Introduction
Glioblastoma are a highly aggressive and the most
common type of primary central nervous system tumor in adults
(1). According to the 2021
classification of the World Health Organization (WHO), glioblastoma
are mainly classified according to the status of isocitrate
dehydrogenase (IDH), IDH1 wildtype and IDH1 mutant, and correspond
to a grade 4 astrocytoma (1).
The development of glioblastoma is often the result
of an accumulation of genetic changes and dysregulation of
signaling cascades, which lead to the uncontrolled proliferation of
tumor cells. Methylation is considered the most common and one of
the most important epigenetic mechanisms (2). It serves an essential role in
embryogenesis, X-inactivation, genomic imprinting, regulation of
gene expression and carcinogenesis (2). Catalysis of the methylation reaction,
in which 5-methylcystocine is formed from cytosine and a methyl
group at position C5 of its pyrimidine ring, is performed by DNA
methyl transferases and S-adenosylmethionine acts as a methyl donor
and cofactor in the reaction (3).
Unmethylated, deaminated cytidines are converted to uracil, which
will lead to both incorrect implantation in the DNA-sequence and
elimination of this error by the DNA repair system. However,
methylated cytidines (5′-methylcytidine) are converted to thymine
by deaminases, which avoids recognition of the error by the DNA
repair system. Consequently, this part of the DNA will be
deactivated, which leads to silencing of the gene and loss of
expression (3).
DNA methylation can be reversed in both active and
passive ways. Passively it can be reversed in the absence of
functional DNA methyltransferase 1 (DNMT1)/UHRF1 proteins during
DNA replication. Actively, there are oxidation-dependent and
oxidation independent DNA methylation mechanisms (4). Regarding the oxidation dependent
mechanism, the enzymatic methylation removal and loss of methylated
cytosines following nucleotide excision repair, and the deamination
of methylated cytosine and the subsequent repair mechanisms can
both lead to demethylation. The oxidation dependent mechanisms
include the oxidation of methylated cytosines by TET proteins and
the subsequent involvement of DNMT1/UHRF1, as well as other ways
including DNA repair mechanisms which remove modified methylated
cytosine. Furthermore, dehydroxymethylation by DNMTs and
decarboxylation of 5caC (5-carboxylcytosine) can lead to the
removal of the methyl group (4). In
humans, the methylation reaction occurs exclusively at the cytosine
nucleotides within CpG dinucleotides in the direction of 5′-CpG-3′.
CpG dinucleotides are particularly abundant in repetitive DNA
sequences or in CpG islands. While 70% of all CpG dinucleotides are
methylated, CpG islands are an exception (2) and are often part of the promoter
regions of genes and are usually unmethylated. Methylations of
promoter regions are associated with gene silencing (3). This is of particular interest when
transcriptional gene silencing prevents the expression of tumor
suppressor genes and thus allows development of a tumor (2).
The RB1 gene is a tumor suppressor gene located on
chromosome 13q14.1-q14.2 (5), which
consists of ~200 kb DNA including 27 exons and encodes
retinoblastoma protein (pRB), a nuclear phosphoprotein, which
consists of 928 amino acid residues. pRB is involved in the
regulation of the cell cycle, cell differentiation and the
initiation of apoptosis (6). The
RB1 gene has been known for a long time and serves an important
role in numerous tumor diseases. The function of RB1 is to
interrupt the cell cycle at the transition of G1 to S phase
(7). Regulation of the activity of
pRB occurs via its phosphorylation status: While pRB is
unphosphorylated in G0 and early G1 phase, it is phosphorylated as
the cell cycle approaches the G1-S transition, it then remains in
this state until it is dephosphorylated again in late M phase
(8). Its role in glioblastoma has
also been previously reported by numerous studies. As a tumor
suppressor gene, RB1 is complexly linked to numerous other genes
and factors in its signaling pathway and modifications of the RB1
signaling pathway also serve an important role in glioblastoma.
It has been previously reported that ~30% of all
malignant astrocytomas had loss of heterozygosity (LOH) of the RB1
gene, whereas this was not observed in low-grade astrocytomas
(9). Thus, loss of function of RB1
contributes to the upgrading from low-malignant to high-malignant
astrocytomas (10). Alteration of
the cyclin dependent kinase inhibitor 2A (CDKN2A), a tumor
suppressor gene located on human chromosome 9p21 which acts as a
negative regulator of the cell cycle (11), or RB1 gene due to mutations or LOH
can often be found in glioblastomas, in most cases only one of the
genes is affected (12,13). Ichimura et al (13) reported that 64% of the 120
glioblastoma cases assessed, had gene alterations, which caused a
disturbance in cell cycle control at the G1-S transition. The same
study reported that in 30% of cases, loss of one allele of the
CDKN2A or RB1 gene occurred, and in only 6% of cases were both
genes unchanged. Alterations in the CDKN2A gene were quite common,
occurring in 40% of all glioblastomas, and the RB1 gene was altered
in 14% of the cases.
The role of RB1 promoter methylation in glioblastoma
is controversial and the literature contains partly contradictory
statements. Numerous studies have been performed previously and
they have yielded different results. To the best of our knowledge,
the present study is the first on this topic which classified
glioblastomas according to the current, 2016 version of the WHO
Classification of Tumors of the Central Nervous System (14). The intention of the present study
was to examine a defined collective of patients with glioblastoma,
which was large as possible, to support a general statement on the
relevance of RB1 methylation in glioblastoma and to evaluate the
impact of methylation on clinical parameters.
Materials and methods
Patients and tissue specimens
All primary glioblastoma tumors (n=85) were obtained
following surgical resection at the Saarland University Medical
Center (Homburg, Germany) between 2003 and 2013 as part of routine
clinical care. Tumors were subsequently examined histologically by
a neuropathologist and the diagnosis was made according to the 2016
WHO classification (14). All
samples were collated in the Institute of Human Genetics at the
Saarland University Medical Center and tissue samples were frozen
in cryo tubes at −80°C directly following surgery. The tissue
samples were removed from −80°C storage from 2013, the beginning of
the present study. The present study was approved by the Ethics
Committee of the General Medical Council of the State Saarland
(43/99) and informed consent for participation was obtained from
all participants involved in the study.
DNA isolation
DNA was extracted from the frozen samples of
glioblastoma. The tumor tissue was minced using forceps and
scissors, according to previously published methods (15). The DNA was dissolved in distilled
water overnight at 4°C and then a photometric concentration
determination was performed using a NanoDrop 2000/2000c
spectrophotometer (Thermo Fisher Scientific, Inc.).
Bisulfite treatment
After the DNA was isolated, double stranded DNA
concentrations were assessed for bisulfite conversion. The isolated
DNA was mixed with sterile water for injection and adjusted to 25
ng/µl. Bisulfite conversion was performed using an EZ DNA
Methylation-Gold kit (Zymo Research Corp.) according to the
manufacturers' instructions. The probes were transferred into the
PTC-100 Thermal Cycler (MJ Research, Inc.; Bio-Rad Laboratories,
Inc.). Afterwards the treatment was continued using the Kit
according to the manufacturers' protocol.
