Introduction
Ischemic cerebrovascular disease, which accounts for
70% of all strokes worldwide, is associated with high morbidity and
mortality rates (1–3). At present, the primary clinical
treatment for ischemic cerebrovascular disease is reperfusion of
the affected area as soon as possible (4). However, when the blood supply is
restored, the resultant damage to the brain tissue and the nervous
system, termed cerebral ischemia/reperfusion (I/R) injury, is a key
issue in the clinical treatment of ischemic cerebrovascular disease
(5). Moreover, it is difficult to
treat cerebral I/R injury due to the complicated pathological
processes and the multiple mechanisms involved (6). At present, clinical interventional
treatment and drugs primarily used in cerebral I/R injury therapy
include thrombolytics, calcium channel antagonists, free-radical
scavengers and excitatory amino acid regulators (7). However, the narrow therapeutic
temporal window and single target approach result in a limited
efficacy of treatment for cerebral I/R injury (8). Therefore, there is an urgent need to
find novel therapies to improve the therapeutic outcomes following
cerebral I/R injury.
Oxysophoridine (OSR) is one of the primary alkaloids
extracted from Sophora alopecuroides L. (9). Previous studies have reported that
OSR has certain pharmacological properties in regard to sedation,
anti-inflammatory effects, arrhythmia and immune regulation
(10,11). Furthermore, OSR has favorable
preventative and therapeutic effects on heart failure, myocardial
infarction, cerebral ischemia and I/R injury and other
cardiovascular and cerebrovascular disease (11). Cao et al (12) reported that OSR protects against
exacerbated spinal cord injury via its anti-inflammatory,
anti-oxidative and anti-apoptotic effects. Another previous study
reported that OSR has a neuroprotective effect on mice with
cerebral I/R injury by suppressing oxidative stress and expression
of the N-methyl-D-aspartate receptor subunit NR1 (13). However, the functional roles and
molecular mechanism of OSR on cerebral I/R injury are incompletely
understood. Therefore, in the present study, the effect of OSR in
cerebral I/R injury was evaluated.
Materials and methods
Animal care and experimental
groups
The present study was approved by the Animal Care
and Use Committee of Hangzhou Red Cross Hospital (Hangzhou, China;
approval no. 20220414) and performed in accordance with Chinese
legislation regarding the use of experimental animals. A total of
30 adult male Sprague-Dawley rats (7 weeks old) weighing 280–320 g
were purchased from the Laboratory Animal Resources, Chinese
Academy of Sciences. The rats were housed at a room temperature of
21±2°C and 55±5% relative humidity with a 12/12-h light/dark cycle
and free access to standard chow and tap water. After one week, the
cerebral I/R injury model was established using improved thread
occlusion of the right middle cerebral artery (MCAO) model as
described previously (14).
Briefly, rats were anesthetized by intraperitoneal administration
of 2.25% pentobarbital sodium (45 mg/kg) for 2 h, a midline
cervical incision was made, the MCA was located and its branches
were ligated. The induction of ischemia was performed by occluding
the right internal carotid artery using a thread. Following
transient MCAO for 2 h, the nylon thread was removed to allow the
return of blood flow through the right internal carotid artery.
Rats in the control group underwent the same surgical procedure but
the right internal carotid artery was not occluded. Rats were
randomly divided into five groups (n=6) as follows: Healthy control
group that was subjected to a sham operation (control); cerebral
I/R injury group that underwent MCAO for 2 h followed by
reperfusion for 24 h (I/R) and I/R + OSR groups which were
administered 60, 120 or 180 mg/kg OSR intraperitoneally once/day
for 7 days before MCAO. The total duration of the experiment was 10
days. All rats were euthanized using intraperitoneal administration
of pentobarbital sodium (200 mg/kg) 24 h after reperfusion. The
humane endpoints of this experiment were as follows: Marked
decrease in food or water intake, labored breathing, inability to
stand or no response to external stimuli. No humane endpoints of
the experiment were reached by any of the rats during the
experiment. Death was verified by lack of heartbeat and a cold
body. No animals were found dead before the end of the
experiment.
Histopathological examination
The brain tissue from rats in five groups was fixed
with 10% neutral formaldehyde buffer at 4°C overnight, dehydrated
in a series of graded concentrations of ethanol, embedded in
paraffin and cut into 4 µm sections. The sections were stained
using hematoxylin for 6 min and 1% eosin for 2 min, both at room
temperature. A light microscope was used to assess the pathological
changes in the samples following I/R treatment at a magnification
of ×400.
2,3,5-triphenyltetrazolium chloride
(TTC) staining
Rat brains were dissected into 2 mm coronal slices
and incubated in 2% TTC (Beijing Solarbio Science & Technology
Co., Ltd.) at 37°C for 10 min. Following TTC staining, the normal
brain tissue stained dark red and the infarcted tissue was
unstained (white). The tissue was fixed in 4% paraformaldehyde
(Beyotime Institute of Biotechnology) for 24 h at 4°C and imaged
using a digital camera (Canon, Inc.). The infarcted volume was
calculated as follows: Infarcted volume (%)=(volume of white
sections/volume of the whole brain) ×100.
Measurement of ATP and Fe2+
levels
ATP levels in brain tissues and HT22 cells were
assessed using the CellTiter-Glo Luminescent Assay kit (Promega
Corporation) according to the manufacturer's protocol.
Fe2+ levels in brain tissue and HT22 cells were
evaluated using an Iron Assay kit (cat. no. ab83366; Abcam)
according to the manufacturer's protocol. The absorbance at 520 nm
was assessed for the determination of iron concentration.
Immunofluorescence staining
Brain sections (10 µm) that were stored at −18°C
were permeabilized using 0.5% Triton X-100 at room temperature for
30 min and blocked using 10% bovine serum albumin (Thermo Fisher
Scientific, Inc.) for 1 h at room temperature. Subsequently, brain
tissue was incubated with primary antibodies against toll-like
receptor (TLR)4 (1:1,000; cat. no. ab22048; Abcam) at 4°C
overnight. The cells were exposed to DAPI (BIOSS) at room
temperature for 5 min were then cultivated with a secondary Alexa
Fluor® 488-conjugated goat anti-mouse antibody (1:1,000;
cat. no. ab150113; Abcam) at room temperature in the dark for 1 h.
Finally, the sections were rinsed with phosphate-buffered saline
(Beyotime Institute of Biotechnology) and imaged using a
fluorescence microscope (magnification, ×400; Leica Microsystems
GmbH).
Cell culture and treatment
The hippocampal HT22 neuronal cell line was
purchased from the Ningbo Mingzhou Biotechnology Co., Ltd. The
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (HyClone, Cytiva), 100 U/ml penicillin
and 100 µg/ml streptomycin (both Gibco; Thermo Fisher Scientific,
Inc.) in a humidified incubator with 5% CO2 at 37°C. To
establish an in vitro oxygen-glucose
deprivation/reoxygenation (OGD/R) model, HT22 cells were cultured
in glucose-free DMEM (Wuhan Procell Life Science & Technology
Co., Ltd.) in an oxygen-free incubator supplied with 5%
CO2 and 95% N2 at 37°C for 2 h. Following
hypoxia treatment, the media was replaced with normoxic
glucose-containing medium and cells were transferred to an
incubator supplied with 95% air and 5% CO2 at 37°C for
24 h. Cells were incubated with OSR (10, 20 or 40 nM) for 48 h at
room temperature before OGD/R challenge. Subsequently, the
indicated cells were treated with 5 µM anisomycin (a novel and
specific p38MAPK activator; MedChemExpress) at room temperature for
1 h or 1 µM erastin (a ferroptosis inducer; Shanghai Aladdin
Biochemical Technology Co., Ltd.) at room temperature for 24 h.
