1
in KYSE-150-CDDP cells. Similar results were also observed in
KYSE-410-CDDP (Fig. 4F-J) and
TE-1-CDDP cells (Fig. 4K-O).
Immunofluorescence analysis was performed to observe the influence
of MCU on MICU2, MICU1, and PD-L1 expression in cisplatin-resistant
ESCC cells. The data confirmed that MCU overexpression markedly
increased MICU2, MICU1, and PD-L1 expression in KYSE-150-CDDP
(Fig. 5A-E), KYSE-410-CDDP
(Fig. 5F-J), and TE-1-CDDP cells
(Fig. 5K-O). In contrast, MCU
knockdown significantly decreased MICU1, MICU2, and PD-L1
expression in cisplatin-resistant ESCC cells. The regulatory
relationship between MCU and PD-L1 was also observed in the
cisplatin-resistant ESCC cells.
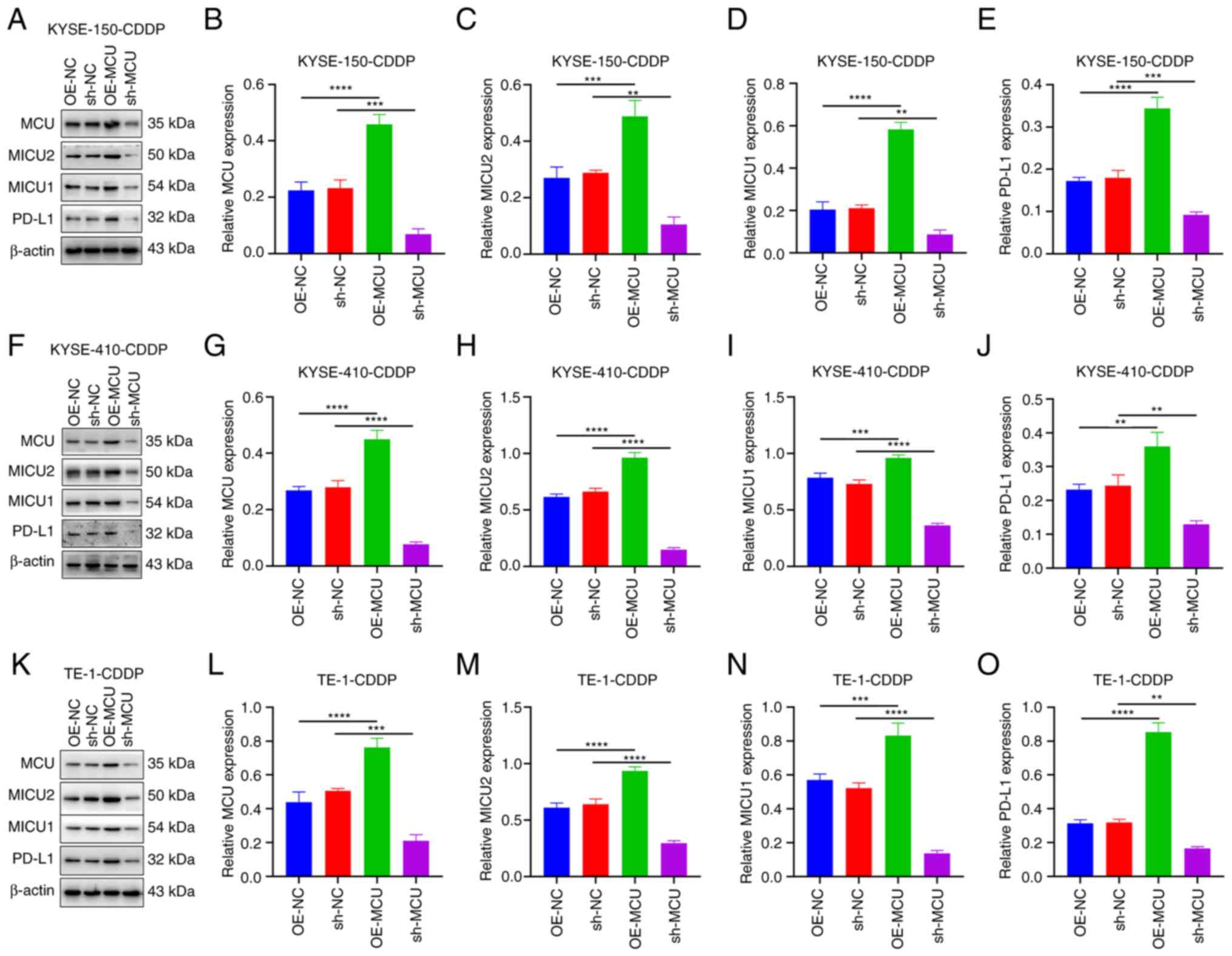 | Figure 4MCU overexpression upregulates MICU1,
MICU2, and PD-L1 expression in cisplatin-resistant ESCC cells.
(A-O) Western blotting was used to determine the expression of MCU,
MICU1, MICU2, and PD-L1 expression in cells overexpressing or
knocked down KYSE-150-CDDP, KYSE-410-CDDP, and TE-1-CDDP cells.
Data are presented as the mean ± SD. **P<0.01,
***P<0.001, ****P<0.0001. MCU,
mitochondrial calcium uniporter; ESCC, esophageal squamous cell
carcinoma; CDDP, cisplatin-resistant; OE, overexpression; shRNA,
short hairpin RNA; NC, negative control. |
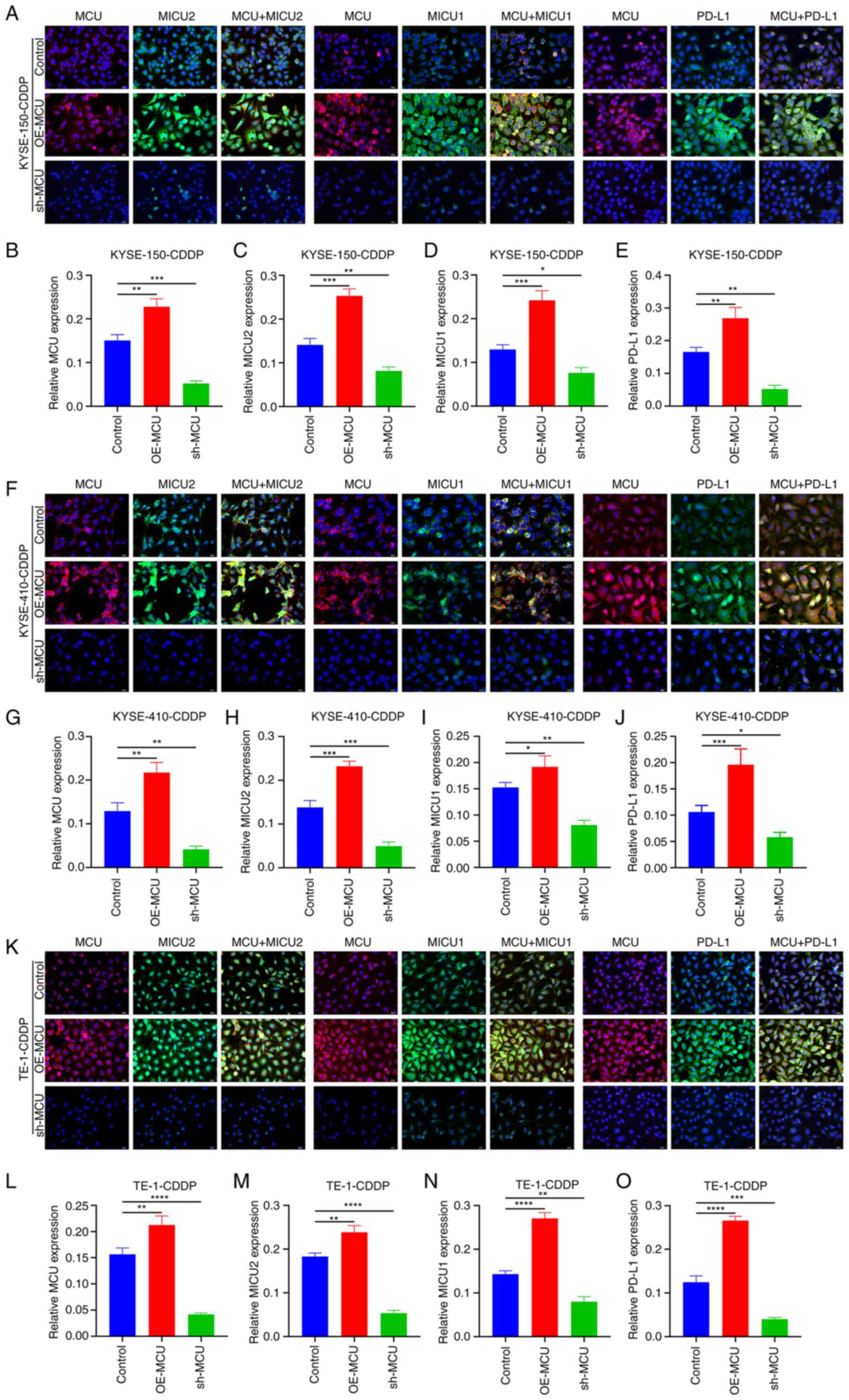 | Figure 5The effects of MCU on MICU1, MICU2,
and PD-L1 expression in cisplatin-resistant ESCC cells. When MCU
was stably overexpressed or knocked down, immunofluorescence
analysis was used to determine the expression of MCU, MICU1, MICU2,
and PD-L1 expression in (A-E) KYSE-150-CDDP, (F-J) KYSE-410-CDDP,
and (K-O) TE-1-CDDP cells. Scale bar, 20 µm. Data are
presented as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. MCU, mitochondrial calcium uniporter;
ESCC, esophageal squamous cell carcinoma; CDDP,