Methylation-specific PCR (MS-PCR)
The MS-PCR method was developed in 1996 by Herman
et al (16). In contrast
with typical PCR, in MS-PCR, two pairs of primers are used instead
of one. The first primer pair is used to amplify the DNA sequence
of a methylated RB1-promoter. The second primer pair is then used
to amplify the DNA sequence of an unmethylated RB1 promoter. For
the analysis of the methylation status of a tumor sample, it is
necessary to set up two PCR assays with tumor DNA, and
amplification of the DNA occurs in only one of the two assays.
Three controls were used, including a positive control which
contained methylated DNA (Zymo Research Corp.) and a negative
control, in which unmethylated DNA from a male donor was used.
According to the manufacturer, these positive controls were Human
HCT116 DKO methylated DNA and were appropriate for use as a
positive control for DNA methylation analysis. As a final control,
distilled water served to test the master mix for contamination.
The sequences of the primers used in the MS-PCR of the RB1 gene
were as follows (17,18): Methylated (m)RB1 forward (F),
5′-GGGAGTTTCGCGGACGTGAC-3′ and reverse (R),
5′-ACGTCGAAACACGCCCCG-3′; and unmethylated (u)RB1 F,
5′-GGGAGTTTTGTGGATGTGAT-3′ and R, 5′-ACATCAAAACACACCCCA-3′. The
sequences of the primers used in the MS-PCR of
O-6-methylguanine-DNA methyl transferase (MGMT), first reported by
Esteller et al (19), were
as follows: uMGMT F, 5′-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3′ and R,
5-AACTCCACACTCTTCCAAAAACAAAACA-3′; and mMGMT F,
5′-TTTCGACGTTCGTAGGTTTTCGC-3′ and R, 5′-GCACTCTTCCGAAAACGAAACG-3′.
Primers were synthesized by Eurofins Genomics Germany GmbH from the
aforementioned, cited sequences.
Each PCR assay consisted of a master mix and DNA (25
ng/µl) in an Eppendorf PCR tube. The master mix comprised distilled
water, taq-buffer (15 mM), forward primer (m/u, 4 pmol), reverse
primer (m/u, 4 pmol), dNTP (10 mM) and taq-polymerase (5 U/µl). A
hot-start PCR with 38 PCR cycles was performed using a PTC-100
thermocycler (MJ Research, Inc.; Bio-Rab Laboratories, Inc.). The
thermocycling conditions were as follows: Preheating at 95°C for 15
min to activate the polymerase, then DNA denaturation at 95°C for
45 sec, primer hybridization at 55°C for 45 sec and elongation at
72°C for 1 min.
In the Neurosurgery Department of the Saarland
University Medical Center in collaboration with the Neuropathology
Department in the Saarland University Medical Center every tumor is
diagnosed according to WHO recommendations; therefore, the 85 tumor
samples were tested for methylation of the MGMT gene as a standard
tool, for all GBM patients (19,20).
DNA isolation was performed using a QIAamp DNA Mini kit 50 DNA
isolation kit (Qiagen N.V.). The methylation status of MGMT was
determined by MS-PCR.
Agarose gel electrophoresis
The reverse transcription MS-PCR products were
evaluated using standard 2.5% agarose gel electrophoresis stained
with ethidium bromide and visualized under ultraviolet illumination
to evaluate the reverse transcription MS-PCR results. To perform
the electrophoresis, an electric field with a potential difference
of 135 V and a current of 260 mA was applied for two h. The bands
were assessed based on their presence on the gel at 93 bp and 89 bp
for unmethylated and methylated MGMT (O-6-Methylguanine-DNA Methyl
transferase) promoter sequence, respectively.
IDH1 immunohistochemistry
Determination of IDH1 status was performed in all
patients, except three who were excluded due to lack of-sample
material. For the determination of the IDH1 mutation status, the
indirect, two-step antibody method with an IDH1 R132H primary
antibody, which could detect the most common IDH1 mutation R132H
was used.
Immunohistochemistry was performed using 3 µm thick
formalin-fixed (fixed in 4% buffered formaldehyde at room
temperature for 24 h), paraffin-embedded (FFPE) tissue sections
were mounted on StarFrost Advanced Adhesive slides (Engelbrecht
Medizin-und Labortechnik GmbH), followed by drying at 80°C for 15
min. Deparaffinisation took place in xylene and sections were
rehydrated in a descending isopropanol series. Sections were
incubated with primary antibodies against R123H mutated
isocitratdehydrogenase 1 (1:50; cat. co. dia-H09; Dianova GmbH) for
30 min. The DAKO REAL kit (cat. no. K5007; Dako; Agilent
Technologies, Inc.) was used according to manufacturers'
instructions, for the visualization of the antigen-antibody
reaction with HRP/DAB.
Statistical analysis
Statistical analysis of the data were performed
using R software R 3.5.2 (www.r-project.org). The R-packages called ‘Survival’,
‘survminer’ and ‘openxlsx’ were used. The Kaplan-Meier method was
used to estimate the survival rates, and the Cox regression was
applied to compare the survival differences among the patients.
Progression-free survival (PFS) was defined as the time from
treatment start date until objective tumor progression or death.
Overall survival (OS) was calculated from the date of initial
surgery until the date of death or last follow-up. OS was defined
as the time from treatment to death, regardless of disease
recurrence. The other potential prognostic variable was the
Karnofsky score (21). The
confidence interval was set at 95% and the significance level was
set at P<0.05.