Cell transfection
TLR4-specific pcDNA overexpression vector (Oe-TLR4)
or pcDNA3.1 empty vector, which served as the negative control
(Oe-NC), was purchased from Shanghai Genechem Co., Ltd. A total of
100 nM plasmids were transfected into HT22 cells using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) for 48 h at 37°C. Following 48 h of transfection,
cells were collected for use in subsequent experiments.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from HT22 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The quality and
concentration of RNA were assessed using NanoDrop 2000 (Shanghai
Aiyan Biotechnology Co., Ltd.) based on the ratio of absorbance at
260 and 280 nm. A PrimeScript™ RT Reagent kit (Takara Bio, Inc.)
was used to reverse-transcribe 2 µg RNA into cDNA using the
thermocycling protocol as follows: 25°C for 5 min, 42°C for 30 min,
85°C for 5 min and then held at 4°C for 5 min. Amplification of the
cDNA was performed using qPCR using an SYBR Premix Ex Taq™ II kit
(Takara Bio, Inc.). The thermocycling protocol was 95°C for 3 min,
followed by 35 cycles of denaturation at 95°C for 30 sec, annealing
at 60°C for 30 sec and extension at 72°C for 1 min. A final
extension step at 72°C for 7 min was performed in each PCR assay.
The primer sequences used for PCR were as follows: TLR4 forward
(F), 5′-CCCATGCATTTGGCCTTAGC-3′ and reverse (R),
5′-AGAGCACTGAACCTCCTTGC-3′; and GAPDH (F),
5′-GTCGTGGAGTCTACTGGCGTCTTCA-3′ and (R),
5′-TCGTGGTTCACACCCATCACAAACA-3′. The relative mRNA levels were
normalized to GAPDH using the 2−ΔΔCq method (15).
Cell Counting Kit-8 (CCK-8) assay
Following various treatments, cells were plated at a
density of 5×103 cells/well in 96-well plates and
cultured in DMEM with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) at 37°C for 24 h. A total of 10 µl CCK-8 solution (Beyotime
Institute of Biotechnology) was added to each well and incubated
for 2 h. The absorbance at 450 nm was assessed using a microplate
reader (Bio-Rad Laboratories, Inc.). All experiments were performed
in triplicate with five independent repeats.
ELISA
IL-1β, TNF-α and IL-10 protein expression levels in
HT22 cells were assessed using ELISA kits (cat. nos. EK0393, EK0526
and EK0418 respectively; Wuhan Boster Biological Technology, Ltd.)
according to the manufacturer's protocols. The absorbance was
assessed at 450 nm using a microplate reader (BioTek Instruments,
Inc.).
Measurement of oxidative stress
To assess reactive oxygen species (ROS) generation,
DCFH-DA staining (cat. no. D6883; MilliporeSigma) was used. HT22
cells were washed with PBS and incubated with 10 µM DCFH-DA in the
dark for 30 min at 37°C. After washing three times with PBS,
fluorescence images were obtained using an Axio-Observer-D1
fluorescence microscope (ZEISS AG; magnification, ×400).
Furthermore, the activity of superoxide dismutase (SOD) and levels
of malondialdehyde (MDA) and catalase (CAT) were assessed using SOD
assay kit (cat. no. S0101M), MDA assay kit (cat. no. S0131S) and
CAT assay kit (cat. no. S0051), all of which were obtained from
Beyotime Institute of Biotechnology. The absorbance at 532 nm was
assessed using a Benchmark microplate reader (Bio-Rad Laboratories,
Inc.).
TUNEL assay
TUNEL assay was performed to evaluate the degree of
apoptosis in tissue and cells. Following fixation with 4%
paraformaldehyde at room temperature for 30 min, tissue sections or
cells were incubated with proteinase K at room temperature for 15
min, placed in 3% H2O2 for 15 min at room
temperature and treated using a TUNEL detection kit for 60 min at
37°C. Following incubation, the PBS-rinsed cells were co-labeled
with 1 µg/ml DAPI working solution for 10 min at 37°C. The positive
cells were mounted with fluorescent mounting media (Beijing
Solarbio Science & Technology Co., Ltd.) and analyzed using
ImageJ 1.8.0 software (National Institutes of Health). More than 10
fields of view/section for each sample were assessed. The labeled
cells were visualized using an Olympus BX53 fluorescence microscope
(magnification, ×100; Olympus Corporation).
Western blotting
Total protein was extracted from tissue and cells
using RIPA lysis buffer (Beyotime Institute of Biotechnology) and
then quantified with bicinchoninic acid (BCA) protein assay kit
(cat. no. P0012S; Beyotime Institute of Biotechnology). An equal
amount of protein (60 µg/lane) was separated on 10% SDS gels and
then transferred to a nitrocellulose blotting membrane (Pall Life
Sciences). The membranes were blocked with 5% non-fat milk
dissolved in 0.1% TBS-T buffer for 2 h at room temperature and
probed with primary antibodies as follows: Bax (1:1,000; ab32503),
Bcl-2 (1:1,000; ab196495), acyl-CoA synthetase long-chain family
member 4 (ACSL4; 1:1,000; ab155282), transferrin 1 (TFR1; 1:1,000;
ab269513), ferritin 1 (FTH1; 1:1,000; ab1837810 glutathione
peroxidase 4 (GPX4; 1:1,000; ab252833), TLR4 (1:1,000; ab217274),
MyD88 (1:1,000; ab219413), phosphorylated (p)-p38 (1:1,000;
ab4822), p38 (1:1,000; ab170099), inducible nitric oxide synthase
(iNOS; 1:1,000; ab178945), cyclooxygenase 2 (COX-2; 1:1,000;
ab179800), p-p65 (1:1,000; ab76302), p65 (1:1,000; ab32536) and
GAPDH (1:1,000; ab8245; all Abcam) overnight at 4°C. The membranes
were incubated with HRP-conjugated anti-mouse or anti-rabbit
secondary antibodies (1:2,000, ab6789 and ab6721, respectively;
Abcam) for 2 h at room temperature. Signals were visualized using
enhanced chemiluminescence reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Densitometry analysis
was performed using ImageJ (Version 1.49; National Institutes of
Health).
Statistical analysis
Statistical analysis was performed using SPSS
version 17.0 (SPSS, Inc.) and data are presented as the mean ± SD.
Comparisons between multiple groups were performed using one-way
ANOVA followed by Bonferroni's post hoc test for multiple
comparisons. All cellular experiments were performed ≥3 times from
three different cultures. P<0.05 was considered to indicate a
statistically significant difference.
