Introduction
Lymphoplasmacytic lymphoma (LPL) is a rare inert
mature B-cell lymphoma, which is known as Waldenström
macroglobulinemia (WM) when it secretes monoclonal IgM. WM
constitutes the majority of LPL cases, with only a small proportion
of LPLs secreting monoclonal IgA, IgG or no monoclonal
immunoglobulin (1,2). Polyneuropathy, organomegaly,
endocrinopathy, monoclonal protein and skin changes (POEMS)
syndrome, also known as Takatsuki syndrome or osteosclerotic
myeloma, is a rare paraneoplastic syndrome caused by an underlying
plasma cell disease (3). The
clinical presentation of POEMS usually involves multisystem damage
characterized by multiple neuropathies, visceral enlargement,
endocrine disorders and changes in skin and M-protein levels
(4). To raise awareness of this
disease, the present study reports a case of biclonal lymphoblastic
lymphoma associated with POEMS syndrome and reviews the relevant
literature.
Case report
A 65-year-old male with a 1-month history of
polydipsia and polyuria with aggravated symptoms for 1 week in
addition to severe peripheral neuropathy was referred to the
Department of Endocrinology, Gansu Provincial Hospital (Lanzhou,
China) on March 8, 2019. One month prior to hospitalization, the
patient presented with symptoms of polydipsia and polyuria without
obvious cause; the patient drank ~3,000 ml liquid daily and the
urine volume was similar to this. The symptoms became significantly
aggravated 1 week prior to admission, when the urine volume
increased to ~5,000 ml per day. The patient had no noteworthy
personal or family health history.
Physical examination revealed a mildly anemic
appearance and decreased muscle strength of the lower limbs with
obvious edema. A routine blood biochemical examination showed the
following, with the normal range presented in parentheses: White
blood cell count, 7.8×109/l (3.5–9.5×109/l);
red blood cell count, 3.62×1012/l
(4.0–5.5×1012/l); hemoglobin (Hb), 113 g/l (120–160
g/l); platelet count 204×109/l
(100–300×109/l); total protein, 54.1 g/l (65–85 g/l);
albumin (ALB), 29.2 g/l (40–55 g/l); globulin, 24.9 g/l (20–40