Results
Real-time MS-PCR
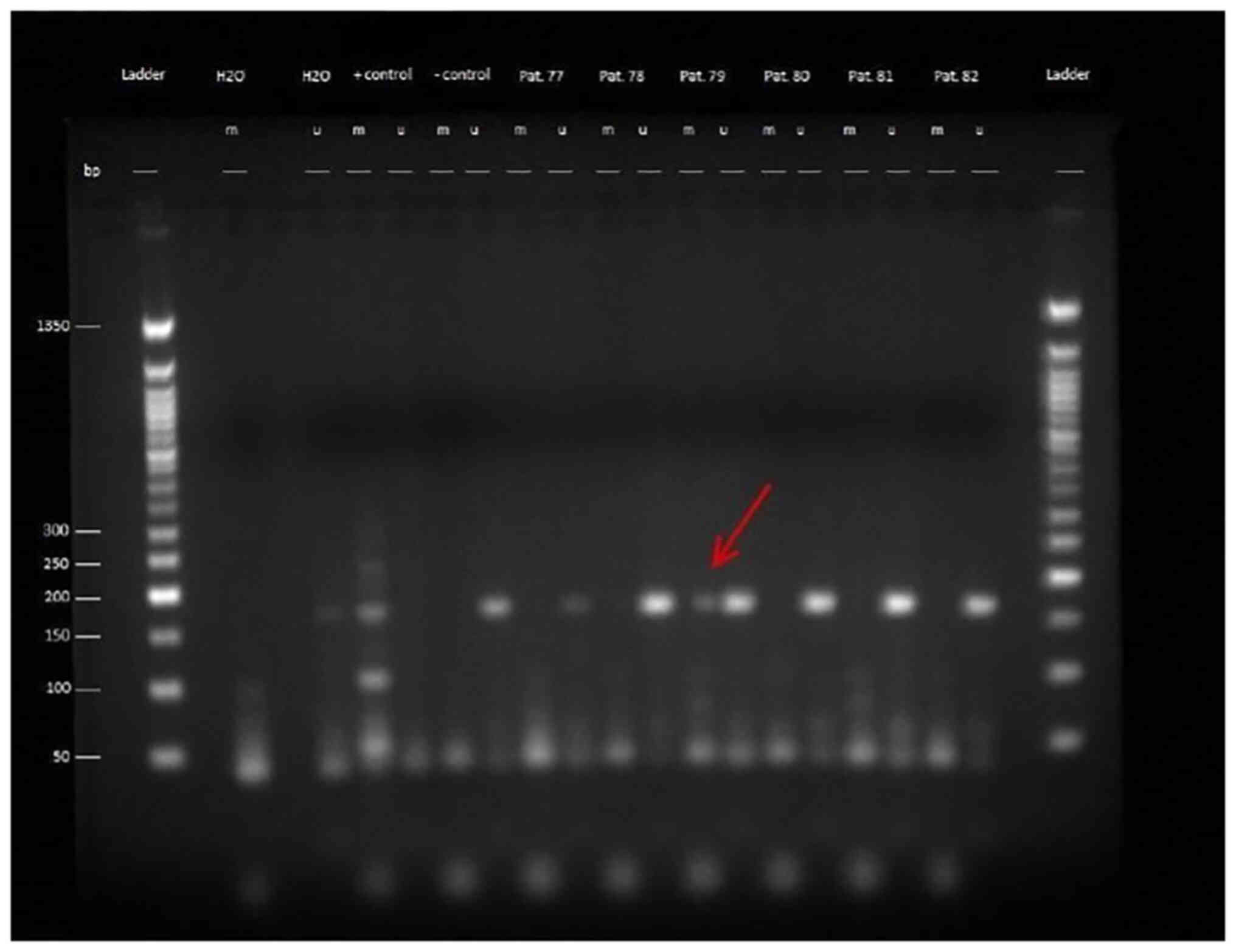
Methylation analysis of the RB1 gene was performed
on glioblastomas isolated from 85 patients. RB1 methylation was
identified in 1/85 patients (1.18%). Of the 85 patients included in
the present study, at the time of the last observation (December
2019), 84 patients had died from the disease, while one patient
(patient 79) was alive and tumor-free. Patient 79 was also the only
patient with an RB1 methylation and the solely sole survivor of the
present study.
The products of the MS-PCR were evaluated by gel
electrophoresis using a 2.5% agarose gel, which was examined under
UV light (Figs. 1 and S1). The length of the PCR products for
the methylated and the unmethylated approach were 172 bp. The bands
in the gel were therefore to be expected between the 150 bp band
and the 200 bp band of the DNA ladder. It was demonstrated that
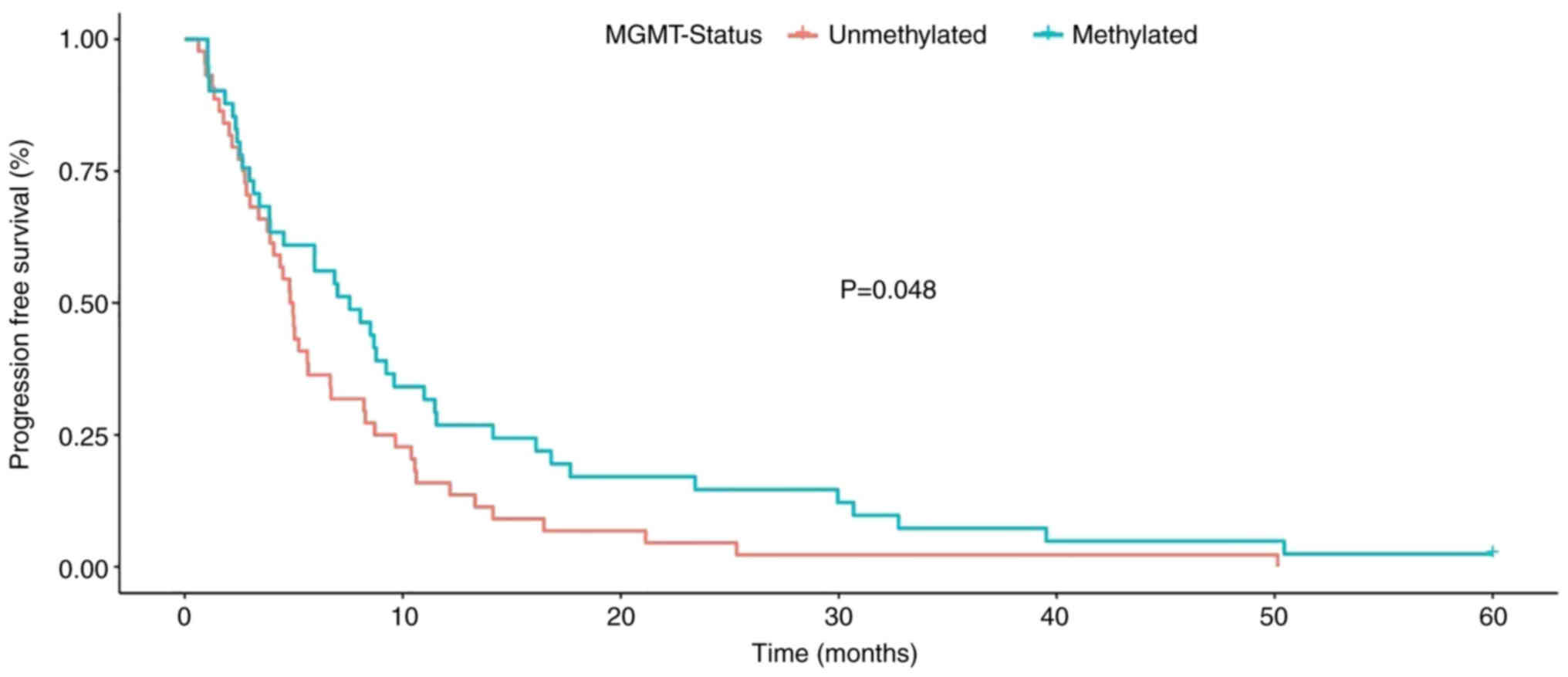
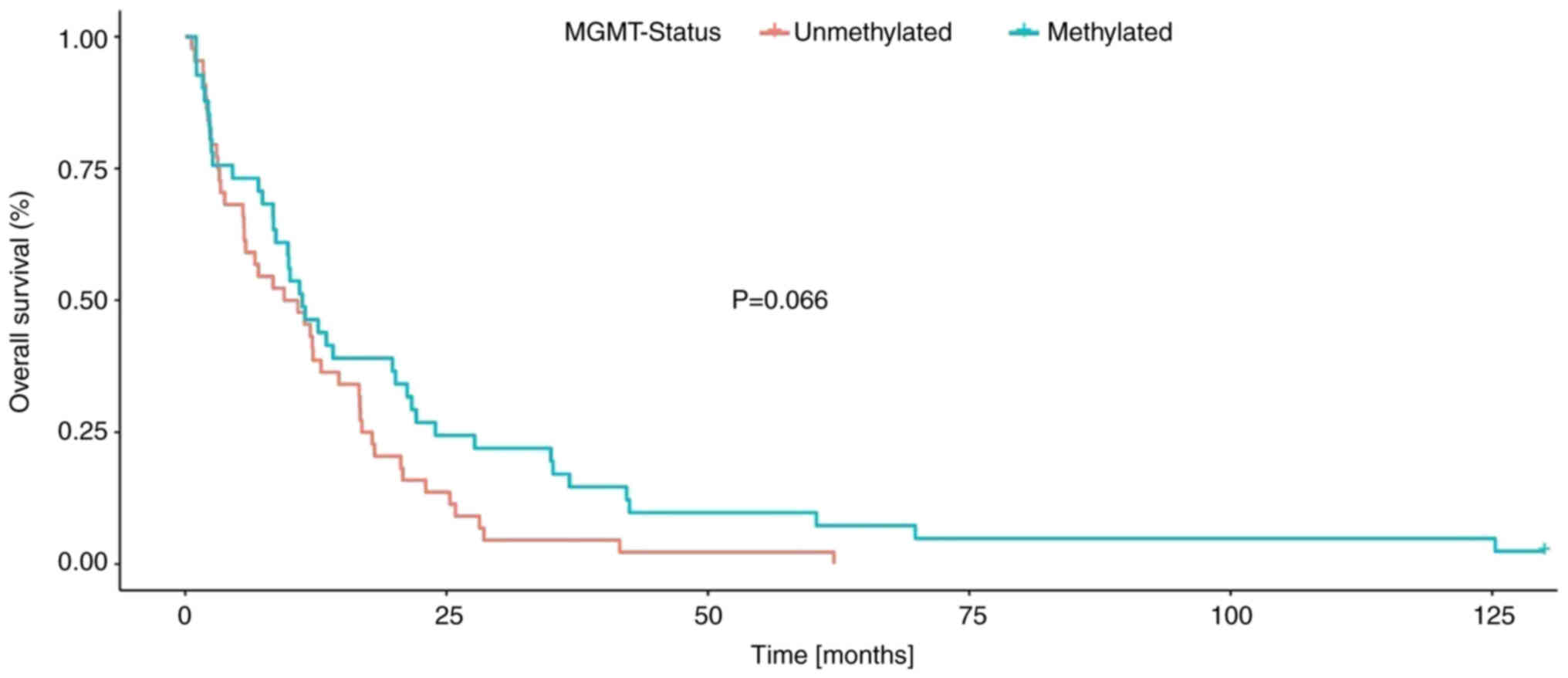
patients with methylated MGMT promoter had a markedly longer
survival. This relationship was statistically significant in
relation to PFS (P=0.048, Fig. 2),
but not in relation to OS (P=0.066, Fig. 3, Table
I).
 | Table I.Summary of the results per
patient. |
Table I.
Summary of the results per
patient.
| Patient | Sex | Age at initial
diagnosis, years | Overall survival,
months | Karnofsky
score | Progression free
survival, months | IDH1mutation
status | MGMT methylation
status | RB1 methylation
status |
|---|
| 1 | F | 81.09 | 0.92 | 70 | 0.92 | 0 | 0 | 0 |
| 2 | M | 80.14 | 2.63 | 70 | 2.63 | 0 | 1 | 0 |
| 3 | M | 75.54 | 1.05 | 80 | 1.05 | 0 | 1 | 0 |
| 4 | F | 72.82 | 22.09 | 90 | 16.11 | 0 | 1 | 0 |
| 5 | F | 69.80 | 1.68 | 80 | 1.12 | 0 | 1 | 0 |
| 6 | F | 69.45 | 9.90 | 90 | 8.05 | 0 | 1 | 0 |
| 7 | F | 52.41 | 20.12 | 100 | 11.54 | 0 | 1 | 0 |
| 8 | F | 52.24 | 25.84 | 100 | 8.71 | 1 | 0 | 0 |
| 9 | F | 39.49 | 18.12 | 90 | 10.55 | 0 | 0 | 0 |
| 10 | M | 74.87 | 7.00 | 90 | 4.83 | 0 | 0 | 0 |