Results
OSR alleviates brain injury and
neuronal apoptosis in I/R-induced rats
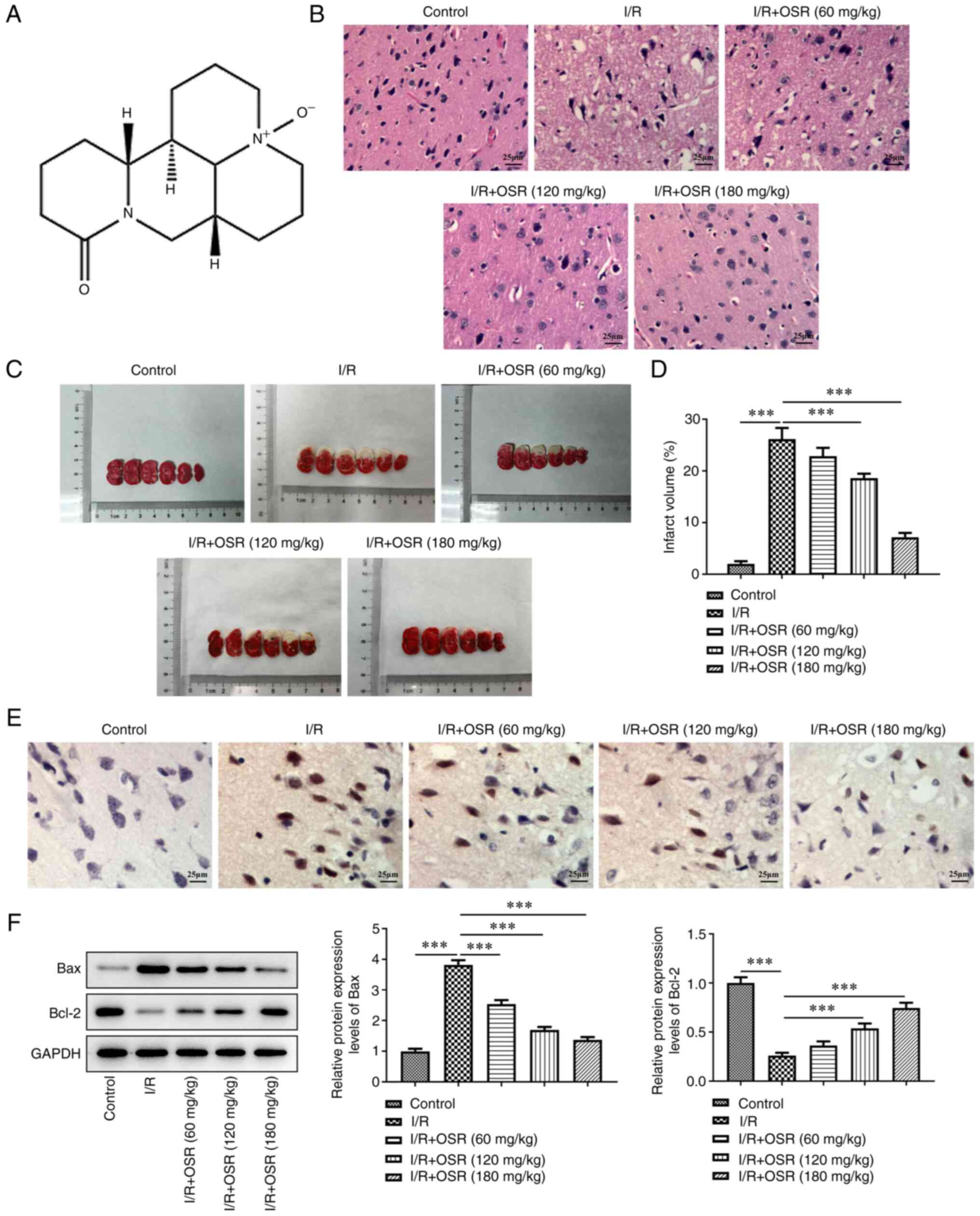
The structure was presented in Fig. 1A. To evaluate the role and
mechanism of OSR in cerebral I/R injury, an in vivo cerebral
I/R injury model was established using MCAO. Control group
demonstrated a clear outline of cortex neurons, a compact structure
and abundant cytoplasm (Fig. 1B).
However, I/R injury resulted in pyknotic and shrunken nuclei,
nuclear loss and numerous vacuolated spaces. These pathological
changes were restored following OSR preconditioning in a
dose-dependent manner. Furthermore, I/R treatment significantly
increased the infarct volume compared with the control while OSR
led to a decrease in cerebral infarct volume compared with the I/R
group (Fig. 1C and D). TUNEL assay
demonstrated that the apoptotic rate of hippocampal neurons in
I/R-induced rats was increased compared with the control; however,
the apoptotic rate was markedly decreased after administration of
OSR compared with the I/R group (Fig.
1E). Western blotting results demonstrated that I/R resulted in
a significant increase in Bax protein expression levels and a
significant decrease in the protein expression levels of Bcl-2
compared with the control (Fig.
1F). Nevertheless, OSR treatment cut down Bax protein and
elevated Bcl-2 content concentration-dependently relative with the
I/R group.
OSR decreases ROS accumulation and
ferroptosis induced by I/R in rat brain tissue
I/R induction elevated ROS levels in brain tissue of
rats; however, OSR treatment markedly decreased accumulation of ROS
(Fig. 2A). Moreover, compared with
the control group, the I/R group demonstrated significantly higher
levels of ATP, whereas decreased levels of ATP were observed in the
OSR intervention group compared with the I/R group (Fig. 2B). Consistently, I/R significantly
increased levels of Fe2+ compared with the control and
OSR pretreatment significantly decreased the increased levels of
Fe2+ (Fig. 2C).
Furthermore, a significant increase in protein expression levels of
ACSL4 and TFR1 and a significant decrease in protein expression
levels of FTH1 and GPX4 were observed in the I/R group compared
with the control group. OSR treatment significantly
dose-dependently reversed the effects of I/R on expression levels
of these proteins in the brain tissue of rats with I/R injury
(Fig. 2D).
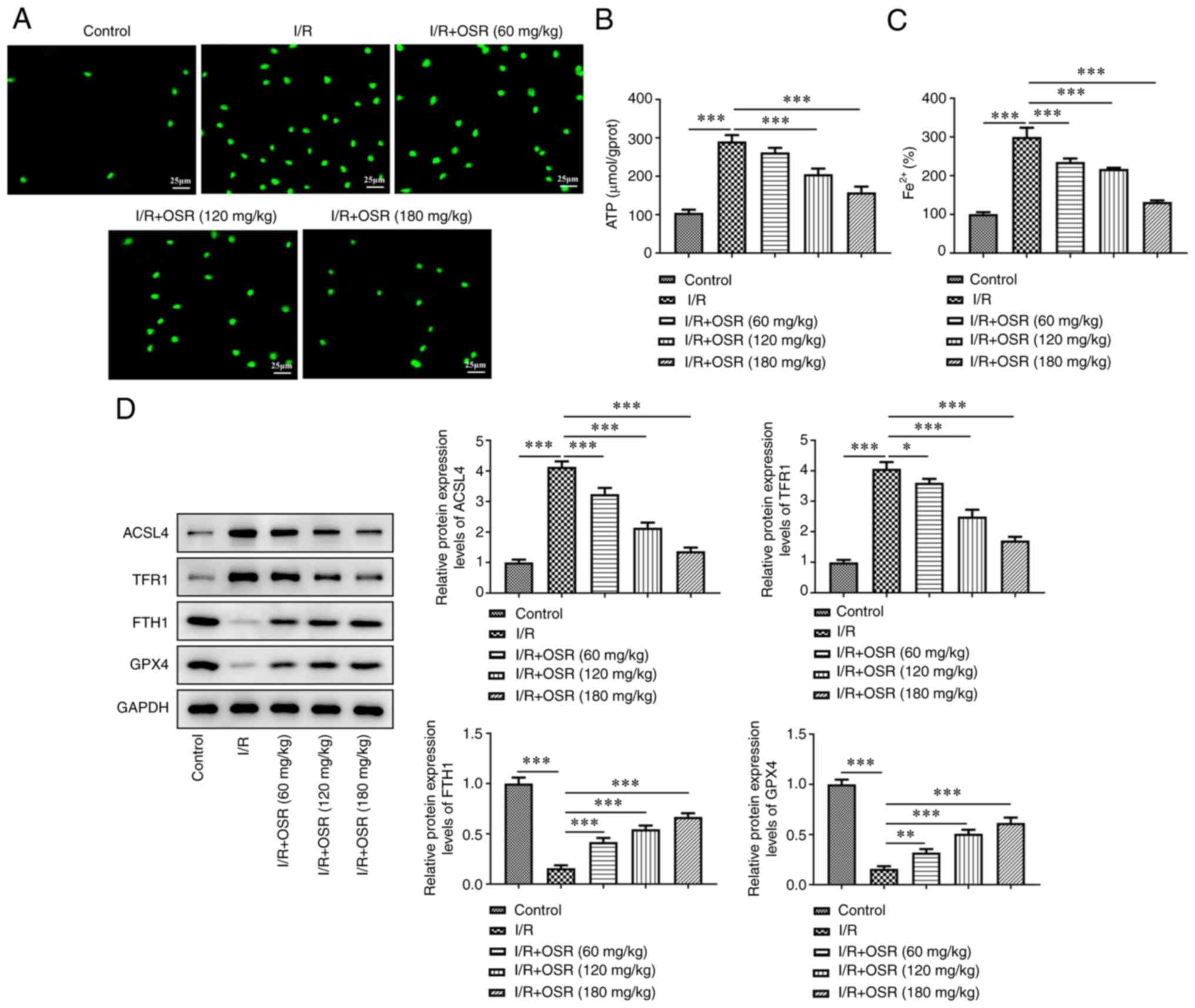 | Figure 2.OSR decreases ROS accumulation and
ferroptosis induced by I/R in rat brain. The levels of (A) ROS, (B)
ATP and (C) Fe2+ were assessed in I/R rat brain tissue.