g/l); creatinine, 121.7 µmol/l (53–106 µmol/l); uric acid, 451
µmol/l (210–440 µmol/l); potassium, 3.3 mmol/l (3.5–5.3 mmol/l);
calcium, 1.84 mmol/l (2.11–2.52 mmol/l); fasting blood glucose
(FBG), 7.0 mmol/l (3.9–6.1 mmol/l); 2-h postprandial plasma glucose
(2 h-PG), 12.0 mmol/l (3.9–7.8 mmol/l); and glycated Hb, 5.9%
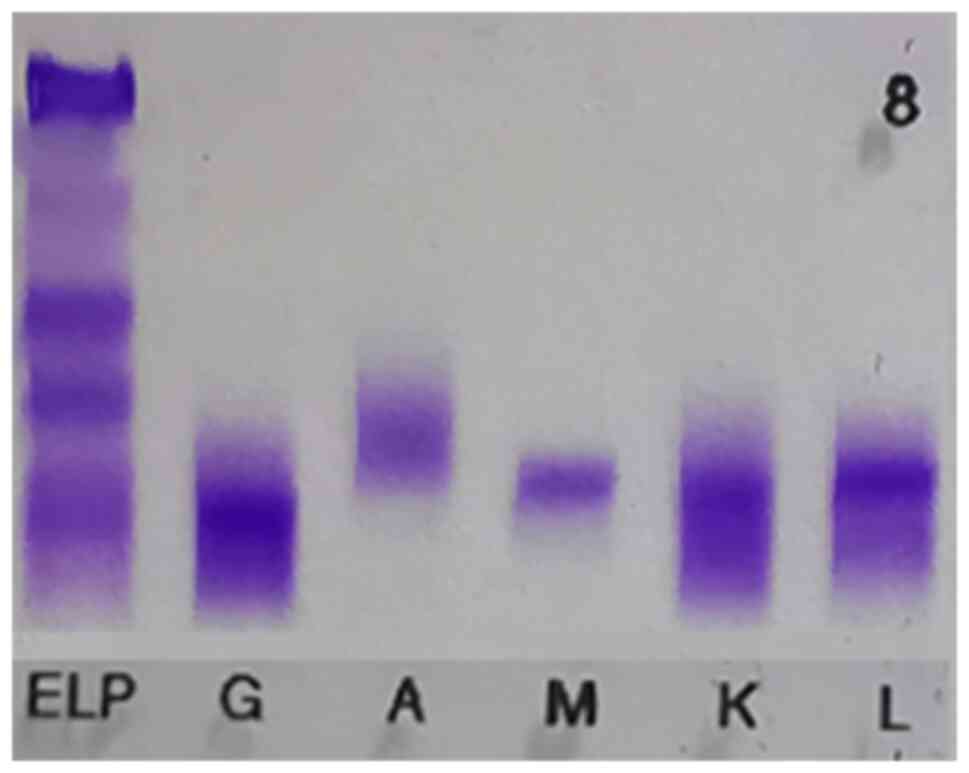
(4.0–6.0%). Serum immunofixation electrophoresis was also performed
(Fig. 1), which provided the
following results: IgG (+), IgA (−), IgM (+), IgG-κ (+) and IgM-λ
(+). In addition, serum protein electrophoresis yielded an M band
(+).
The case was considered a plasma cell disease and
transferred to the Hematology Department on March 12, 2019. Further
laboratory examination was performed, the results of which were as
follows: Ferritin, 350.62 ng/ml (21.84–274.66 ng/ml);
β2-microglobulin, 4.3 mg/l (0.8–1.8 mg/l); erythrocyte
sedimentation rate, 23 mm/h (0–15 mm/h); cardiac troponin T, 28.8
ng/l (<14 ng/l); N-terminal-pro B-type natriuretic peptide,
1,153 pg/ml (<125 pg/ml); urine protein, 0.304 g/24 h (0–0.141
g/24 h); creatinine clearance, 56.82 ml/min (80–120 ml/min); IgG,
9.22 g/l; IgA, 2.06 g/l; IgM, 1.36 g/l; complement C3, 0.31 g/l
(0.79–1.52 g/l); complement C4, 0.10 g/l (0.16–0.38 g/l); κ, 7.78
mg/l; λ, 3.15 mg/l; and vascular endothelial growth factor (VEGF),
449.06 pg/ml (0–142 pg/ml). An ultrasound examination revealed
hemangioma in the left lobe of the liver and multiple lymph node
echoes in the neck, armpit and groin. The largest lymph node was
27×7 mm, with no evident abnormal lymph node structure. A molecular
biology test was positive for rearrangement of the gene
immunoglobulin heavy locus (IGH), and testing for the fusion gene
myeloid differentiation factor 88 (MyD88)-L265P was also positive.
Examination of molecular cytogenetics using fluorescence in situ
hybridization showed that the cells were fibroblast growth factor
receptor 3/IGH and MAF/IGH negative. Bone marrow chromosome
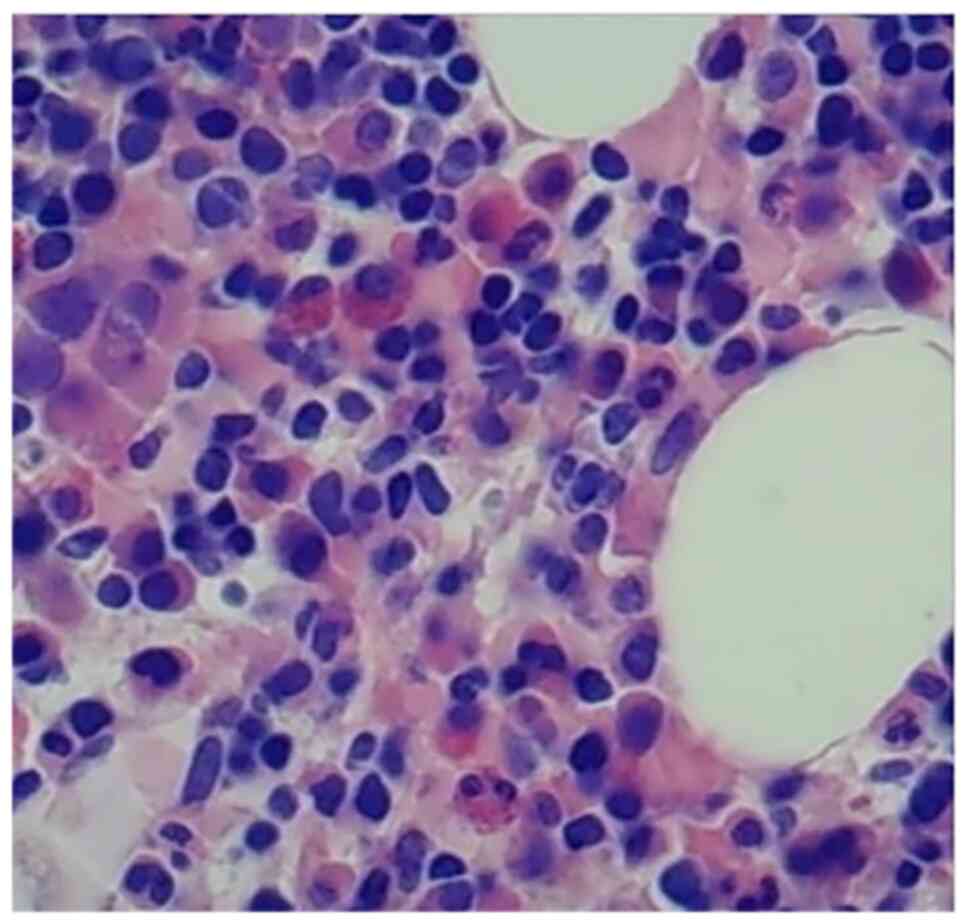
karyotyping revealed 10 normal metaphase karyotypes. Bone marrow
biopsies (Fig. 2) showed highly