cisplatin-resistant; Ns, not significant; OE, overexpression;
shRNA, short hairpin RNA; OE, overexpression; shRNA, short hairpin
RNA. |
MCU knockdown decreases the proliferation
and MMP of cisplatin-resistant ESCC cells
The role of MCU on cisplatin resistance was further
investigated in cisplatin-resistant ESCC cells. The results of the
colony formation assays showed that MCU overexpression
significantly enhanced the proliferative capacity of
cisplatin-resistant KYSE-150, KYSE-410, and TE-1 cells (Fig. 6A-D). Conversely, MCU knockdown
significantly reduced the proliferative ability of KYSE-150-CDDP,
KYSE-410-CDDP, and TE-1-CDDP cells. As shown in Fig. 6E-H, the MMP of KYSE-150-CDDP,
KYSE-410-CDDP, and TE-1-CDDP cells was markedly increased by MCU
overexpression. In contrast, MCU knockdown significantly reduced
the MMP of cisplatin-resistant ESCC cells.
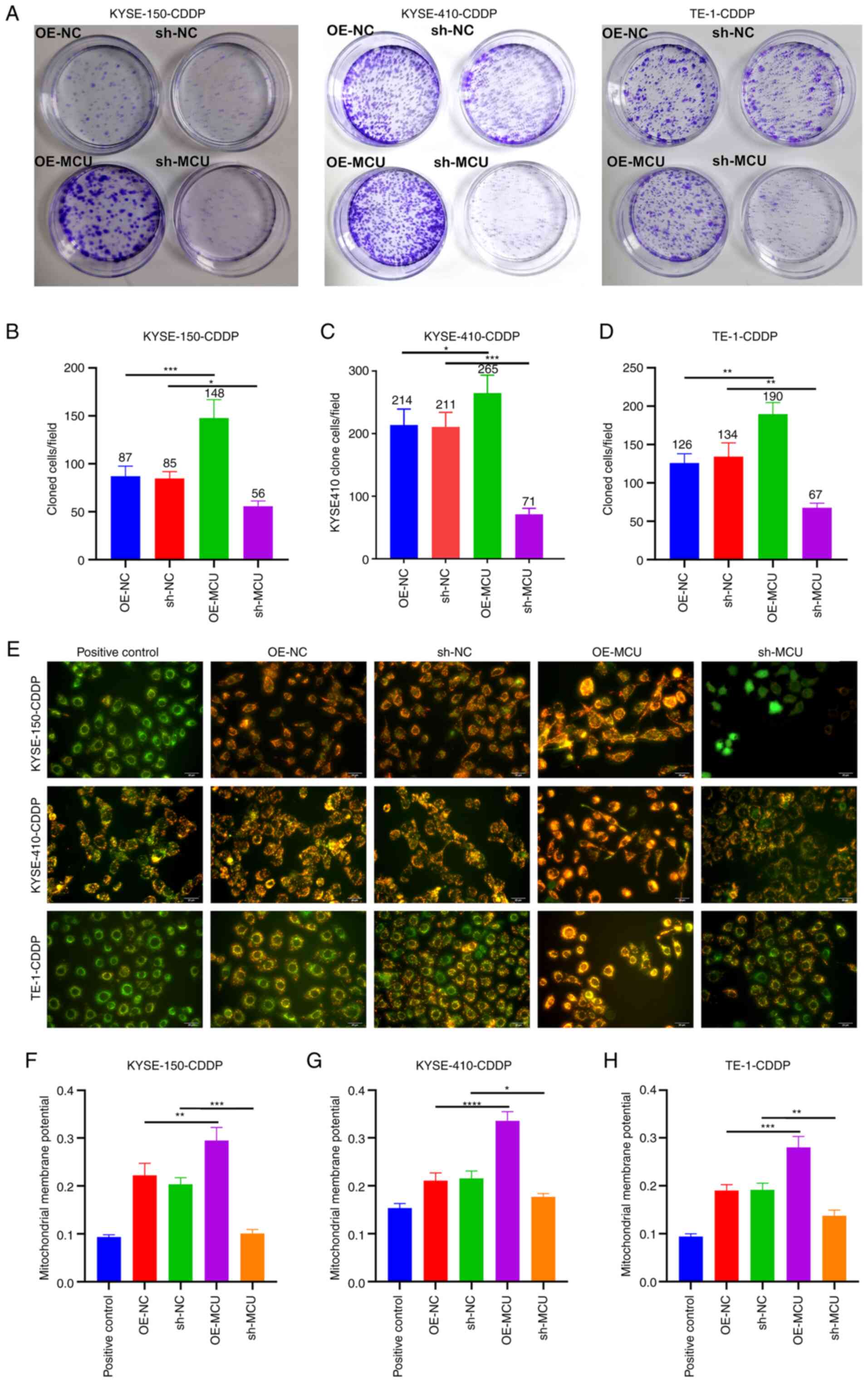 | Figure 6MCU inhibition reduces the
proliferation and MMP of cisplatin-resistant ESCC cells. (A-D) MCU
was stably overexpressed or knocked down, and the proliferative
capacities of KYSE-150-CDDP, KYSE-410-CDDP, and TE-1-CDDP cells
were evaluated using colony formation assays. (E-H) The
mitochondrial membrane potential was determined in KYSE-150-CDDP,
KYSE-410-CDDP, and TE-1-CDDP cells in which MCU had been
overexpressed or knocked down. Scale bar, 20 µm. Data are
presented as the mean ± SD of three repeats. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. MCU, mitochondrial calcium uniporter;
ESCC, esophageal squamous cell carcinoma; CDDP,
cisplatin-resistant; OE, overexpression; shRNA, short hairpin
RNA. |
MCU inhibition reduces the migration of
cisplatin-resistant ESCC cells
The effects of MCU on the migratory capacity of
cisplatin-resistant ESCC cells were observed using wound healing
and Transwell assays. As shown in Fig.