The control was set as 100%. Scale bar, 25 µm. (D) Protein
expression levels of ACSL4, TFR1, FTH1 and GPX4 were
semi-quantified using western blotting. Data are presented as mean
± SD. Comparisons between multiple groups were performed using
one-way ANOVA followed by Bonferroni's post hoc test for multiple
comparisons. *P<0.05, **P<0.01 and ***P<0.001. ROS,
reactive oxygen species; OSR, oxysophoridine; I/R,
ischemia/reperfusion; TFR1, transferrin 1; FTH1, ferritin 1; GPX4,
glutathione peroxidase 4; ACSL4, acyl-CoA synthetase long-chain
family member. |
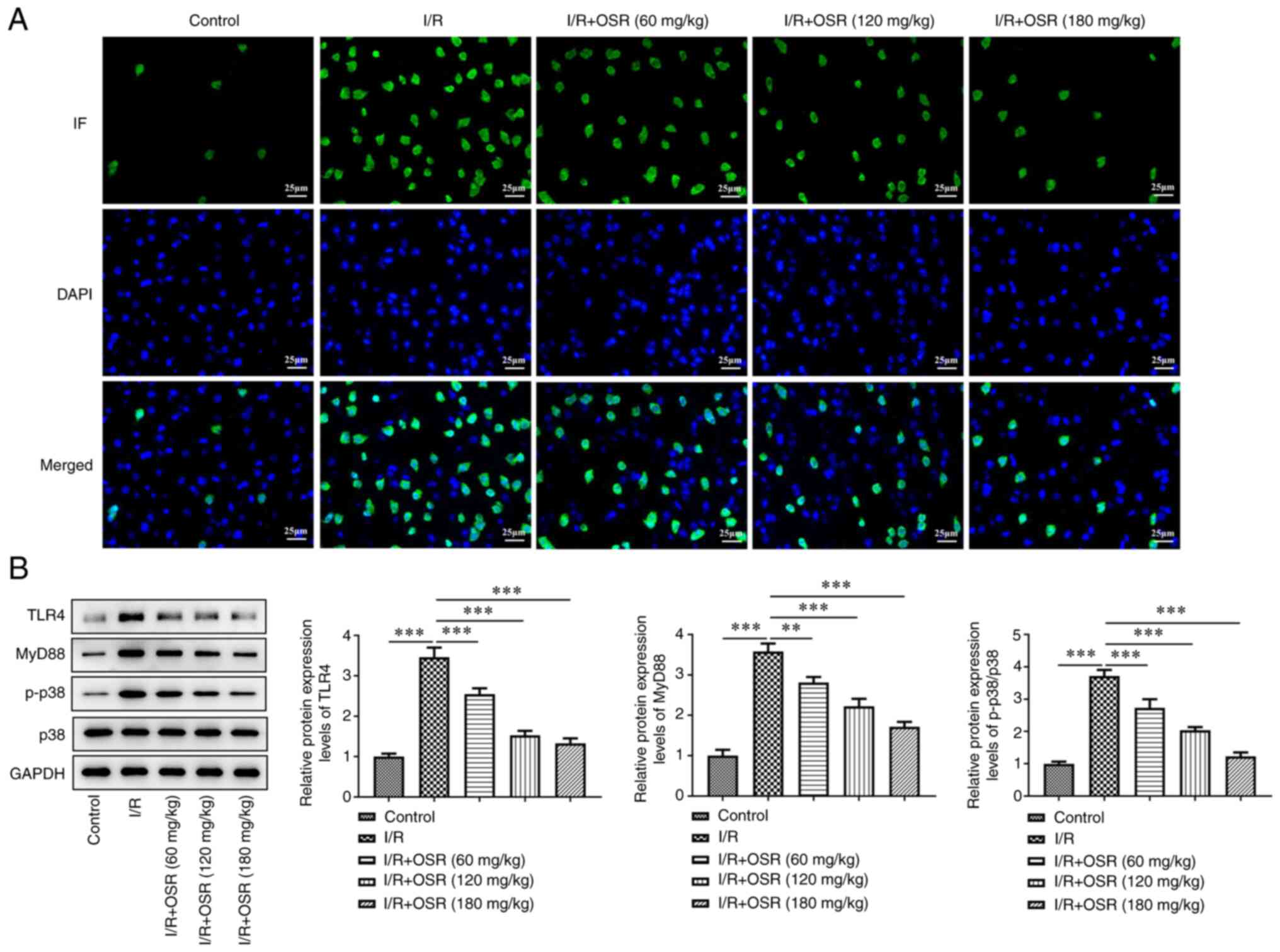
OSR inhibits TLR4/p38MAPK signaling in
brain tissue of I/R-induced rats
To evaluate the mechanism of action of OSR in
I/R-induced rats, the activity of the TLR4/p38MAPK signaling
pathway was assessed. Immunofluorescence staining demonstrated that
the number of positive cells was markedly higher in the I/R group
compared with the control group and that OSR markedly decreased the
number of positive cells compared with the I/R group, which
indicated that levels of TLR4 decreased following OSR treatment
(Fig. 3A). Furthermore, western
blotting demonstrated that I/R significantly enhanced protein
expression levels of TLR4, MyD88 and p-p38 compared with the
control and the changes were significantly reversed by OSR
treatment (Fig. 3B).
OSR suppresses OGD/R-induced neuronal
ferroptosis by inhibiting TLR4/p38MAPK signaling
To evaluate the role of TLR4/p38MAPK signaling in
the regulation of OSR in cerebral I/R injury, an in vitro
cerebral I/R injury model was established using OGD/R. The protein
expression levels of TLR4, MyD88 and p-p38 in HT22 cells were
significantly increased following I/R exposure compared with the
control group and OSR significantly decreased expression levels of
these proteins compared with the OGD/R group (Fig. 4A). TLR4 was overexpressed in HT22
cells and transfection efficiency was assessed using RT-qPCR and
western blotting. As depicted in Fig.