active bone marrow hyperplasia (50–60%) and a low ratio of
granulocytes to red blood cells. Cells of all stages of the
granulocyte lineage were visible, with the majority of the cells
being in the middle and lower stages. Cells at all stages of the
red blood cell lineage were observed, which were mainly middle- and
late-stage erythrocytes. Plasma cells and lymphocytes were abundant
and were either scattered or clustered. The reticular fiber
staining result was grade MF-1, indicating early-stage primary
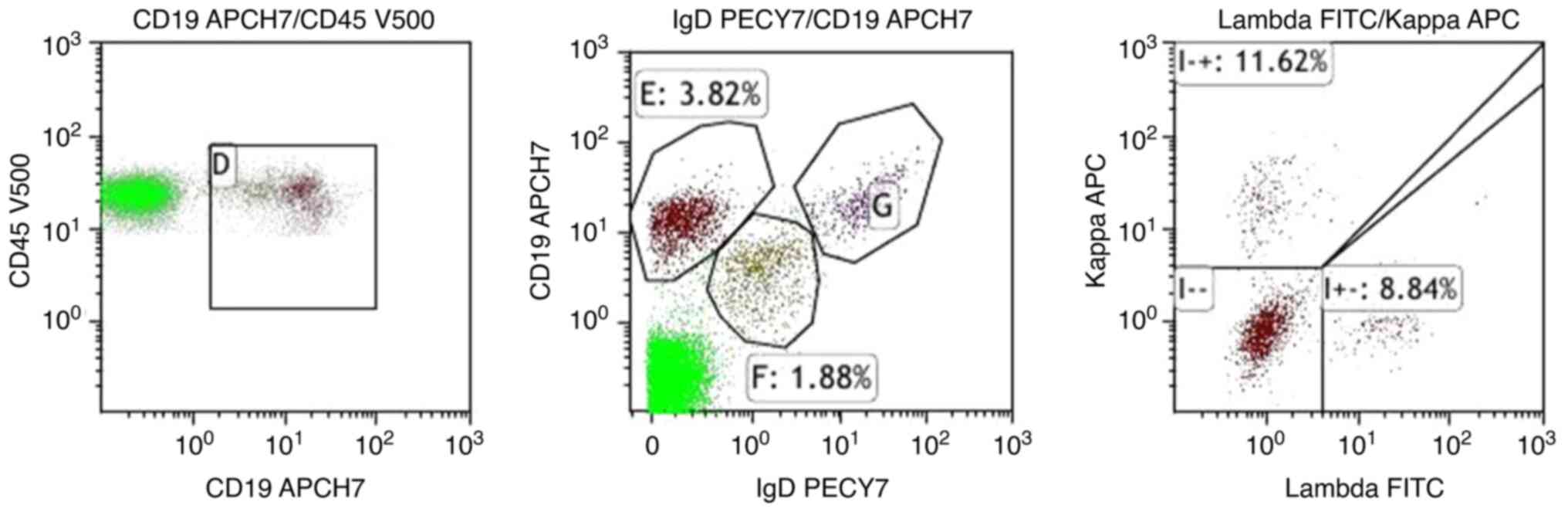
myelofibrosis. For flow cytometry, 1.5-2.0 ml EDTA bone marrow
sample was taken, single nucleated cells were isolated, and
immunofluorescent labelling was applied. (APCH7, APD, PECY7, FITC,
CD45 V500 Beckman Coulter Commercial Enterprise Detection by BC
NAVIOS flow cytometry (Beckman Coulter); the medullary phenotype
was normal (Fig. 3). Most
granulocytes were mature, and no abnormal phenotypes were observed
for red blood cells, monocytes, T-lymphocytes and natural killer
cells. However, there were two groups of abnormal B cells. One
group of abnormal B-lymphocytes constituted 3.82% of the nuclear
cell population and were CD19 and CD20 positive, and CD5, CD10,
CD103, IgD, IgM, CD23 and FMC7 negative, similar to CD5 and CD10
negative monoclonal B-lymphocytes. The other group of abnormal
B-lymphocytes, which constituted 1.88% of the nuclear cell
population, had restricted λ expression, were CD19 and CD20
positive, IgM positive, IgD dim, and κ, CD5, CD10, CD103, CD23 and
FMC7 negative, also similar to CD5 and CD10 negative monoclonal
B-lymphocytes. Plasma cells accounted for 0.20% of the nuclear
cells. Furthermore, the serum κ level was 4,025.0 mg/l (3.3–19.4
mg/l), serum λ level was 92.0 mg/l (5.71–26.3 mg/l) and serum κ/λ
ratio was 43.75 (0.26–1.65), while the urine κ level was 194.0 mg/l
(0.39~15.1 mg/l), urine λ level was 16.7 mg/l (0.81–10.10 mg/l) and
urine κ/λ ratio was 11.62 (0.461–4.0). Bone density tests indicated
reduced bone mass. A vibration perception threshold test (VPT)
indicated mild-to-moderate sensory impairment of the foot sensory
nerve and severe peripheral neuropathy, which was classified as