7A-D, MCU upregulation significantly increased wound closure of
KYSE-150-CDDP, KYSE-410-CDDP, and TE-1-CDDP cells. Conversely, MCU
knockdown significantly decreased wound closure in the
cisplatin-resistant ESCC cells. The results of the Transwell assays
also showed that MCU overexpression significantly increased the
number of KYSE-150-CDDP, KYSE-410-CDDP, and TE-1-CDDP cells that
had migrated (Fig. 7E-H); the
opposite results were observed when MCU was stably knocked down in
the cisplatin-resistant ESCC cells. The migratory and invasive
abilities of cisplatin-resistant and non-cisplatin-resistant ESCC
cells were compared, and the results showed that the invasive and
migration capacity of cisplatin-resistant ESCC cells was not
significantly increased, but decreased compared with the
non-cisplatin-resistant cells. Interestingly, overexpression or
knockdown of MCU also regulated the invasive and migration capacity
of cisplatin-resistant cells (Fig.
S4). These results further suggest that MCU plays an important
role in the metastasis of cisplatin-resistant and
non-cisplatin-resistant ESCC cells.
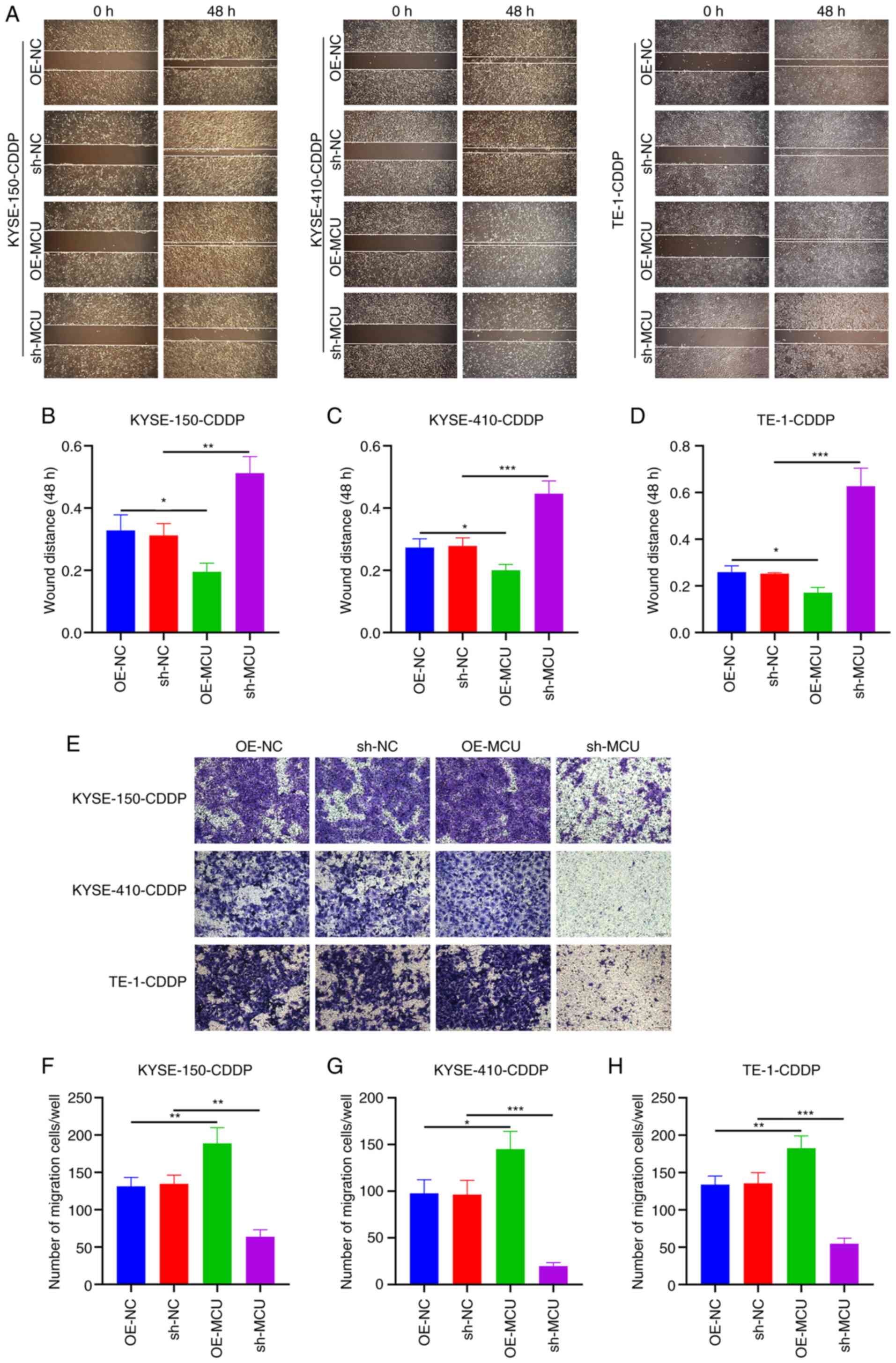 | Figure 7MCU knockdown decreased the migratory
capacity of cisplatin-resistant ESCC cells. (A-D) MCU was stably
overexpressed or knocked down, and wound healing was measured in
KYSE-150-CDDP, KYSE-410-CDDP, and TE-1-CDDP cells. Scale bar, 200
µm. (E-H) Transwell migration assays were performed on the
MCU overexpressing or MCU knocked down KYSE-150-CDDP,
KYSE-410-CDDP, and TE-1-CDDP cells. Scale bar, 50 µm. Data
are presented as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001. MCU,
mitochondrial calcium uniporter; ESCC, esophageal squamous cell
carcinoma; CDDP, cisplatin-resistant; OE, overexpression; shRNA,
short hairpin RNA. |
MCU knockdown suppresses tumor growth and
cisplatin resistance in ESCC xenograft mice
BALB/c nude mice were subcutaneously injected with
sh-NC-transfected KYSE-150 cells, sh-NC-transfected KYSE150-CDDP
cells, sh-MCU-transfected KYSE-150 cells, or sh-MCU-transfected
KYSE-150-CDDP cells (2×106 cells in 0.1 ml) in the back
(n=5 per group). The tumor volumes were determined on days 7, 14,
and 21. Mice were euthanized on day 21 after inoculation of cells.
The results showed that MCU knockdown significantly reduced the
tumor volume both in KYSE-150 and KYSE-150-CDDP cell xenograft mice
(Fig. 8A-E). The expression of
proliferation markers CyclinD1 and Ki-67 was examined in the
dissected tumors using western blotting. Expression of CyclinD1 and
Ki-67 was significantly increased in cisplatin-resistant ESCC
xenograft tumors compared with the ESCC xenograft tumors (Fig. 8F-H). MCU knockdown substantially
reduced the expression of CyclinD1 and Ki-67 both in ESCC xenograft
tumors and cisplatin-resistant ESCC xenograft tumors. Similar
findings were observed regarding Ki-67 expression suing IHC
(Fig. 8I and J). Thus, MCU
knockdown significantly reduced tumor growth in cisplatin-resistant
and non-cisplatin-resistant ESCC cell xenograft mice.