4B and C, the mRNA and protein expressions of TLR4 were
significantly increased in Oe-TLR4 group when compared with those
in Oe-NC group. Cells were treated with OSR (40 µM) and 5 µM
p38MAPK agonist anisomycin was added to cells. OGD/R treatment
markedly elevated the levels of ROS compared with the control and
OSR markedly counteracted these effects (Fig. 4D). However, TLR4 overexpression and
anisomycin both abated the effects of OSR treatment. Furthermore,
levels of ATP and Fe2+ were significantly increased by
OGD/R while OSR significantly suppressed the OGD/R-induced increase
in ATP and Fe2+ levels in the OGD/R + OSR group compared
with the OGD/R group. TLR4 overexpression and anisomycin
significantly reversed the effects of OSR on the levels of ATP and
Fe2+ compared with the OGD/R + OSR + Oe-NC and OGD/R +
OSR groups, respectively (Fig. 4E and
F). OGD/R was demonstrated to significantly increase protein
expression levels of ACSL4 and TFR1 and significantly decrease
protein expression levels of FTH1 and GPX4 compared with the
control and OSR significantly reversed these trends compared with
the OGD/R group. Moreover, TLR4 overexpression and anisomycin
treatment both significantly reversed the effects of OSR on these
ferroptosis-associated proteins compared with the OGD/R + OSR +
Oe-NC and OGD/R + OSR groups, respectively (Fig. 4G and H).
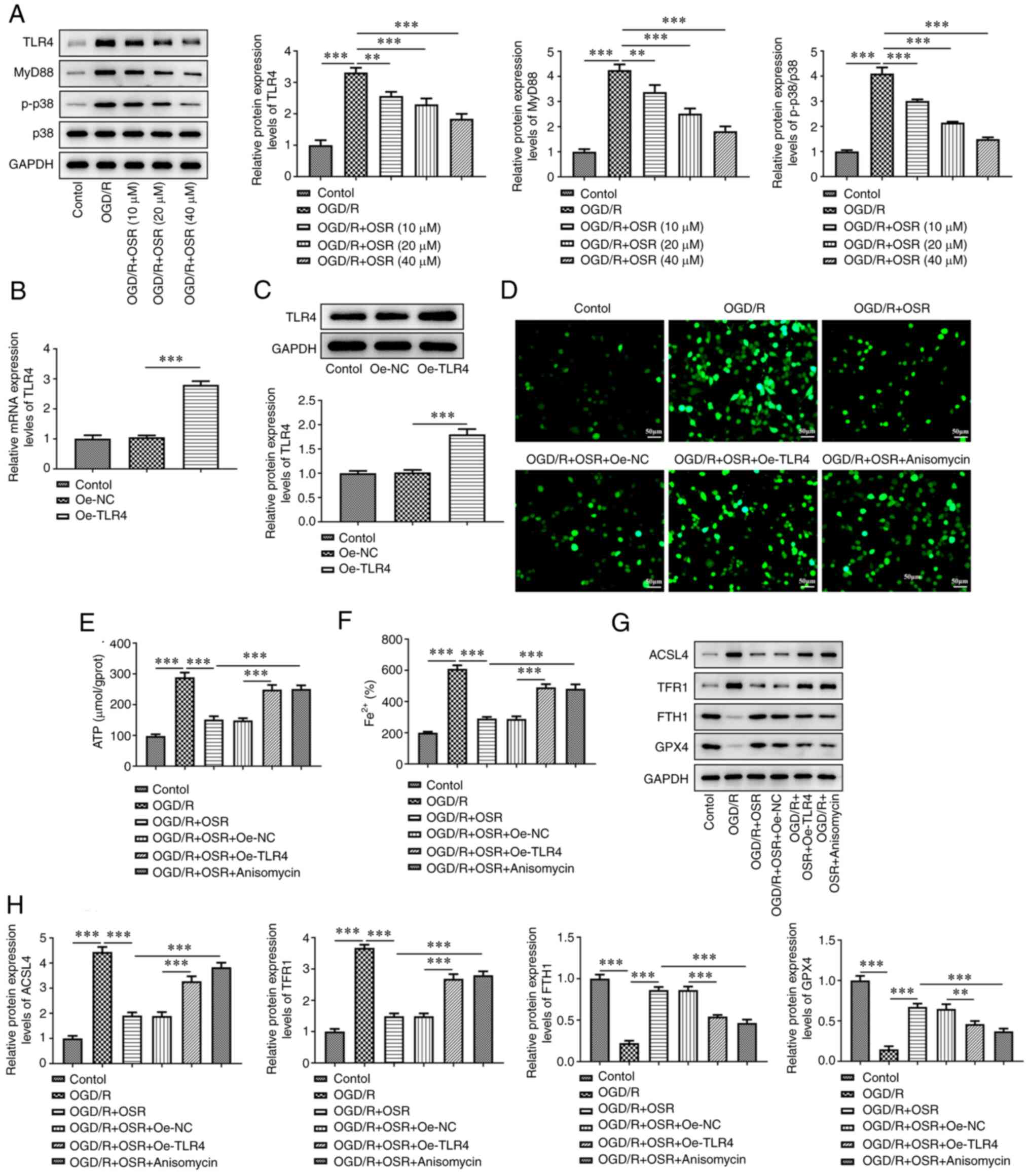 | Figure 4.OSR suppresses OGD/R-induced neuronal
ferroptosis by inhibiting TLR4/p38MAPK signaling. (A) Protein
expression levels of TLR4, MyD88, p-p38 and p38 were
semi-quantified using western blotting. (B) mRNA and (C) protein
expression levels of TLR4 in OGD/R-induced cells were assessed
using reverse transcription-quantitative PCR and western blotting,
respectively. The levels of (D) reactive oxygen species, (E) ATP
and (F) Fe2+ were assessed in OGD/R-induced cells. Scale
bar, 50 µm. (G) Protein expression levels of ACSL4, TFR1, FTH1 and
GPX4 were (H) semi-quantified using western blotting. Data are
presented as mean ± SD. Comparisons between multiple groups were
performed using one-way ANOVA followed by Bonferroni's post hoc
test for multiple comparisons. **P<0.01 and ***P<0.001.