| 11 | M | 72.14 | 2.47 | 90 | 2.47 | 0 | 0 | 0 |
| 12 | M | 69.50 | 21.24 | 100 | 8.78 | 0 | 1 | 0 |
| 13 | M | 63.10 | 5.79 | 80 | 4.37 | 0 | 0 | 0 |
| 14 | M | 53.60 | 62.04 | 80 | 50.14 | 0 | 0 | 0 |
| 15 | M | 57.93 | 11.21 | 90 | 9.24 | 0 | 1 | 0 |
| 16 | M | 55.88 | 16.73 | 100 | 4.80 | 0 | 0 | 0 |
| 17 | M | 52.38 | 16.70 | 90 | 12.16 | 0 | 0 | 0 |
| 18 | M | 52.72 | 42.48 | 100 | 16.80 | 0 | 1 | 0 |
| 19 | M | 45.59 | 12.16 | 100 | 4.08 | 0 | 0 | 0 |
| 20 | M | 43.54 | 5.65 | 80 | 5.65 | 1 | 0 | 0 |
| 21 | M | 44.54 | 28.14 | 100 | 16.47 | 0 | 0 | 0 |
| 22 | F | 54.08 | 69.83 | 90 | 39.52 | 0 | 1 | 0 |
| 23 | M | 67.05 | 3.78 | 90 | 3.78 | 0 | 0 | 0 |
| 24 | M | 71.00 | 1.08 | 70 | 1.08 | 0 | 1 | 0 |
| 25 | F | 54.05 | 8.45 | 80 | 5.95 | 0 | 1 | 0 |
| 26 | M | 75.32 | 5.52 | 90 | 1.35 | 0 | 0 | 0 |
| 27 | F | 69.17 | 2.99 | 80 | 2.99 | 0 | 0 | 0 |
| 28 | M | 44.62 | 36.76 | 90 | 32.75 | 0 | 1 | 0 |
| 29 | M | 73.47 | 2.33 | 80 | 2.33 | 0 | 1 | 0 |
| 30 | M | 50.57 | 12.23 | 100 | 5.03 | 0 | 0 | 0 |
| 31 | M | 70.61 | 1.97 | 90 | 1.58 | 0 | 0 | 0 |
| 32 | M | 59.36 | 23.01 | 100 | 9.67 | 0 | 0 | 0 |
| 33 | M | 64.29 | 16.64 | 90 | 10.39 | 0 | 0 | 0 |
| 34 | F | 50.23 | 10.03 | 70 | 7.56 | 0 | 1 | 0 |
| 35 | M | 45.54 | 11.97 | 90 | 2.76 | 0 | 0 | 0 |
| 36 | F | 82.90 | 1.05 | 70 | 1.05 | 0 | 1 | 0 |
| 37 | F | 83.02 | 0.62 | 80 | 0.62 | 0 | 0 | 0 |
| 38 | M | 71.64 | 27.68 | 70 | 6.87 | 0 | 1 | 0 |
| 39 | F | 73.74 | 2.40 | 90 | 2.40 | 0 | 1 | 0 |
| 40 | F | 70.99 | 2.04 | 90 | 2.04 | 0 | 0 | 0 |
| 41 | F | 68.24 | 2.17 | 80 | 2.17 | 0 | 0 | 0 |
| 42 | F | 52.56 | 10.78 | 80 | 5.23 | 0 | 0 | 0 |
| 43 | M | 54.72 | 7.00 | 70 | 7.00 | 0 | 1 | 0 |
| 44 | F | 35.97 | 60.36 | 90 | 50.43 | 0 | 1 | 0 |
| 45 | M | 72.40 | 3.39 | 70 | 3.39 | 0 | 0 | 0 |
| 46 | M | 72.30 | 1.84 | 80 | 1.84 | 0 | 1 | 0 |
| 47 | M | 68.22 | 13.51 | 100 | 3.42 | 0 | 1 | 0 |
| 48 | M | 66.78 | 41.56 | 90 | 21.14 | 0 | 0 | 0 |
| 49 | M | 58.87 | 4.54 | 90 | 4.54 | 0 | 1 | 0 |