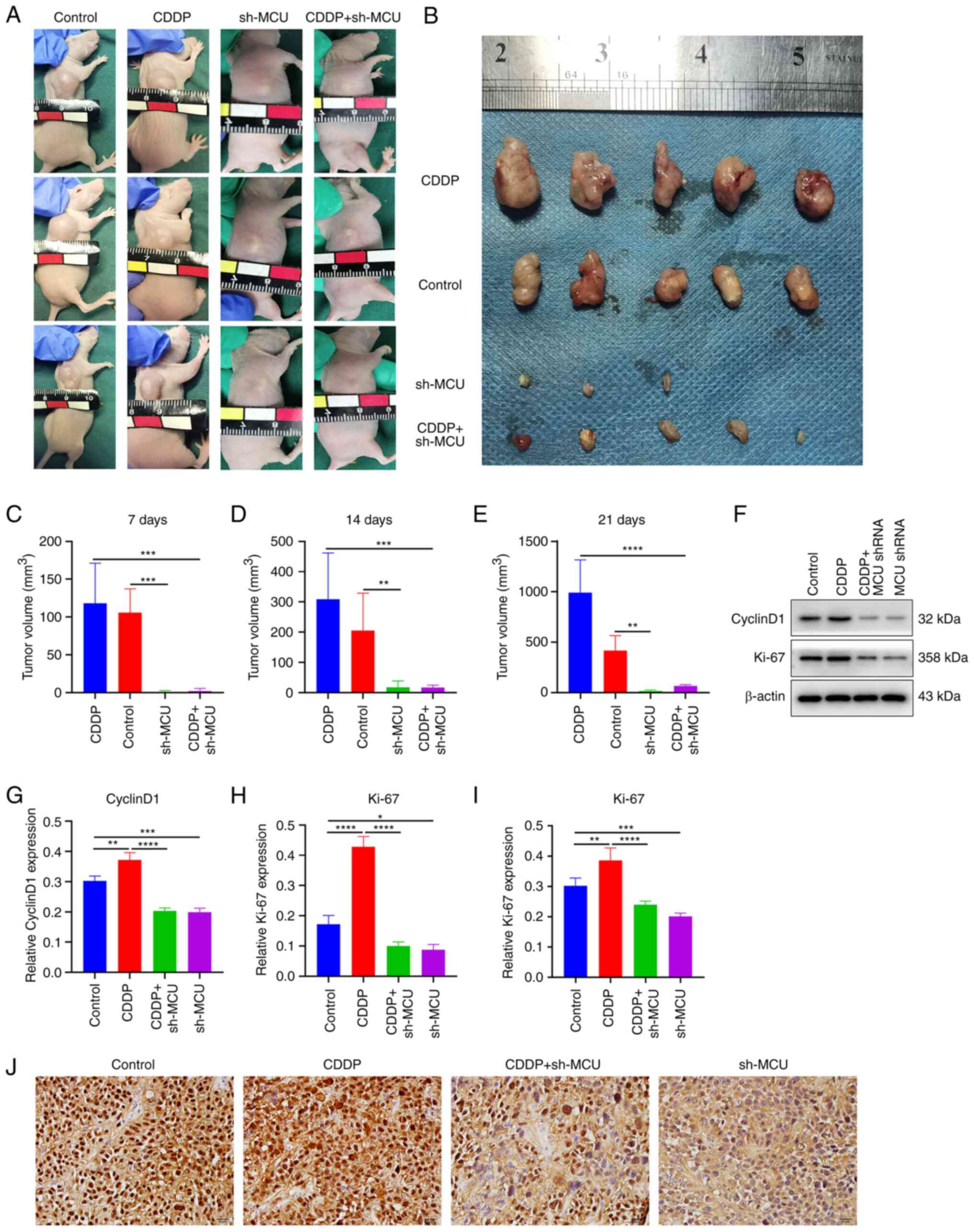 | Figure 8MCU knockdown reduced tumor growth
and cisplatin resistance in ESCC xenograft mouse models. (A) BALB/c
nude mice were subcutaneously injected with sh-NC-transfected
KYSE-150 cells (Control), sh-NC-transfected KYSE-150-CDDP cells
(CDDP), sh-MCU-transfected KYSE-150 cells (sh-MCU), or
sh-MCU-transfected KYSE-150-CDDP cells (CDDP+sh-MCU) (n=5 each
group). Images of xenograft mice were acquired 21 days following
the injection of cells. (B) Representative images of tumors removed
after 21 days. (C-E) Tumor volume was measured on days 7, 14, and
21 after the injection of cells. (F-H) CyclinD1 and Ki-67
expression was detected in the dissected tumors using western
blotting. (I and J) Ki-67 expression was examined in the dissected
tumors by immunohistochemistry. Scale bar, 20 µm. Data are
presented as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. MCU, mitochondrial calcium uniporter;
ESCC, esophageal squamous cell carcinoma; OE, overexpression;
shRNA, short hairpin RNA. |
MCU knockdown reduces MICU2, MICU1, and
PD-L1 expression and EMT in cisplatin-resistant ESCC xenograft
mice
Using western blotting, the expression of MCU,
MICU2, MICU1, PD-L1, and EMT markers (Vimentin and β-catenin) was
assessed in xenograft tumors. Compared with the ESCC xenograft
tumors, MCU, MICU2, MICU1, PD-L1, Vimentin, and β-catenin exhibited
higher expression in cisplatin-resistant ESCC xenograft tumors
(Fig. 9A-G). MCU inhibition
reduced the expression of MCU, MICU2, MICU1, PD-L1, Vimentin, and
β-catenin both in ESCC xenograft tumors and cisplatin-resistant
ESCC xenograft tumors (Fig. 9A-G).
IHC analysis also showed that the expression of MCU, MICU2, MICU1,
PD-L1, and Vimentin expression was significantly increased, whilst
E-cadherin expression was significantly decreased in
cisplatin-resistant ESCC xenograft tumors compared with the ESCC
xenograft tumors (Fig. 9H-N). MCU
inhibition significantly reduced the expression of MCU, MICU2,
MICU1, PD-L1, and Vimentin, but increased E-cadherin expression
both in ESCC xenograft tumors and cisplatin-resistant ESCC
xenograft tumors. These results were confirmed using
immunofluorescence analysis (Fig.
10). Thus, MCU knockdown reduced MICU2, MICU1, and PD-L1
expression, and abrogated EMT in cisplatin-resistant and
non-cisplatin-resistant ESCC xenografts.