OGD/R, oxygen-glucose deprivation/reoxygenation; I/R,
ischemia/reperfusion; p, phosphorylated; Oe, overexpression; NC,
negative control; OSR, oxysophoridine; TLR, toll-like receptor;
TFR1, transferrin 1; FTH1, ferritin 1; GPX4, glutathione peroxidase
4; ACSL4, acyl-CoA synthetase long-chain family member. |
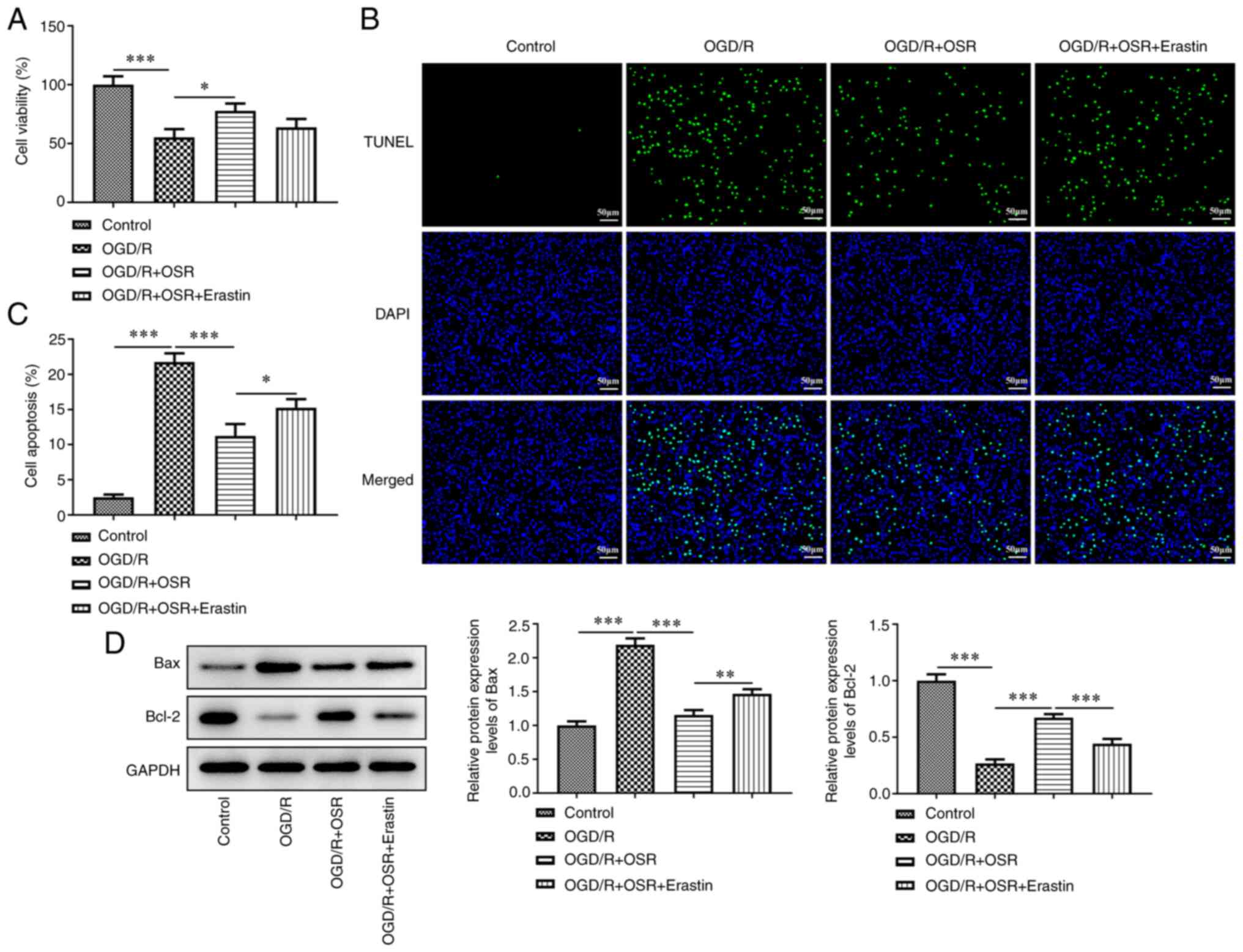
Erastin decreases the protective
effect of OSR on viability in OGD/R-induced cells
To study the role of ferroptosis in OGD/R induced
cells, the ferroptosis inducer erastin was used. OGD/R
significantly decreased cell viability compared with the control,
whereas OSR significantly reversed this effect compared with OGD/R
group and cell viability was markedly decreased by erastin
(Fig. 5A). TUNEL assay
demonstrated a marked increase in the rate of cell apoptosis
following OGD/R treatment compared with the control and OSR
markedly inhibited apoptosis in OGD/R-induced cells compared with
the OGD/R group; however, erastin markedly accelerated cell
apoptosis rate (Fig. 5B and C).
Furthermore, OGD/R significantly increased Bax protein expression
levels and decreased Bcl-2 protein expression levels compared with
the control, whereas OSR significantly reversed the expression
levels of these two proteins compared with the OGD/R group. Erastin
demonstrated the inverse effect on these protein expression levels,
evidenced by increased Bax protein expression level and decreased
Bcl-2 protein expression level in OGD/R + OSR + Erastin group
compared with the OGD/R + OSR group (Fig. 5D).
Erastin decreases the inhibitory
effect of OSR on OGD/R-induced oxidative stress and inflammatory
response
OGD/R led to a significant decrease in SOD activity
and CAT levels, but significantly increased MDA levels compared
with the control. OSR treatment significantly enhanced the levels
of SOD and CAT and significantly reduced the levels of MDA compared
with the OGD/R group; however, this was significantly reversed by
the addition of erastin compared with the OGD/R + OSR group
(Fig. 6A). Furthermore, protein
expression levels of the proinflammatory factors IL-1β and TNF-α
were significantly elevated and the protein expression levels of
anti-inflammatory factor IL-10 were significantly decreased
following OGD/R exposure compared with the control. OSR
pretreatment significantly reversed the effect of OGD/R on the
protein expression levels of inflammatory factors compared with the
OGD/R group (Fig. 6B). However,
erastin significantly reversed the effects of OSR on the
aforementioned inflammatory factors compared with the OGD/R + OSR
group. Furthermore, OGD/R significantly increased protein
expression levels of iNOS, COX-2 and p-p65 compared with the
control. However, OSR significantly inhibited the expression levels
of these proteins compared with the OGD/R group and this inhibitory
effect was significantly reversed following treatment with erastin
compared with the OGD/R + OSR group (Fig. 6C).
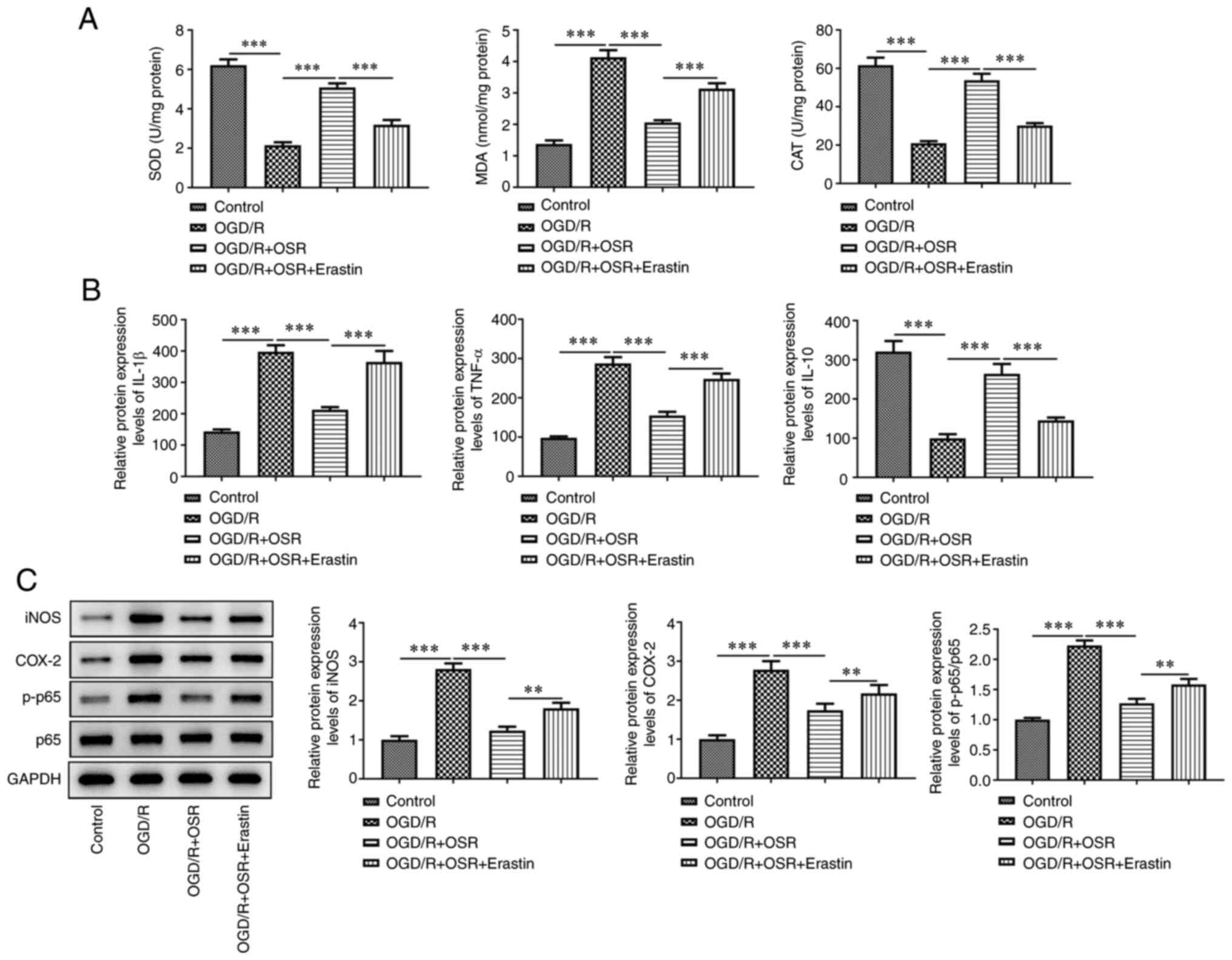 | Figure 6.Erastin decreases the inhibitory
effect of OSR on OGD/R-induced oxidative stress and inflammatory
response. (A) Levels of SOD, MDA and CAT were assessed using
specific detection kits. (B) Protein expression levels of IL-1β,
TNF-α and IL-10 were assessed using ELISA. (C) Protein expression
levels of iNOS, COX-2, p-p65 and p65 were semi-quantified using
western blotting. Data are presented as mean ± SD. Comparisons
between multiple groups were performed using one-way ANOVA followed
by Bonferroni's post hoc test for multiple comparisons. **P<0.01
and ***P<0.001. OSR, oxysophoridine; OGD-R, oxygen-glucose
deprivation/reoxygenation; SOD, superoxide dismutase; MDA,
malondialdehyde; CAT, catalase; iNOS, inducible nitric oxide
synthase; COX-2, cyclooxygenase 2; p, phosphorylated. |
Discussion
Cerebrovascular disease is one of the primary
conditions associated with notable risk to human health and
survival (16). Cerebral ischemia
results in decreased cerebral blood flow and insufficient blood
oxygen supply to brain tissue due to vascular thrombosis, resulting
in brain cell damage (17,18). Therefore, it is of importance to
restore blood flow to the ischemic area as soon as possible using
thrombolytics or mechanical recanalization (19). However, while recanalizing
occlusive cerebrovascular arteries, these treatments often
aggravate the pathological damage to ischemic tissue and the
nervous system and worsen the clinical symptoms, a well-established
concept termed cerebral I/R injury (20). In the present study, the effect of
OSR on protecting against cerebral I/R injury was assessed using a
cerebral I/R injury model in vivo and in vitro and it
was demonstrated that the protective role of OSR may be associated
with inhibition of ferroptosis mediated by the TLR4/p38MAPK
signaling pathway.