clinical neuropathy grade 2. These observations in combination with
the aforementioned laboratory results led to the patient being
diagnosed with LPL/WM associated with POEMS syndrome.
Based on this diagnosis, the patient was treated
with rituximab, intravenous immunoglobulin and ALB support
treatments to relieve symptoms. After two courses of treatment, the
symptoms were in remission and the patient was discharged. The
patient accepted routine follow-up appointments, and after 6 months
the condition of the patient remained stable.
Discussion
LPL/WM is a proliferative disease of
lymphoplasmacytic cells characterized by the secretion of
monoclonal IgM in the bone marrow, which is classified by the World
Health Organization as a non-Hodgkin's LPL (3). The diagnostic criteria for LPL/WM
include three elements: i) The presence of monoclonal IgM
immunoglobulin in the serum of patients with WM; ii) the presence
of typical lymphoplasmacytic cells in the bone marrow space; and
iii) the presence of associated clinical symptoms, including fever,
weight loss and enlargement of the liver, spleen and superficial
lymph nodes. Lin and Medeiros (4)
suggested that when IgM protein levels are higher than 30–40 g/l,
the increase in concentration of IgM may lead to systemic
amyloidosis and cryoglobulinemia. Furthermore, Baehring et
al (5) identified peripheral
neuropathy in 20% of patients with LPL/WM. The most common clinical
presentation of LPL/WM is symmetrical slow bilateral numbness of
the lower extremities with demyelination, and
anti-myelin-associated glycoprotein antibodies are often detected
in the serum of these patients. The neuropathological features of
POEMS syndrome are typically demyelination, with polyneuropathy and
monoclonal plasma cell proliferative disorder being the two
mandatory criteria for POEMS syndrome. In addition, for the
diagnosis of POEMS syndrome, at least one other major and one minor
criterion are required (6,7); polyneuropathy with monoclonal protein
is the primary diagnosis and organomegaly, endocrinopathy with skin
changes is the secondary diagnosis.