| 50 | M | 54.02 | 17.88 | 70 | 5.00 | 0 | 0 | 0 |
| 51 | F | 54.73 | 20.61 | 90 | 14.14 | 0 | 0 | 0 |
| 52 | M | 52.03 | 34.98 | 90 | 23.41 | 0 | 1 | 0 |
| 53 | M | 49.77 | 14.70 | 90 | 3.91 | n.a. | 0 | 0 |
| 54 | M | 44.46 | 21.67 | 90 | 17.69 | 0 | 1 | 0 |
| 55 | M | 50.00 | 8.42 | 80 | 2.96 | n.a. | 1 | 0 |
| 56 | M | 41.51 | 12.72 | 80 | 9.60 | 0 | 1 | 0 |
| 57 | M | 37.25 | 125.29 | 90 | 5.95 | 1 | 1 | 0 |
| 58 | F | 59.64 | 2.20 | 30 | 2.20 | 0 | 1 | 0 |
| 59 | F | 68.97 | 10.95 | 90 | 8.52 | 0 | 1 | 0 |
| 60 | F | 71.48 | 2.33 | 90 | 0.95 | 0 | 0 | 0 |
| 61 | F | 51.92 | 8.68 | 80 | 8.68 | 0 | 1 | 0 |
| 62 | F | 76.13 | 35.18 | 70 | 29.95 | 0 | 1 | 0 |
| 63 | M | 65.19 | 7.40 | 60 | 3.16 | n.a. | 1 | 0 |
| 64 | F | 46.74 | 42.21 | 90 | 30.67 | 0 | 1 | 0 |
| 65 | F | 69.15 | 11.41 | 70 | 6.71 | 0 | 0 | 0 |
| 66 | M | 50.24 | 9.83 | 70 | 3.88 | 0 | 1 | 0 |
| 67 | M | 72.79 | 14.14 | 90 | 14.14 | 1 | 1 | 0 |
| 68 | M | 56.76 | 9.47 | 70 | 8.22 | 0 | 0 | 0 |
| 69 | F | 65.80 | 6.67 | 80 | 6.67 | 0 | 0 | 0 |
| 70 | M | 56.14 | 3.12 | 80 | 2.83 | 0 | 0 | 0 |
| 71 | M | 42.29 | 20.81 | 80 | 4.96 | 0 | 0 | 0 |
| 72 | F | 63.01 | 3.29 | 80 | 2.66 | 0 | 0 | 0 |
| 73 | F | 55.46 | 12.99 | 90 | 8.28 | 0 | 0 | 0 |
| 74 | M | 48.40 | 28.57 | 100 | 13.32 | 0 | 0 | 0 |
| 75 | M | 75.39 | 16.90 | 90 | 10.62 | 0 | 0 | 0 |
| 76 | F | 57.38 | 11.47 | 90 | 11.47 | 0 | 1 | 0 |
| 77 | F | 63.87 | 2.53 | 90 | 2.53 | 0 | 1 | 0 |
| 78 | M | 56.19 | 23.93 | 100 | 10.98 | 1 | 1 | 0 |
| 79 | M | 54.89 | alive | 100 | no relapse | 0 | 1 | 1 |
| 80 | M | 73.78 | 1.78 | 90 | 1.78 | 0 | 0 | 0 |
| 81 | F | 67.53 | 8.42 | 100 | 4.50 | 0 | 0 | 0 |
| 82 | M | 64.05 | 5.62 | 90 | 5.62 | 0 | 0 | 0 |
| 83 | M | 74.50 | 1.71 | 90 | 1.25 | 0 | 0 | 0 |
| 84 | M | 48.69 | 25.32 | 100 | 25.32 | 0 | 0 | 0 |
| 85 | M | 65.99 | 19.82 | 90 | 3.91 | 0 | 1 | 0 |
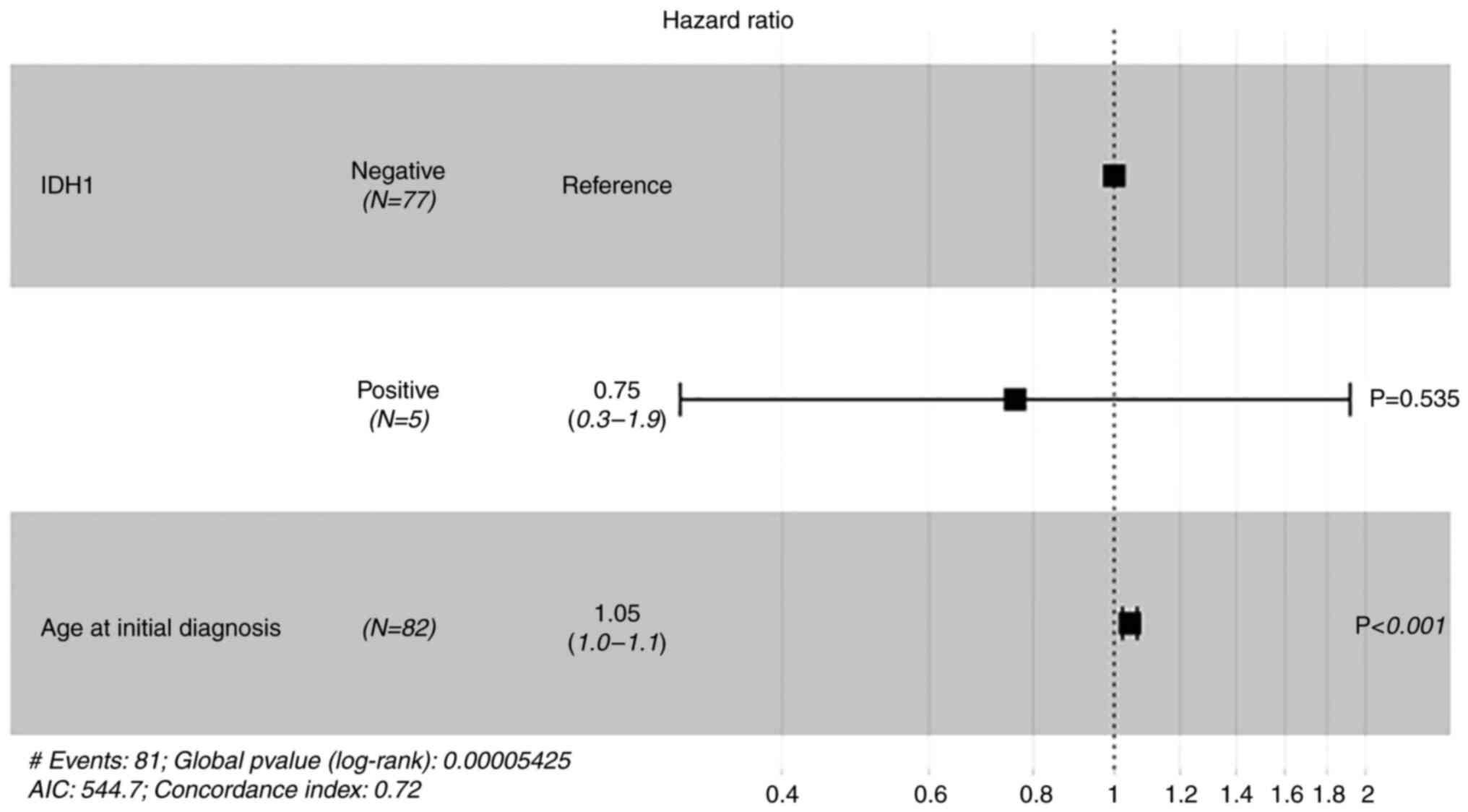
An R132H mutation of IDH1 (Figs. 4 and 5) was identified in five patients (6.10%).