Studies have reported that Traditional Chinese
Medicine can intervene in the pathological process of cerebral I/R
injury by affecting multiple targets and/or pathways. For example,
galangin represses the ferroptosis in hippocampal tissue of gerbils
by activating the SLC7A11/GPX4 axis, thus protecting against
cerebral I/R injury (21). Wang
et al hypothesized that schizandrin protects neurons from
cerebral ischemia by regulating the AMPK-mTOR pathway (22). The above findings suggested TCM
possesses unique advantages and potential in the treatment of
cerebral I/R injury. OSR is an alkaloid found in Sophora
alopecuroides L, that has extensive anti-inflammatory and
anti-apoptotic properties and has been reported to exhibit
protective effects in spinal cord, brain and hippocampal neuron
injury induced by I/R (12,23).
Rui et al (24) reported
that OSR markedly decreases neurological deficit score, neuronal
damage and apoptosis and is therefore regarded as a potential
neuroprotective agent for cerebral ischemia injury. Wang et
al (25) evaluated the effects
of OSR in decreasing ischemic cerebral injury using an MCAO mouse
model and reported that the neuroprotective effect of OSR was
associated with inhibition of oxidative stress and apoptosis. In
the present study, it was demonstrated using hematoxylin and eosin
and TTC staining that OSR decreased brain injury and inhibited
neuronal apoptosis based on significantly decreased Bax protein
expression levels and significantly increased Bcl-2 protein
expression levels in I/R-induced rats and hippocampal neuronal HT22
cells, which was consistent with previous reports (24,25).
The activation of innate immune receptors such as
TLRs serves an important role in induction of inflammatory
responses; TLR4 was the first mammalian TLR recognized (26). Previous studies have reported that
TLR4 expression is upregulated following cerebral I/R and that this
is alleviated in TLR4-deficient mice (27,28).
A case of previous study has also reported that OSR exerts
antioxidant and anti-inflammatory effects in Alzheimer's disease by
targeting the TLR4/NF-κB signaling pathway and TLR4-specific
inhibitor TAK-242 effectively enhances the effects of OSR (23). Another study reported that TAK-242
serves a neuroprotective and anti-ferroptotic role by inhibiting
TLR4/p38MAPK signaling in hypoxic-ischemic brain injury (29). In the present study, I/R or OGD/R
treatment induced production of TLR4, MyD88 and p-p38, but OSR
significantly decreased expression levels of these proteins, which
suggested that OSR exerted an inhibitory effect on the TLR4/p38MAPK
signaling pathway in cerebral I/R injury.
Ferroptosis is a modulated form of cell death
characterized by lethal iron-dependent accumulation of lipid
peroxides and is involved in several types of brain disease,
including cerebral I/R (30). Guan
et al (31) reported that
carvonol exerts a neuroprotective effect on I/R-induced hippocampal
neuronal injury by alleviating ferroptosis. Guo et al
(32) reported that carthamin
yellow protects rats against cerebral I/R injury by inhibiting
inflammation and ferroptosis in a MCAO model. In the present study,
I/R and OGD/R significantly increased ROS, ATP, Fe2+,
ACSL4 and TFR1 levels and significantly decreased FTH1 and GPX4
levels, thereby inducing neuronal ferroptosis. OSR inhibited
expression of ferroptosis-associated proteins, attenuated oxidative
stress, decreased the pro-inflammatory response and ameliorated
activation of hippocampal neuronal ferroptosis. However, TLR4
overexpression and treatment with the p38MAPK activator anisomycin
both significantly exacerbated OGD-induced oxidative stress and
ferroptosis, which suggested that the TLR/p38MAPK signaling pathway
mediated the ferroptotic process following I/R and OGD/R exposure
and that this signaling pathway was regulated by OSR. In the study,
the effects of TLR4/p38MAPK overexpression on OGD-induced
ferroptosis in OSR-treated cells were evaluated but the effects of
this pathway on OGD/R-induced viability, oxidative stress and
inflammatory response in OSR-treated cells was not assessed.
Moreover, there was a lack of validation at the mitochondrial level
and there was no detection of lipid peroxidation products. Further
work is required to evaluate the effect of TLR4/p38MAPK on other
functional roles in OGD/R-induced neuronal cells with or without
OSR, verify the results of the present study at the mitochondrial
level and detect lipid peroxidation products.
In conclusion, the results of the present study
indicated that OSR attenuated brain injury and neuronal apoptosis,
oxidative stress and inflammatory response in cerebral I/R injury
by inhibiting ferroptosis. Moreover, OSR decreased OGD/R-induced
neuronal ferroptosis by inactivation of the TLR4/p38MAPK signaling
pathway.
Acknowledgements
Not applicable.