Physical examination findings of the patient in the
present study included a mildly anemic appearance, mild lid edema,
palpable superficial lymph nodes and severe edema of both lower
extremities. Serum immunofixation electrophoresis results were
positive for IgG-κ and IgM-λ, along with globulin M. Other
molecular biology tests were positive for IGH gene rearrangement
and the fusion gene MyD88-L265P. Flow cytometry revealed that
plasma cells accounted for 0.20% of the nuclear cells. Two sets of
abnormal B lymphocytes (CD5 and CD10 negative, CD19 and CD20
positive) were detected, indicating the presence of a plasma cell
phenotype. Based on these findings, the patient was diagnosed with
LPL/WM (7,8). VPT of the foot showed mild to moderate
profound sensory deficits with severe peripheral nerve disease, and
the FBG and 2h-PG levels were above the normal range. VEGF levels
were also markedly elevated, and the patient presented with
multiple lymphadenitis, edema and abnormal plasma cell
proliferation. Therefore, he was considered to have LPL/WM with
POEMS syndrome based on a retrospective analysis.
The clinical features of previously reported
patients were analyzed and compared with the present case. The four
patients (9,10) reported in the previous literature
comprises three males and one female with an age of onset between
54 and 73 years, with a mean age of 64.3 years. Three patients had
higher IgM levels than IgG levels and the other patient had lower
IgM levels than IgG levels. The patient in the present case was a
65-year-old male, who was identified as having both IgG-κ and Ig
M-λ according to immunofixation electrophoresis analysis, with IgM
levels lower than those of IgG, as shown in Table I. Cases of LPL with coexisting IgM
and IgG are very rare, and their presence in combination with POEMS
syndrome have not previously been reported, to the best of our
knowledge. Therefore, it is not possible to explain the differences
or similarities between the present case and the previously
reported cases, nor the etiology of IgM and IgG coexistence.
 | Table I.Clinical data of five cases of
biclonal lymphoplasmacytic lymphoma/Waldenström
macroglobulinemia. |
Table I.
Clinical data of five cases of
biclonal lymphoplasmacytic lymphoma/Waldenström
macroglobulinemia.
| First author,
year | Age, years | Sex | Symptoms | Type | IgM (g/l) | IgG (g/l) | (Refs.) |
|---|
| He, 2019 | 73 | F | Lymphadenopathy,
weakness, lymphadenopathy | IgG-λ, IgM-κ | 8.3 | 9.6 | (9) |
|
| 54 | M | Weakness, loss of
weight | IgG-κ, IgM-κ | 7.2 | 1.2 |
|
|
| 64 | F | Anemia, weakness | IgG-κ, IgM-κ | 23.5 | 9.7 |
|
|
| 66 | M | Anemia, fever | IgG-κ, IgM-κ | 36.6 | 14.5 |
|
| Present study | 65 | M | Anemia, polydipsia,
polyuria | IgG-κ, IgM-λ | 1.35 | 9.22 | - |
MyD88 and CXC motif chemokine receptor 4 (CXCR4)
mutational activations are the most common molecular genetic
alterations that occur in LPL/WM. MyD88 is an adaptor that binds to
Toll-like receptor 4, as well as to interleukin-1 and interleukin-2
receptors. When MyD88 binds to these receptors, they can be
activated directly or indirectly through interactions with TIR
domain-containing adaptor protein and Bruton's tyrosine kinase,
thereby activating the NF-κB signaling pathway (11). Hunter et al (12) found that methylation of the PR
domain-containing protein 5 and WNK lysine deficient protein kinase
2 genes led to inhibition of the NF-κB signaling pathway, which
suggests that hypomethylating drugs may be effective in the
treatment of LPL/WM. The L265P mutation in MyD88 occurs in 90% of
patients with WM, making this mutation useful for differentiating
LPL/WM from marginal zone lymphoma (13). CXCR4 is a G-protein-coupled receptor
that plays an important role in cytokine release and chemotaxis.