A Cox regression analysis was performed with the variables age,
IDH1 status and overall survival (Fig.
6). This demonstrated that there was a significant (P<0.001)
association of low age at initial diagnosis and longer survival
however, no significant association with IDH1 was demonstrated.
With a case number of n=5 for IDH1 mutation, further statistical
analysis was considered to not be meaningful.
Clinical results
Kaplan-Meier analysis and Cox regression analysis
were performed to analyze the clinical parameters. When evaluating
the values using the Kaplan-Meier curve, the log-rank test was used
to determine the P-value. Numerous factors and their influence on
patient survival were assessed in terms of both PFS and OS. The
median OS in the study population was 15.7 months. The shortest OS
was 0.62 months and the longest, with the exception of the patient
who was still alive, was 125.29 months. The 95% confidence interval
was 15±3.83 months with a standard deviation of 18.73 months. The
median PFS was 9.0 months; the shortest PFS was 0.62 months, and
the longest PFS was 50.43 months. The 95% confidence interval was
9±2.13 months with a standard deviation of 10.12 months. The median
age of the patient population at initial diagnosis was 59.6 years
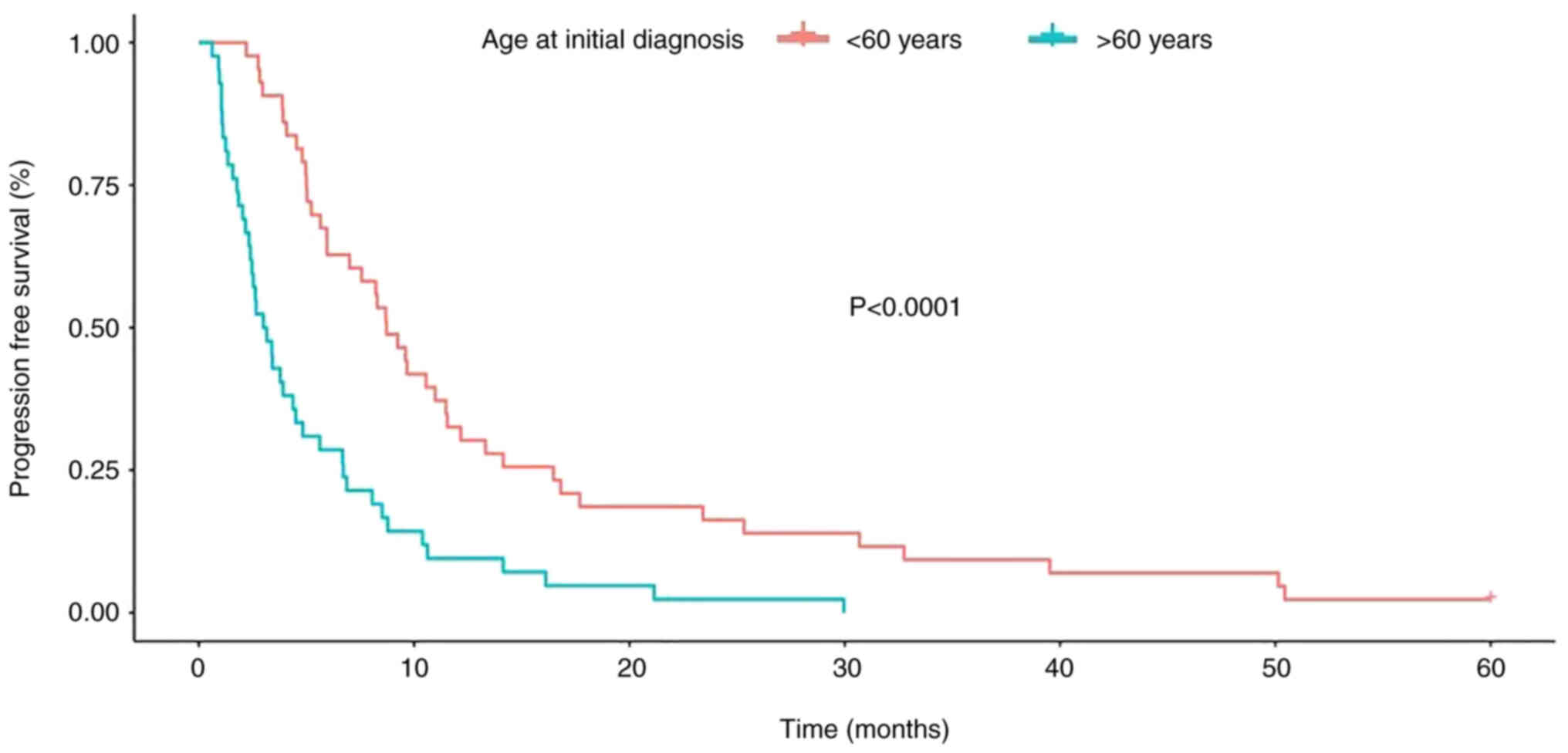
(range, 35–82 years). Kaplan-Meier analysis demonstrated a
significant reduction in both OS and PFS with an increased age
(>60) at initial diagnosis. With a calculated median age at
diagnosis of 59.6 years, a cutoff of 60 years of age was used to
compare the survival of patients >60 years (n=42) with those
<60 years (n=43). In both the analysis of OS and PFS, a worse
prognosis was demonstrated in the group of patients >60 years
(Figs. 7 and 8).