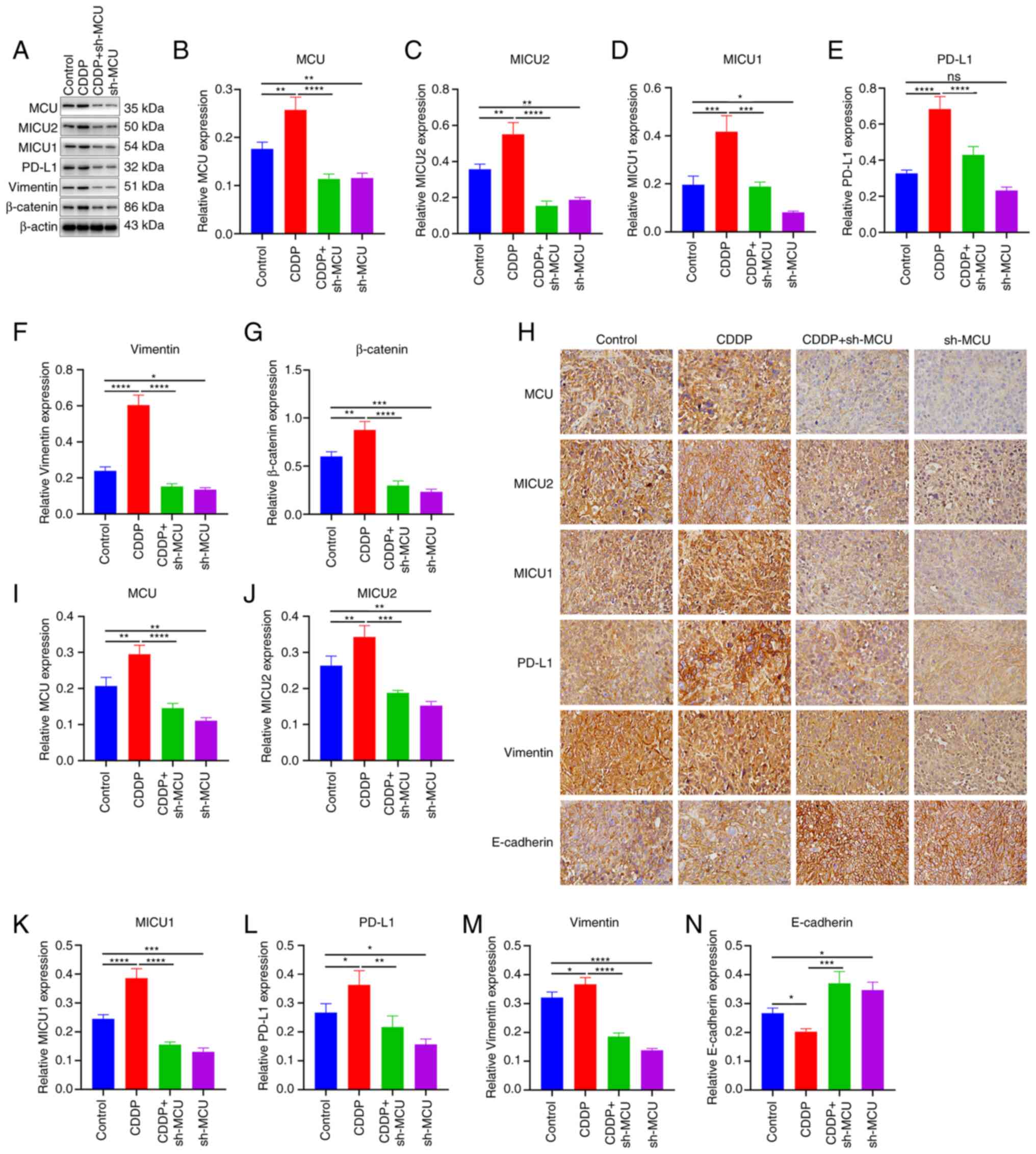 | Figure 9MCU knockdown decreased MICU1, MICU2,
and PD-L1 expression, and abrogated EMT in cisplatin-resistant ESCC
xenograft mice. (A-G) MCU, MICU1, MICU2, PD-L1, Vimentin, and
β-catenin expression was detected in sh-NC-transfected KYSE-150
(Control), sh-NC-transfected KYSE-150-CDDP (CDDP),
sh-MCU-transfected KYSE-150 (sh-MCU), or sh-MCU-transfected
KYSE-150-CDDP (CDDP+sh-MCU) cell-xenograft tumors using western
blotting. (H-N) MCU, MICU1, MICU2, PD-L1, Vimentin, and E-cadherin
expression was determined in Control, CDDP, sh-MCU, and CDDP+sh-MCU
cell-xenograft tumors by IHC. Scale bar, 20 µm. Data are
presented as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001
****P<0.0001. MCU, mitochondrial calcium uniporter;
ESCC, esophageal squamous cell carcinoma; OE, overexpression;
shRNA, short hairpin RNA. |
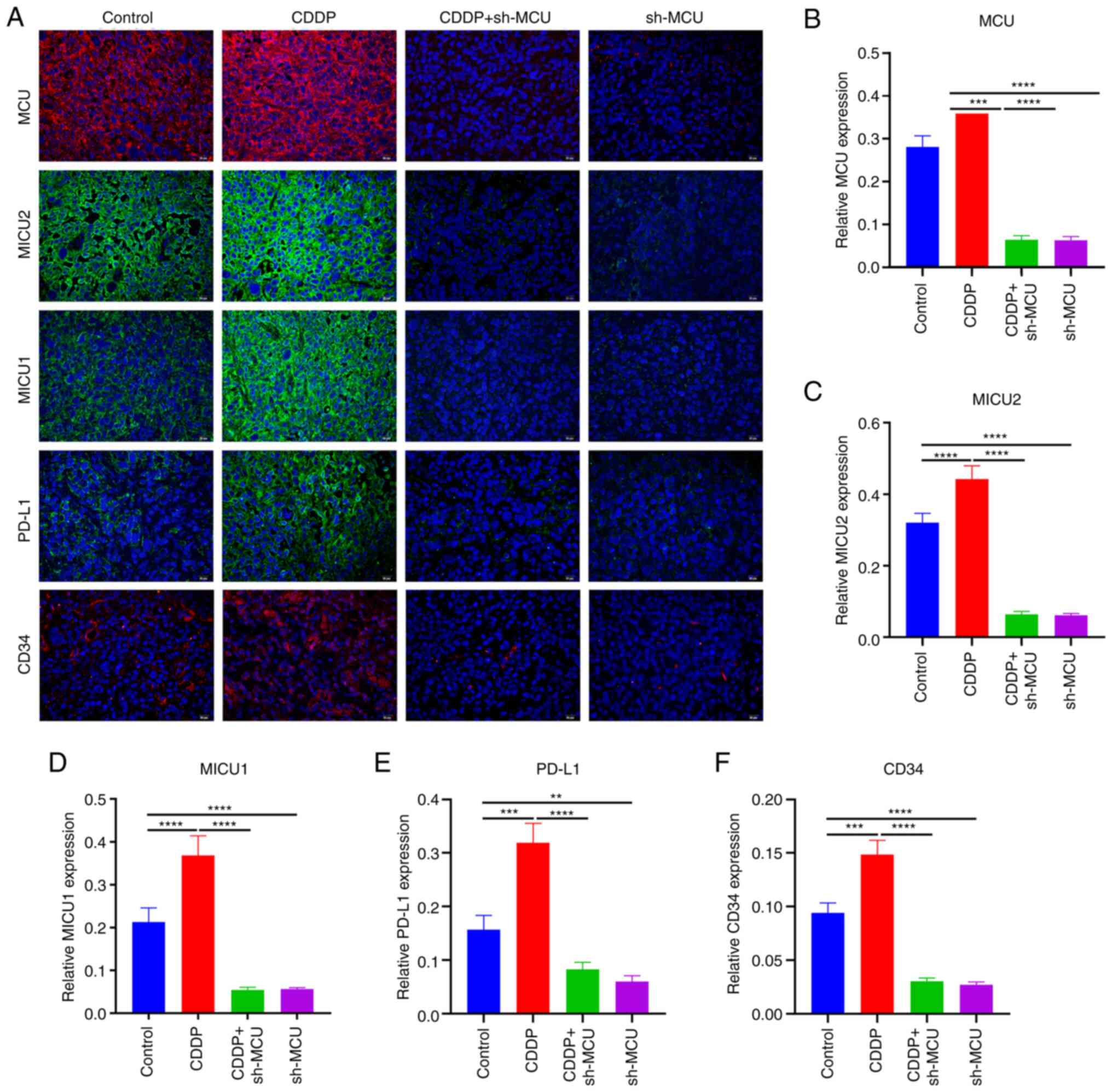 | Figure 10MCU knockdown decreases MICU1, MICU2,
PD-L1, and CD34 expression in cisplatin-resistant ESCC xenograft
mice. (A-F) MCU, MICU1, MICU2, PD-L1, and CD34 expression were
determined using immunofluorescence analysis. Scale bar, 20
µm Data are presented as the mean ± SD.