Kristinsson and Landgren (14)
found that LPL/WM cells express the chemokine receptor SDF-1a.
Knockout of the CXCR4 gene or the use of CXCR4 inhibitors and G
protein inhibitors to target SDF-1a inhibits the migration and
adhesion of WM cells. LPL/WM cells also express another chemokine
receptor, a4b1 integrin very late antigen-4 (VLA-4), which
interacts directly with CXCR4, thereby activating the AKT and MAPK
signaling pathways, which promotes LPL/WM cell survival and
resistance to apoptosis (15,16).
Varettoni et al (17)
suggested that mutation of CXCR4 may be an independent risk factor
for a shorter treatment-free survival as patients progressed from
asymptomatic to symptomatic LPL/WM. The CXCR4/SDF-1 axis interacts
with VLA-4 and regulates the migration and adhesion of WM cells to
the basement membrane, while CXCR4 regulates migration, in
particular transcortical migration, as well as adhesion to stromal
and endothelial cells (18). We
hypothesize that CXCR4 mutations are a risk factor for promoting
the transition from asymptomatic WM to symptomatic WM.
As an inert and incurable disease, it must be
emphasized that the clinical treatment LPL/WM should be initiated
only after patients have been identified as having the relevant
indications. Current treatment is primarily aimed at relieving
symptoms, rather than achieving complete hematological remission.
Since patients also express high levels of CD20, the application of
rituximab is a promising treatment option. Rituximab inhibits
lymphocyte proliferation and induces apoptosis through
antibody-dependent cytotoxicity. Ghobrial et al (19) suggested that IgM expression
increases in response to treatment during the early phase of
rituximab therapy, which is referred to as an IgM burst. Current
treatment regimens are mainly based on rituximab combinations,
including rituximab combined with alkylating agents, such as
chlorambucil or cyclophosphamide, or rituximab combined with
nucleoside analogs, for example, fludarabine. Simon et al
(20) noted that, although
fludarabine is used as a first-line treatment,
fludarabine-containing regimens are more appropriate for use as a
treatment option for patients with relapsed or refractory WM due to
the high risk of secondary malignancies. Rituximab can also be used
in combination with bortezomib and dexamethasone, and a study on
relapsed or refractory WM (21)
suggested that bortezomib should be used in the treatment regimen
when herpes zoster prophylaxis is a priority. Rituximab has also
been used in combination with immunomodulatory agents, including
lenalidomide or thalidomide, and a study showed that R-FC
combination chemotherapy containing rituximab resulted in improved
overall objective disease response rates and faster optimal
response times in patients with LPL/WM (22). Brown et al (23) reported no significant differences in
clinical characteristics, treatment response or prognosis between
patients with biclonal immunoglobulinemia and those with
traditional LPL/WM. Due to the limited number of cases described,
however, the issue of treatment response and patient prognosis
requires further study to determine the best treatment option(s).
Souchet et al (24)
described the use of rituximab to treat WM with POEMS syndrome.
Studies have also shown that rituximab or low doses of chlorambucil
are safe and effective treatment options for elderly patients
(25–27). In the present case, the patient was
a 65-year-old male, and rituximab was selected as a monotherapy.
The patient was discharged with remission of symptoms following two
courses of treatment. After 6 months of follow-up, the condition of
the patient was stable, and the treatment was subsequently
abandoned due to financial issues.
Acknowledgements
Not applicable.