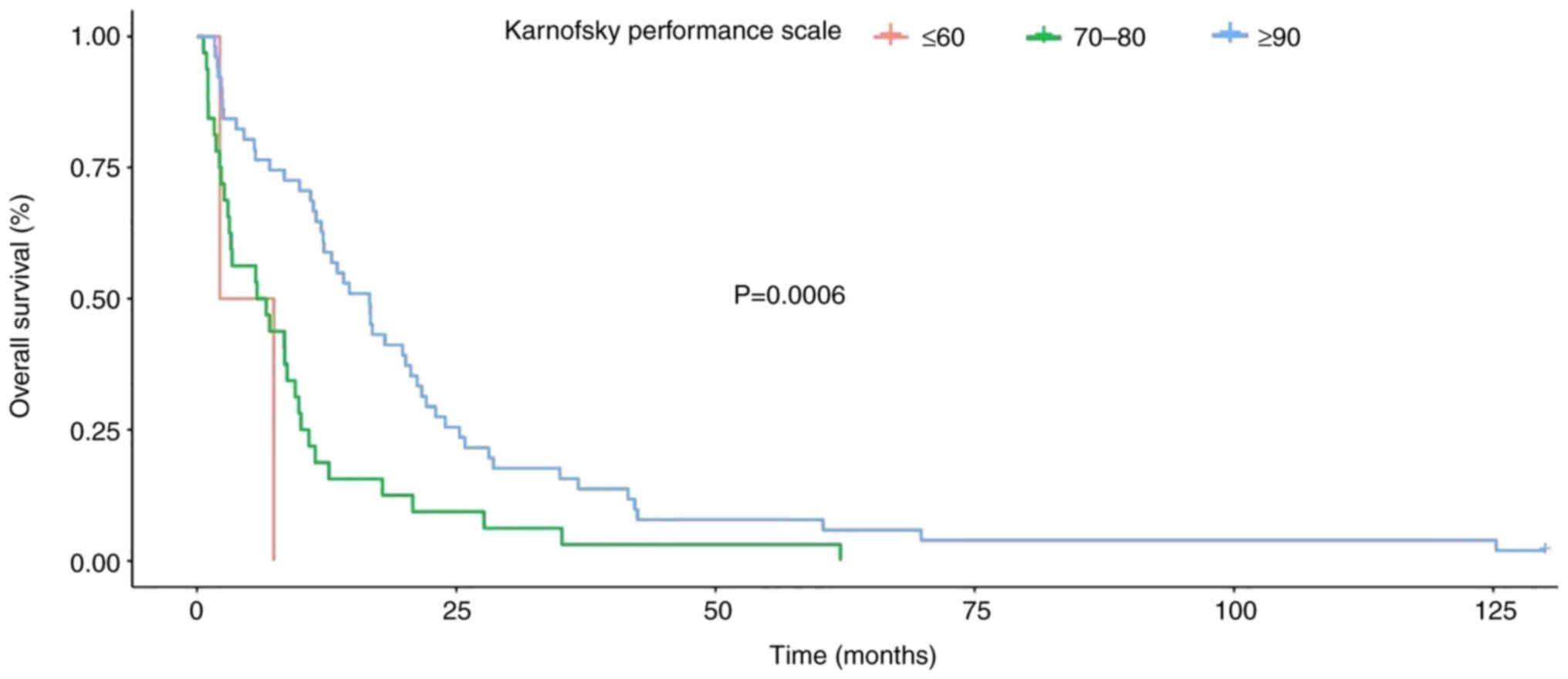
It was demonstrated that patients with a high
Karnofsky score at the time of initial diagnosis had a
significantly better OS rate and there was a significant
prolongation of both OS and PFS (Fig.
9).
Illustrative case
A statistical evaluation of RB1 methylation was not
reasonable with only a single case, therefore the case in which a
methylation of the RB1-promoter was detected is presented as a case
report.
Patient 79 was the only patient in the present study
whose tumor demonstrated RB1-promoter methylation. Patient 79, born
in 1958, was diagnosed in December 2012 following progressive
right-sided headache, occasional nausea and photosensitivity.
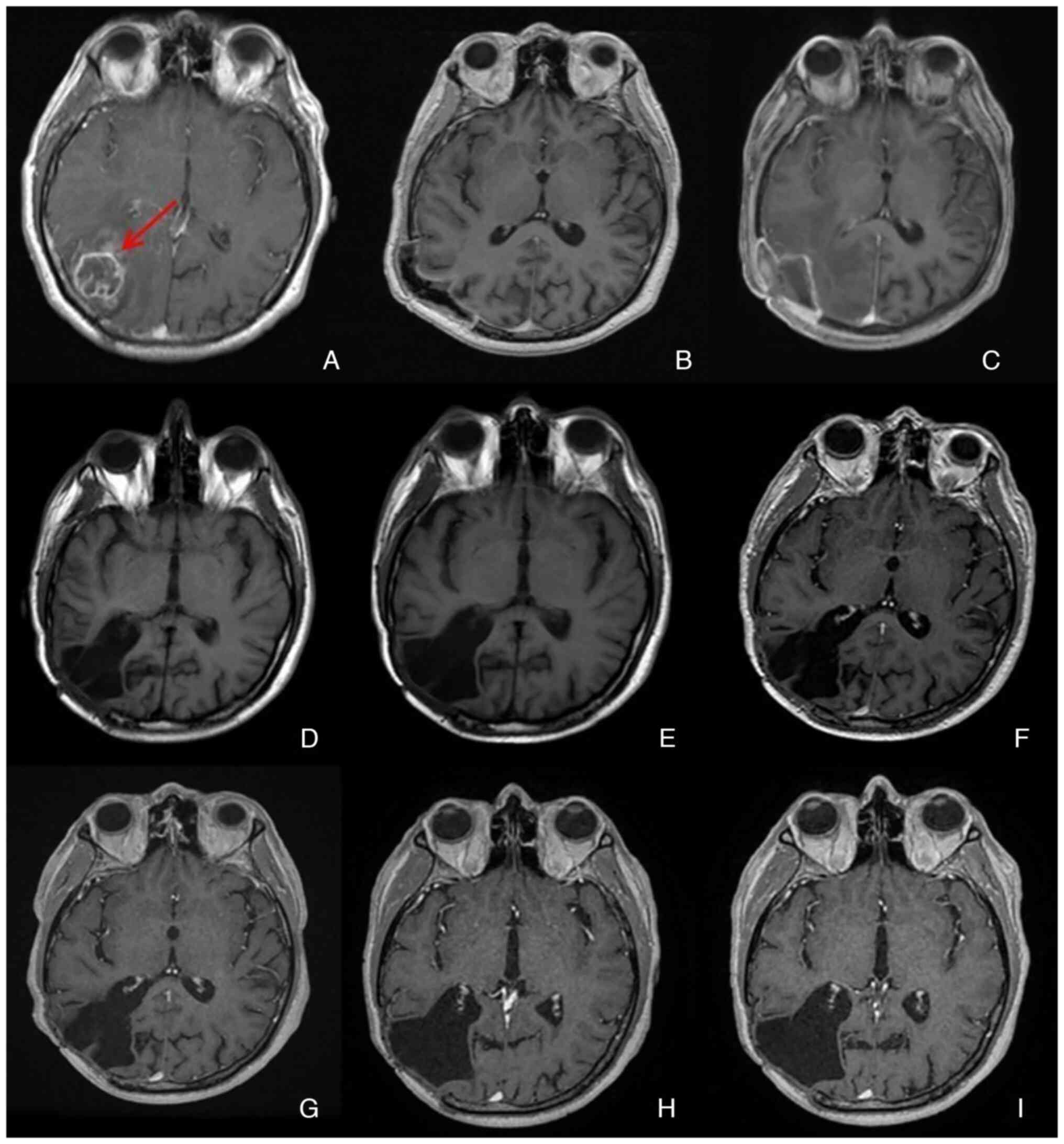
Radiological imaging was performed using computed tomography. A
parieto-occipital mass was identified and glioblastoma was
suspected. Subsequent magnetic resonance imaging revealed a
multi-cystic lesion with extensive perifocal edema extending to the
frontal and occipital regions. The mass showed a strong, marginal
enhancement after contrast application. There was also a midline
shift of approximately 7 mm, to the left. Locally, the outer
cerebrospinal fluid spaces were effaced. The patient did not have
any known history of tumor disease. A Karnofsky score of 100 was
calculated (21). Neurologic
examination demonstrated a slightly smaller right pupil compared
with the left; however, both pupils were indirectly and directly
light reactive. Assessment of the visual field showed a hemianopsia
on the left side, the remaining cranial nerve status was
unremarkable. There were no sensory motor deficits in the
extremities and, the intrinsic muscle reflexes were side-to-side
and unremarkable. Bladder and rectum function was intact. A right
occipital osteoclastic craniotomy and tumor resection was performed
using a microsurgical technique under neuronavigational guidance
including an insertion of Carmustine wafers into the tumor cavity.