Funding
This work was supported by Zhejiang TCM Science and Technology
Project (grant no. 2023004198).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and MM designed the study and drafted and revised
the manuscript. LL and GF analyzed the data and reviewed the
literature. JZ and MM confirmed the authenticity of all the raw
data. All authors performed the experiments. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
All procedures using animals were approved by the
Animal Care and Use Committee of Hangzhou Red Cross Hospital
(approval no. 20220414) and performed in accordance with Chinese
legislation regarding experiment animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu W, Wong A, Law AC and Mok VC:
Cerebrovascular disease, amyloid plaques, and dementia. Stroke.
46:1402–1407. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lanzino G and Brown RD Jr: Introduction:
Management of ischemic cerebrovascular disease. Neurosurg Focus.
36:1–2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang L, Jia J, Hong Z, Zhang L and Zhang
J: Effects of chemerin and homocysteine levels and their
associations with occurrence and development of ischemic
cerebrovascular disease. Lipids Health Dis. 20:1082021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J and Wang DM: Update in endovascular
therapy of ischemic cerebrovascular disease. Zhonghua Wai Ke Za
Zhi. 59:192–195. 2021.(In Chinese). PubMed/NCBI
|
|
5
|
Lim S, Kim TJ, Kim YJ, Kim C, Ko SB and
Kim BS: Senolytic Therapy for cerebral ischemia-reperfusion injury.
Int J Mol Sci. 22:119672021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang Z, Weian C, Susu H and Hanmin W:
Protective effects of mangiferin on cerebral ischemia-reperfusion
injury and its mechanisms. Eur J Pharmacol. 771:145–151. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Banz Y and Rieben R: Role of complement
and perspectives for intervention in ischemia-reperfusion damage.
Ann Med. 44:205–217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Mufti F, Amuluru K, Roth W, Nuoman R,
El-Ghanem M and Meyers PM: Cerebral ischemic reperfusion injury
following recanalization of large vessel occlusions. Neurosurgery.
82:781–789. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao XQ, Zhang YH, Long W and Liu PX:
Oxysophoridine suppresses the growth of hepatocellular carcinoma in
mice: In vivo and cDNA microarray studies. Chin J Integr Med.
18:209–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang R, Deng X, Gao Q, Wu X, Han L, Gao X,
Zhao S, Chen W, Zhou R, Li Z and Bai C: Sophora
alopecuroides L.: An ethnopharmacological, phytochemical, and
pharmacological review. J Ethnopharmacol. 248:1121722020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meng C, Liu C, Liu Y and Wu F:
Oxysophoridine attenuates the injury caused by acute myocardial
infarction in rats through anti-oxidative, anti-inflammatory and
anti-apoptotic pathways. Mol Med Rep. 11:527–532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao Z, Chen L, Liu Y and Peng T:
Oxysophoridine rescues spinal cord injury via anti-inflammatory,
anti-oxidative stress and anti-apoptosis effects. Mol Med Rep.
17:2523–2528. 2018.PubMed/NCBI
|
|
13
|
Wang H, Li Y, Jiang N, Chen X, Zhang Y,
Zhang K, Wang T, Hao Y, Ma L, Zhao C, et al: Protective effect of
oxysophoridine on cerebral ischemia/reperfusion injury in mice.
Neural Regen Res. 8:1349–1359. 2013.PubMed/NCBI
|
|
14
|
Zhai Z and Feng J: Left-right asymmetry
influenced the infarct volume and neurological dysfunction
following focal middle cerebral artery occlusion in rats. Brain
Behav. 8:e011662018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mehanna R and Jankovic J: Movement
disorders in cerebrovascular disease. Lancet Neurol. 12:597–608.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sveinsson OA, Kjartansson O and
Valdimarsson EM: Cerebral ischemia/infarction-epidemiology, causes
and symptoms. Laeknabladid. 100:271–279. 2014.(In Icelandic).
PubMed/NCBI
|
|
18
|
Suzuki H, Kanamaru H, Kawakita F, Asada R,
Fujimoto M and Shiba M: Cerebrovascular pathophysiology of delayed
cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol
Histopathol. 36:143–158. 2021.PubMed/NCBI
|
|
19
|
Qin Y, Zhang Q and Liu Y: Analysis of
knowledge bases and research focuses of cerebral
ischemia-reperfusion from the perspective of mapping knowledge
domain. Brain Res Bull. 156:15–24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kawadkar M, Mandloi AS, Saxena V,
Tamadaddi C, Sahi C and Dhote VV: Noscapine alleviates cerebral
damage in ischemia-reperfusion injury in rats. Naunyn Schmiedebergs
Arch Pharmacol. 394:669–683. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guan X, Li Z, Zhu S, Cheng M, Ju Y, Ren L,
Yang G and Min D: Galangin attenuated cerebral ischemia-reperfusion
injury by inhibition of ferroptosis through activating the
SLC7A11/GPX4 axis in gerbils. Life Sci. 264:1186602021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang G, Wang T, Zhang Y, Li F, Yu B and
Kou J: Schizandrin protects against OGD/R-induced neuronal injury
by suppressing autophagy: Involvement of the AMPK/mTOR pathway.
Molecules. 24:36242019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen R, Wang Z, Zhi Z, Tian J, Zhao Y and
Sun J: Targeting the TLR4/NF-κB pathway in β-amyloid-stimulated
microglial cells: A possible mechanism that oxysophoridine exerts
anti-oxidative and anti-inflammatory effects in an in vitro model
of Alzheimer's disease. Brain Res Bull. 175:150–157. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rui C, Yuxiang L, Ning J, Ningtian M,
Qingluan Z, Yinju H, Ru Z, Lin M, Tao S and Jianqiang Y:
Anti-apoptotic and neuroprotective effects of oxysophoridine on
cerebral ischemia both in vivo and in vitro. Planta Med.
79:916–923. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang TF, Lei Z, Li YX, Wang YS, Wang J,
Wang SJ, Hao YJ, Zhou R, Jin SJ, Du J, et al: Oxysophoridine
protects against focal cerebral ischemic injury by inhibiting
oxidative stress and apoptosis in mice. Neurochem Res.
38:2408–2417. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Iadecola C and Anrather J: The immunology
of stroke: From mechanisms to translation. Nat Med. 17:796–808.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu J, Chen Q, Jian Z, Xiong X, Shao L,
Jin T, Zhu X and Wang L: Daphnetin protects against cerebral
ischemia/reperfusion injury in mice via inhibition of TLR4/NF-κB
signaling pathway. Biomed Res Int. 2016:28160562016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Caso JR, Pradillo JM, Hurtado O, Lorenzo
P, Moro MA and Lizasoain I: Toll-like receptor 4 is involved in
brain damage and inflammation after experimental stroke.
Circulation. 115:1599–1608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu K, Zhu X, Sun S, Yang W, Liu S, Tang
Z, Zhang R, Li J, Shen T and Hei M: Inhibition of TLR4 prevents
hippocampal hypoxic-ischemic injury by regulating ferroptosis in
neonatal rats. Exp Neurol. 345:1138282021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guan X, Li X, Yang X, Yan J, Shi P, Ba L,
Cao Y and Wang P: The neuroprotective effects of carvacrol on
ischemia/reperfusion-induced hippocampal neuronal impairment by
ferroptosis mitigation. Life Sci. 235:1167952019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo H, Zhu L, Tang P, Chen D, Li Y, Li J
and Bao C: Carthamin yellow improves cerebral ischemia-reperfusion
injury by attenuating inflammation and ferroptosis in rats. Int J
Mol Med. 47:522021. View Article : Google Scholar : PubMed/NCBI
|