Funding
This study was supported by the Key Research Program of Gansu
Province (grant no. 21YF5FA131) and Gansu Province Higher Education
Innovation Fund Project (grant no. 2021A-084).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QW wrote the manuscript and made a significant
contribution to the conception and design of the study. WG and HL
collected data. HL interpreted data. QL revised the manuscript
critically for important intellectual content WG analyzed data. QL
and QW confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics and Research
Committee of Gansu Provincial People's Hospital.
Patient consent for publication
Informed written consent was obtained from the
patient for publication of this report and the accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD and Jaffe ES: The 2016 revision of the world health organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Polyatskin IL, Artemyeva AS and Krivolapov
YA: Revised WHO classification of tumors of hematopoietic and
lymphoid tissues, 2017 (4th edition): Lymphoid tumors. Arkh Patol.
81:59–65. 2019.(In Russian). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Swerdlow SH, Campo E and Harris NH: WHO
classification of tumors of hematopoietic and lymphoid tissues. Int
Agency Res Cancer. 68–71. 2008.
|
|
4
|
Lin P and Medeiros LJ: Lymphoplasmacytic
lymphoma/waldenstrom macroglobulinemia: An evolving concept. Adv
Anat Pathol. 12:246–255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baehring JM, Hochberg EP, Raje N,
Ulrickson M and Hochberg FH: Neurological manifestations of
waldenstrom macroglobulinemia. Nat Clin Pract Neurol. 4:547–556.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yun S, Johnson AC, Okolo ON, Arnold SJ,
McBride A, Zhang L, Baz RC and Anwer F: Waldenström
macroglobulinemia: Review of pathogenesis and management. Clin
Lymphoma Myeloma Leuk. 17:252–262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Niu T: The interpretation of version
1.2018 of the NCCN guidelines for waldenstr(o)m's
macroglobulinemia/lymphoplasmacytic lymphoma. West China Med J.
33:393–397. 2018.(In Chinese).
|
|
8
|
Hematology Oncology Committee of China
Anti-Cancer Association, . Leukemia & Lymphoma Group Society of
Hematology at Chinese Medical Association; Union for China Lymphoma
Investigators: The consensus of the diagnosis and treatment of
lymphoplasmacytic lymphoma/Walderström macroglobulinemia in China
(2016 version). Zhonghua Xue Ye Xue Za Zhi. 37:729–734. 2016.(In
Chinese),. PubMed/NCBI
|
|
9
|
He J, Hu LW and Gu XK: Lymphoplasmacytic
lymphoma with the coexistence of monoclonal IgM-kappa and
IgG-kappa: A case report and literature review. J Clin Hematol.
32:219–221. 2019.(In Chinese).
|
|
10
|
Zhang M, Hong F and Fu X: Three cases of
lymphoplasmacytic lymphoma were reported and reviewed in the
literature. Chinese Tumor Clin. 37:1190–1191. 2010.
|
|
11
|
Ngo VN, Young RM, Schmitz R, Jhavar S,
Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al:
Oncogenically active MYD88 mutations in human lymphoma. Nature.
470:115–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hunter ZR, Xu L, Yang G, Tsakmaklis N, Vos
JM, Liu X, Chen J, Manning RJ, Chen JG, Brodsky P, et al:
Transcriptome sequencing reveals a profile that corresponds to
genomic variants in waldenström macroglobulinemia. Blood.
128:827–838. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shin SY, Lee ST, Kim HY, Park CH, Kim HJ,
Kim JW, Kim SJ, Kim WS and Kim SH: Detection of MYD88 L256P in
patients with lymphoplasmacytic lymphoma/waldedstrom
macorglobulinemia and other B-cell non-hodgkin lyphomas. Blood Res.