**P<0.01, ***P<0.001,
****P<0.0001. OE, overexpression; shRNA, short
hairpin RNA; MCU, mitochondrial calcium uniporter; CDDP,
cisplatin-resistant. |
MCU knockdown reduces angiogenesis in
cisplatin-resistant ESCC in vivo
Using immunofluorescence analysis, the expression of
CD34 (an indicator of microvessel density in xenograft tumors) was
detected. The results showed that CD34 expression was increased in
cisplatin-resistant ESCC xenograft tumors when compared with normal
ESCC xenograft tumors (Fig. 10A and
F). MCU inhibition substantially decreased the expression of
CD34 both in ESCC xenograft tumors and cisplatin-resistant ESCC
xenograft tumors. Thus, the results data indicated that MCU
knockdown may inhibit angiogenesis in cisplatin-resistant and
non-cisplatin-resistant ESCC xenograft mice.
Discussion
Cisplatin-based regimens have been routinely applied
for the treatment of patients with ESCC (19). However, the administration of
cisplatin is typically accompanied by the development of cisplatin
resistance (23). Understanding
the mechanisms underlying the acquisition of cisplatin resistance
may enable the development of more effective treatment schemes,
thereby overcoming chemotherapy resistance and improving ESCC
clinical outcomes (24-26). A tumor is a heterogeneous cell
mixture that harbors multiple genetic variations and gene
expression patterns (27,28). In the present study, it was
demonstrated that targeting MCU suppressed tumor growth and
decreased PD-L1 expression in both cisplatin-resistant and
non-cisplatin-resistant ESCC cells. In future studies, additional
ESCC cell lines and subcutaneous tumor models of esophageal cancer
cells in nude mice will be used to elucidate the molecular
regulatory mechanism of reversing cisplatin resistance through MUC
knockout.
In three ESCC cell lines, MCU overexpression
markedly enhanced proliferation, migration, and MMP, and the
opposite results were observed when MCU was stably knocked down.
The carcinogenic effects of MCU have been reported in various types
of cancer. As an example, MCU facilitates migration, invasion,
angiogenesis, and tumor growth of gastric cancer (29). Here, the therapeutic effects of MCU
inhibition were also confirmed in cisplatin-resistant ESCC cells,
indicating that targeting MCU may overcome cisplatin resistance.
The expression levels of cyclinD1 and Ki-67 represent the changes
in the G1 and G2 phases, which are closely related to the
malignancy of the tumor and the treatment and prognosis of the
patients (30). In the present
study, an ESCC xenograft tumor mouse model and a
cisplatin-resistant ESCC xenograft tumor mouse model were
constructed. Tumor growth was significantly enhanced by cisplatin
resistance. Targeting MCU substantially reduced ESCC and
cisplatin-resistant ESCC tumor growth. As reflected by the
expression of tumor proliferation markers (CyclinD1 and Ki-67),
tumor proliferative capacities were markedly increased in
cisplatin-resistant ESCC tumors, and this was reversed by MCU
inhibition.
Mitochondrial Ca2+ homeostasis is pivotal
in the regulation of aerobic metabolism and cell survival and plays
an important role in the prevention and treatment of cancer
metastasis. When the MCU regulates mitochondrial calcium ions,
MICU1 and MICU2 can be used as the 'switch' of calcium ions
entering mitochondria mediated by MCU. MCU forms a complex
structure with MICU1 and MICU2 to regulate the intake of
mitochondrial calcium ions, affect the MMP, and participate in the
metastasis of cancer cells (31-33).
However, the best-known mechanism for cancer metastasis is EMT. In
this study, the effect of MCU overexpression or knockout on the
expression of EMT-related markers (Vimentin and β-catenin) was
assessed. MCU could reverse the occurrence of EMT and reduce the
expression of related markers, indicating the role of MCU in the
process of cancer metastasis. As previously mentioned, MCU may
affect the expression of PD-L1 through MICU1 and MICU2 as well as
regulating MMP, highlighting novel possibilities for clinical
combination therapy.
Monoclonal antibodies targeting PD-1/PD-L1 have
exhibited durable therapeutic responses among patients with ESCC
(34). Nevertheless, only a few
patients may achieve clinical benefits (35). Previously, neoadjuvant chemotherapy
was shown to increase PD-L1 expression in ESCC (36). Cisplatin enhances the antitumor
immunity of PD-1/PD-L1 inhibitors in ESCC but upregulates PD-L1
expression in cancer cells (37).
Here, the results showed that PD-L1 expression was significantly
upregulated in cisplatin-resistant ESCC xenograft tumors compared
to ESCC xenograft tumors, indicating that PD-L1 was associated with
cisplatin resistance. A recent study found that PD-L1 could enhance
cisplatin resistance in gastric cancer (38). MCU knockdown substantially reduced
PD-L1 expression in ESCC and cisplatin-resistant ESCC xenograft
tumors. The above findings indicated that the interaction between
MCU and PD-L1 may promote cisplatin resistance in ESCC. It was
hypothesized that MCU may affect the expression of PD-L1 by
regulating MICU1, MICU2, and the MMP.
The EMT process may induce cisplatin resistance by
transforming relatively motionless epithelial cells into mobile
mesenchymal cells and altering cell-cell adhesion and the cellular
extracellular matrix, thereby promoting the migration of cancer
cells (39,40). The EMT process is reversible and
may be modulated through various molecular signals (41). Here, vimentin and β-catenin
expression was significantly increased and E-cadherin was markedly
decreased in cisplatin-resistant ESCC xenograft tumors, indicating
the activation of the EMT process during cisplatin resistance.
However, MCU knockdown distinctly impaired the EMT process in
cisplatin-resistant ESCC xenograft tumors. This indicated that
targeting MCU could mitigate cisplatin resistance in ESCC by
suppressing the EMT process.
Angiogenesis during carcinogenesis supplies oxygen
and nutrients to proliferative tumor cells, which serves as a
therapeutic target against ESCC (42). Silencing oncogenes involved in
angiogenesis is required for the treatment of ESCC (43). The results of the present study
demonstrated that CD34-labeled angiogenesis was markedly enhanced
in cisplatin-resistant ESCC xenograft tumors and that targeting MCU
substantially reduced angiogenesis in ESCC and cisplatin-resistant
ESCC xenograft tumors. The results also showed the morphological
alterations of microvessels in cisplatin-resistant ESCC and the
therapeutic effects of MCU inhibition in targeting angiogenesis in
ESCC.
The present study reported for the first time the
oncogenic role of MCU in ESCC and demonstrated the inhibitory
effect of MCU on tumor growth in cisplatin-resistant and
non-cisplatin-resistant ESCC, indicating that MCU knockdown may
play an anticancer role by reducing the expression of PD-L1 and EMT
markers. Thus, targeting MCU may be a promising therapeutic
strategy for the treatment of ESCC.
In conclusion, the results of the present study
suggested that knockdown of MCU reduced the proliferation,
migration, and MMP in ESCC cell lines and cisplatin-resistant ESCC
cell lines. Furthermore, targeting MCU suppressed tumor growth and
alleviated cisplatin resistance in ESCC xenograft mouse models.
Collectively, these results highlighted a pharmacological strategy
in targeting MCU that may be utilized for mitigating cisplatin
resistance in ESCC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YM, XW, FZ, and SY conceived the study, and wrote,
revised and edited the manuscript. YL analyzed the data, and wrote,
revised, and edited the manuscript. YH and HY performed the
experiments and analyzed the data. XM performed the experiments and
drafted the manuscript. HL performed the experiments and data
analysis. RH and WL performed the experiments and revised the
manuscript for intellectual content. XZ performed the experiments
and drafted the manuscript. XZ and BCC analyzed the data and
revised the manuscript. YM and XW confirm the authenticity of all
the raw data. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The protocols used in the present study strictly
adhered to the Guidelines for the Care and Use of Laboratory
Animals and was approved by the Ethics Committee of Ningxia Medical
University (approval no. 2020880).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ESCC
|
esophageal squamous cell carcinoma
|
|
MCU
|
mitochondrial calcium uniporter
|
|
shRNA
|
short hairpin RNA
|
|
NC
|
negative control
|
|
GFP
|
green fluorescent protein
|
|
HRP
|
horseradish peroxidase
|
|
MMP
|
mitochondrial membrane potential
|
|
IHC
|
immunohistochemistry
|
Acknowledgments
Not applicable.
Funding
This study was supported by funding from the Natural Science
Foundation of Ningxia (grant no. 2021AAC03343), Ningxia Hui
Autonomous Region Key Research and Development Plan Project
Specification (grant nos. 2020BEG03001 and 2018BEG02007), and the
Science Foundation of Inner Mongolia Autonomous Region (grant no.
2020MS08179).
References
|
1
|
Arnold M, Ferlay J, van Berge Henegouwen
MI and Soerjomataram I: Global burden of oesophageal and gastric
cancer by histology and subsite in 2018. Gut. 69:1564–1571. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2017 Oesophageal Cancer Collaborators:
The global, regional, and national burden of oesophageal cancer and
its attributable risk factors in 195 countries and territories,
1990-2017: A systematic analysis for the global burden of disease
study 2017. Lancet Gastroenterol Hepatol. 5:582–597. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Laarhoven HW: Is chemotherapy for
advanced or metastatic oesophageal squamous cell carcinoma no
longer needed? Lancet Oncol. 21:743–745. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu Y, Xie C, Yang H, Ho JWK, Wen J, Han L,
Lam KO, Wong IYH, Law SYK, Chiu KWH, et al: Computed
tomography-based deep-learning prediction of neoadjuvant
chemoradiotherapy treatment response in esophageal squamous cell
carcinoma. Radiother Oncol. 154:6–13. 2021. View Article : Google Scholar
|
|
5
|
Leng XF, Daiko H, Han YT and Mao YS:
Optimal preoperative neoadjuvant therapy for resectable locally
advanced esophageal squamous cell carcinoma. Ann N Y Acad Sci.
1482:213–224. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsu PK, Yeh YC, Chien LI, Huang CS and Hsu
HS: Clinicopathological significance of pathologic complete lymph
node regression after neoadjuvant chemoradiotherapy in esophageal
squamous cell carcinoma. Ann Surg Oncol. 28:2048–2058. 2021.
View Article : Google Scholar
|
|
7
|
Chen S, Yang M, Wang C, Ouyang Y, Chen X,
Bai J, Hu Y, Song M, Zhang S and Zhang Q: Forkhead box D1 promotes
EMT and chemoresistance by upregulating lncRNA CYTOR in oral
squamous cell carcinoma. Cancer Lett. 503:43–53. 2021. View Article : Google Scholar
|
|
8
|
Chang WM, Chang YC, Yang YC, Lin SK, Chang
PM and Hsiao M: AKR1C1 controls cisplatin-resistance in head and
neck squamous cell carcinoma through cross-talk with the STAT1/3
signaling pathway. J Exp Clin Cancer Res. 38:2452019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oshimori N, Oristian D and Fuchs E: TGF-β
promotes heterogeneity and drug resistance in squamous cell
carcinoma. Cell. 160:963–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mohapatra P, Shriwas O, Mohanty S, Ghosh
A, Smita S, Kaushik SR, Arya R, Rath R, Das Majumdar SK, Muduly DK,
et al: CMTM6 drives cisplatin resistance by regulating Wnt
signaling through the ENO-1/AKT/GSK3β axis. JCI Insight.
6:e1436432021.
|
|
11
|
Liu Z, Gu S, Lu T, Wu K, Li L, Dong C and
Zhou Y: IFI6 depletion inhibits esophageal squamous cell carcinoma
progression through reactive oxygen species accumulation via
mitochondrial dysfunction and endoplasmic reticulum stress. J Exp
Clin Cancer Res. 39:1442020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Wang C, Su J, Xie Q, Ma L, Zeng L,
Yu Y, Liu S, Li S, Li Z and Sun L: Tolerance to endoplasmic
reticulum stress mediates cisplatin resistance in human ovarian
cancer cells by maintaining endoplasmic reticulum and mitochondrial
homeostasis. Oncol Rep. 34:3051–3060. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu
Y, Guo X, Ji L, Huang Q, Zhang H, et al: MCU-dependent
mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2
pathway to promote ROS production and metastasis of HCC cells.
Oncogene. 36:5897–5909. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui C, Yang J, Fu L, Wang M and Wang X:
Progress in understanding mitochondrial calcium uniporter
complex-mediated calcium signalling: A potential target for cancer
treatment. Br J Pharmacol. 176:1190–1205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vultur A, Gibhardt CS, Stanisz H and
Bogeski I: The role of the mitochondrial calcium uniporter (MCU)
complex in cancer. Pflugers Arch. 470:1149–1163. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Jin M, Wang Y, Zhu J, Tan R, Zhao
J, Ji X, Jin C, Jia Y, Ren T and Xing J: MCU-induced mitochondrial
calcium uptake promotes mitochondrial biogenesis and colorectal
cancer growth. Signal Transduct Target Ther. 5:592020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tosatto A, Sommaggio R, Kummerow C,
Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I,
Szabadkai G, et al: The mitochondrial calcium uniporter regulates
breast cancer progression via HIF-1α. EMBO Mol Med. 8:569–585.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stoica SI, Onose G, Pitica IM, Neagu AI,
Ion G, Matei L, Dragu LD, Radu LE, Chivu-Economescu M, Necula LG,
et al: Molecular aspects of hypoxic stress effects in Chronic
ethanol exposure of neuronal cells. Curr Issues Mol Biol.