51:181–186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kristinsson SY and Landgren O: What causes
Waldenström's macroglobulinemia: Genetic or immune-related factors,
or a combination? Clin Lymphoma Myeloma Leuk. 11:85–87. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cao Y, Hunter ZR, Liu X, Xu L, Yang G,
Chen J, Patterson CJ, Tsakmaklis N, Kanan S, Rodig S, et al: The
WHIM-like CXCR4 (S338X) somatic mutation activates AKT and ERK, and
promotes resistance to Ibrutinib and other agents used in the
treatment of Waldenström macroglobulinemia. Leukemia. 29:169–176.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ngo HT, Leleu X, Lee J, Jia X, Melhem M,
Runnels J, Moreau AS, Burwick N, Azab AK, Roccaro A, et al:
SDF-1/CXCR4 and VLA-4 interaction regulates homing in Waldenström
macroglobulinemia. Blood. 112:150–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Varettoni M, Zibellini S, Defrancesco I,
Ferretti VV, Rizzo E, Malcovati L, Gallì A, Porta MGD, Boveri E,
Arcaini L, et al: Pattern of somatic mutations in patients with
Waldenström macroglobulinemia or IgM monoclonal gammopathy of
undetermined significance. Haematologica. 102:2077–2085. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Konoplev S, Medeiros LJ, Bueso-Ramos CE,
Jorgensen JL and Lin P: Immunophenotypic profile of
lymphoplasmacytic lymphoma/Waldenström macroglobulinemia. Am J Clin
Pathol. 124:414–420. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghobrial IM, Fonseca R, Greipp PR, Blood
E, Rue M, Vesole DH and Gertz MA; Eastern Cooperative Oncology
Group, : Initial immunoglobulin M ‘flare’ after rituximab therapy
in patients diagnosed with Waldenström macroglobulinemia: An
eastern cooperative oncology group study. Cancer. 101:2593–2598.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Simon L, Fitsiori A, Lemal R, Dupuis J,
Carpentier B, Boudin L, Corby A, Aurran-Schleinitz T, Gastaud L,
Talbot A, et al: Bing-Neel syndrome, a rare complication of
Waldenström macroglobulinemia: Analysis of 44 cases and review of
the literature. A study on behalf of the French Innovative Leukemia
Organization (FILO). Haematologica. 100:1587–1594. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ghobrial IM, Hong F, Padmanabhan S, Badros
A, Rourke M, Leduc R, Chuma S, Kunsman J, Warren D, Harris B, et
al: Phase II trial of weekly bortezomib in combination with
rituximab in relapsed or relapsed and refractory Waldenström
macroglobulinemia. J Clin Oncol. 28:1422–1428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haider S, Latif T, Hochhausler A, Lucas F
and Abdel Karim N: Waldenstrom's macroglobulinemia and peripheral
neuropathy, organomegaly, endocrinopathy, monoclonal gammopathy,
and skin changes with a bleeding diathesis and rash. Case Rep Oncol
Med. 2013:8908642013.PubMed/NCBI
|
|
23
|
Brown R and Ginsberg L: POEMS syndrome:
Clinical update. J Neurol. 266:268–277. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Souchet L, Levy V, Ouzegdouh M, Tamburini
J, Delmer A, Dupuis J, Le Gouill S, Pégourié-Bandelier B,
Tournilhac O, Boubaya M, et al: Efficacy and long-term toxicity of
the rituximab-fludarabine-cyclophosphamide combination therapy in
Waldenstrom's macroglobulinemia. Am J Hematol. 91:782–786. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feugier P, Van Hoof A, Sebban C,
Solal-Celigny P, Bouabdallah R, Fermé C, Christian B, Lepage E,
Tilly H, Morschhauser F, et al: Long-term results of the R-CHOP
study in the treatment of elderly patients with diffuse large
B-cell lymphoma: A study by the Groupe d'Etude des Lymphomes de
l'Adulte. J Clin Oncol. 23:4117–4126. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Coiffier B, Thieblemont C, Van Den Neste
E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M,
Sebban C, et al: Long-term outcome of patients in the LNH-98.5
trial, the first randomized study comparing rituximab-CHOP to
standard CHOP chemotherapy in DLBCL patients: A study by the Groupe
d'Etudes des Lymphomes de l'Adulte. Blood. 116:2040–2045. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Foà R, Del Giudice I, Cuneo A, Del Poeta
G, Ciolli S, Di Raimondo F, Lauria F, Cencini E, Rigolin GM,
Cortelezzi A, et al: Chlorambucil plus rituximab with or without
maintenance rituximab as first-line treatment for elderly chronic
lymphocytic leukemia patients. Am J Hematol. 89:480–486. 2014.
View Article : Google Scholar : PubMed/NCBI
|