45:1655–1680. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xue W, Shen Z, Li L, Zheng Y, Yan D, Kan Q
and Zhao J: Long non-coding RNAs MACC1-AS1 and FOXD2-AS1 mediate
NSD2-induced cisplatin resistance in esophageal squamous cell
carcinoma. Mol Ther Nucleic Acids. 23:592–602. 2020. View Article : Google Scholar
|
|
20
|
Cui W, Fang T, Duan Z, Xiang D, Wang Y,
Zhang M, Zhai F, Cui X and Yang L: Dihydroartemisinin sensitizes
esophageal squamous cell carcinoma to cisplatin by inhibiting sonic
hedgehog signaling. Front Cell Dev Biol. 8:5967882020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Ji H, Zhu Q, Yu X, Du J and Jiang
Z: Co-inhibition of BMI1 and Mel18 enhances chemosensitivity of
esophageal squamous cell carcinoma in vitro and in vivo. Oncol
Lett. 17:5012–5022. 2019.PubMed/NCBI
|
|
22
|
Barra GB, Santa Rita TH, Almeida ALSC,
Jácomo RH and Nery LFA: Serum has higher proportion of Janus kinase
2 V617F mutation compared to paired EDTA-whole blood sample: A
model for somatic mutation quantification using qPCR and the 2
method. Diagnostics (Basel). 10:1532020. View Article : Google Scholar
|
|
23
|
Sugimura K, Yamasaki M, Yasuda T, Yano M,
Hirao M, Fujitani K, Kimura Y, Miyata H, Motoori M, Takeno A, et
al: Long-term results of a randomized controlled trial comparing
neoadjuvant Adriamycin, cisplatin, and 5-fluorouracil vs docetaxel,
cisplatin, and 5-fluorouracil followed by surgery for esophageal
cancer (OGSG1003). Ann Gastroenterol Surg. 5:75–82. 2020.
View Article : Google Scholar
|
|
24
|
Liu H, Zhang J, Luo X, Zeng M and Xu L,
Zhang Q, Liu H, Guo J and Xu L: Overexpression of the long
noncoding RNA FOXD2-AS1 promotes cisplatin resistance in esophageal
squamous cell carcinoma through the miR-195/Akt/mTOR axis. Oncol
Res. 28:65–73. 2020. View Article : Google Scholar
|
|
25
|
Suo D, Wang L, Zeng T, Zhang H, Li L, Liu
J, Yun J, Guan XY and Li Y: NRIP3 upregulation confers resistance
to chemoradiotherapy in ESCC via RTF2 removal by accelerating
ubiquitination and degradation of RTF2. Oncogenesis. 9:752020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou T, Fu H, Dong B, Dai L, Yang Y, Yan W
and Shen L: HOXB7 mediates cisplatin resistance in esophageal
squamous cell carcinoma through involvement of DNA damage repair.
Thorac Cancer. 11:3071–3085. 2020. View Article : Google Scholar :
|
|
27
|
Ben-David U, Siranosian B, Ha G, Tang H,
Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R, et
al: Genetic and transcriptional evolution alters cancer cell line
drug response. Nature. 560:325–330. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
PCAWG Transcriptome Core Group; Calabrese
C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, Lehmann
KV, Liu F, Shiraishi Y, et al: Genomic basis for RNA alterations in
cancer. Nature. 578:129–136. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Song X, Cheng G, Zhang J, Dong L,
Bai J, Luo D, Xiong Y, Li S, Liu F, et al: The regulatory mechanism
and biological significance of mitochondrial calcium uniporter in
the migration, invasion, angiogenesis and growth of gastric cancer.
Onco Targets Ther. 13:11781–11794. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu P, Zhang X, Ni W, Fan H, Xu J, Chen Y,
Zhu J, Gu X, Yang L, Ni R, et al: Upregulated HOXC8 expression is
associated with poor prognosis and oxaliplatin resistance in
hepatocellular carcinoma. Dig Dis Sci. 60:3351–3363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kamer KJ, Jiang W, Kaushik VK, Mootha VK
and Grabarek Z: Crystal structure of MICU2 and comparison with
MICU1 reveal insights into the uniporter gating mechanism. Proc
Natl Acad Sci USA. 116:3546–3555. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Patron M, Checchetto V, Raffaello A,
Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De
Stefani D and Rizzuto R: MICU1 and MICU2 finely tune the
mitochondrial Ca2+ uniporter by exerting opposite effects on MCU
activity. Mol Cell. 53:726–737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang C, Jacewicz A, Delgado BD, Baradaran
R and Long SB: Structures reveal gatekeeping of the mitochondrial
Ca2+ uniporter by MICU1-MICU2. Elife. 9:e599912020.
View Article : Google Scholar
|
|
34
|
Lee J, Kim B, Jung HA, La Choi Y and Sun
JM: Nivolumab for esophageal squamous cell carcinoma and the
predictive role of PD-L1 or CD8 expression in its therapeutic
effect. Cancer Immunol Immunother. 70:1203–1211. 2021. View Article : Google Scholar
|
|
35
|
McBride S, Sherman E, Tsai CJ, Baxi S,
Aghalar J, Eng J, Zhi WI, McFarland D, Michel LS, Young R, et al:
Randomized phase II trial of nivolumab with stereotactic body
radiotherapy versus nivolumab alone in metastatic head and neck
squamous cell carcinoma. J Clin Oncol. 39:30–37. 2021. View Article : Google Scholar :
|
|
36
|
Fukuoka E, Yamashita K, Tanaka T, Sawada
R, Sugita Y, Arimoto A, Fujita M, Takiguchi G, Matsuda T, Oshikiri
T, et al: Neoadjuvant chemotherapy increases PD-L1 expression and
CD8+ tumor-infiltrating lymphocytes in esophageal
squamous cell carcinoma. Anticancer Res. 39:4539–4548. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tran L, Allen CT, Xiao R, Moore E, Davis
R, Park SJ, Spielbauer K, Van Waes C and Schmitt NC: Cisplatin
alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition
in head and neck squamous cell carcinoma. Cancer Immunol Res.
5:1141–1151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu L, Cai S, Deng Y, Zhang Z, Zhou X, Su Y
and Xu D: PD-1/PD-L1 enhanced cisplatin resistance in gastric
cancer through PI3K/AKT mediated P-gp expression. Int
Immunopharmacol. 94:1074432021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhu X, Chen L, Liu L and Niu X:
EMT-mediated acquired EGFR-TKI resistance in NSCLC: Mechanisms and
strategies. Front Oncol. 9:10442019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ashrafizadeh M, Zarrabi A, Hushmandi K,
Kalantari M, Mohammadinejad R, Javaheri T and Sethi G: Association
of the epithelial-mesenchymal transition (EMT) with cisplatin
resistance. Int J Mol Sci. 21:40022020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding
Q, Xu Z and Chen Y: Inhibition of ATM reverses EMT and decreases
metastatic potential of cisplatin-resistant lung cancer cells
through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 38:1492019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Y, Wang D, Peng H, Chen X, Han X, Yu
J, Wang W, Liang L, Liu Z, Zheng Y, et al: Epigenetically
upregulated oncoprotein PLCE1 drives esophageal carcinoma
angiogenesis and proliferation via activating the PI-PLCε-NF-κB
signaling pathway and VEGF-C/Bcl-2 expression. Mol Cancer.
18:12019. View Article : Google Scholar
|
|
43
|
Mao Y, Wang Y, Dong L, Zhang Y, Zhang Y,
Wang C, Zhang Q, Yang S, Cao L, Zhang X, et al: Hypoxic exosomes
facilitate angiogenesis and metastasis in esophageal squamous cell
carcinoma through altering the phenotype and transcriptome of
endothelial cells. J Exp Clin Cancer Res. 38:3892019. View Article : Google Scholar : PubMed/NCBI
|