The intraoperative course was completed without complications.
During which, glioblastoma was confirmed histopathologically.
Further pathologic diagnosis resulted in the detection of the cell
proliferation markers, Microtubule-Associated Protein 2,
Phophorylated Histone H and Glial Fibrillary Acidic Protein. The
Ki67 proliferation index was 60% (high index ≥20%). Methylation of
the MGMT-promoter was present. IDH-1 mutation status was negative.
Postoperative computed tomography scan of the head demonstrated
good tumor resection with no evidence of postoperative bleeding.
The patient received analgesia as needed and antithrombotic
prophylaxis. Postoperatively, the neurological status was
unchanged, with no evidence of new neurological deficits. Due to
the Carmustine wafer insertion into the tumor cavity, the patient
received anti-edematous therapy with 8 mg dexamethasone 4 times
daily for 4 weeks, as recommended (22). In January 2013, the bone flap became
infected and had to be removed. With cerebrospinal fluid leaking
due to a dura defect, an external ventricular drain had to be
implanted. In April 2013 an epidural abscess was detected and an
abscess removal had to be performed. Treatment was performed
according to the Stupp protocol (23) from February 2013 and was
discontinued in September 2013 after completion of the 6th cycle of
Temozolomide chemotherapy. Adjuvant radiotherapy of the tumor
region according to the protocol amounted to a total radiation dose
of 60 Gy with simultaneous chemotherapy with Temozolomide (75
mg/m2 body surface area). Subsequently, scheduled
follow-up examinations were performed at 3 month intervals.
Including the last follow-up in 12/2019, the clinical condition has
been stable since then. MRI scans performed for radiological
control also demonstrated stable findings without tumor remnant or
tumor recurrence (Fig. 10).
Discussion
Gliomas are characterized by global changes in DNA
methylation. They are usually triggered by IDH mutation and this
mutation can affect both DNA methylation and histone modifications
(24). Changes in DNA methylation
in gliomas have been previously reported to modify the binding
affinity of numerous transcription factors such as HOX and
CCCTC-binding factor (CTCF) (24).
IDH-1 methylations are most likely early events in the tumor
genesis and can lead to DNA methylation and therefore silencing of
genes, such as tumor suppressors, including Rb1 (25). Furthermore, the R132H-mutation in
IDH-1 can lead to increased formation of the D-2-Hydorxygultarate
(inhibitor) and decreased levels of α-ketoglutarate (cofactor for
histone and DNA demethylases) by inactivation of the isocitrate
dehydrogenase (26). In summary,
this is defined by three main steps. First, mutant IDH1 produces
2-Hydroxyglutatrate from α-ketoglutarate, then 2-Hydroxyglutatrate
inhibits histone and DNA demethylases, which leads to an increase
in methylation levels. Finally, methylation to CTCF in the DNA
inhibits CTCF binding and induces rearrangement of the
topologically associating domains (26).
The RB1 tumor suppressor gene has been implicated in
the pathogenesis of numerous neoplastic diseases such as
retinoblastoma and small cell lung cancers (27). However, to the best of our
knowledge, there has not previously been a conclusive statement
about the role of RB1 promoter methylation in Glioblastoma, due to
partly contradictory results. In the present study, a methylation
analysis of the RB1 promoter was performed using bisulfite
conversion, MS-PCR and gel electrophoresis on a total of 85 primary
glioblastoma samples from 85 patients. Methylation of the promoter
of RB1 was identified in only one patient, which corresponded to a
methylation rate of 1.18%.
Epigenetic modifications of the genetic material are
part of the genesis of most tumors and also serve an important role
in glioblastoma (28). RB1,
together with E2F, p53, CDK4/6, and CDKN2A, is part of a regulatory
circuitry that controls cell division, differentiation, senescence
and apoptosis (28,29). In the present study, methylation of
the RB1-promoter was demonstrated (1/85; 1.18%) which indicated
that RB1 promoter methylation did not appear to be a relevant event
in the development and progression of glioblastoma. Previous
studies reported partially divergent results in this regard. The
two studies with the largest numbers of cases of RB1 methylation in
glioblastoma, are by Nakamura et al (30) from 2001 (n=56) and Gonzalez-Gomez
et al (31) in 2003 (n=42).
Nakamura et al evaluated the methylation status of the RB1
promoter in 56 patients and identified a methylated promoter in a
total of 14 patients, which corresponded to 25% of the cases
examined, markedly higher than in the present study. They also
subdivided glioblastomas into primary and secondary according to
the older WHO classification (30)
and reported that promoter methylation was significantly more
common in secondary (43%) than primary (14%) glioblastomas
(30). A lower proportion of
detected RB1 methylation was consistent with the results of
Nakamura et al; however, the proportion in the present study
was markedly reduced at 1.18%. The WHO classification (2016) was
used in the present study, which no longer classifies glioblastomas
as primary or secondary. Rather, glioblastomas are classified
according to IDH-1 mutation status. According to the 2016 WHO
classification, all of the tumors included in the present study
were classified as glioblastomas, however it is possible that some
of the included samples in the study by Nakamura et al,
classified according to the 2000 WHO classification, would no
longer be classified as glioblastomas. Methodologically, Nakamura
et al used MS-PCR following bisulfite conversion, which was
performed using the CpGenome DNA Modification Kit (Intergen
Health). The primers used were the same as those used in the
present study. Gel electrophoresis was also performed as in the
present study with a 3% agarose gel using ethidium bromide for
visualization (30). It can be
summarized that, except for the use of a different kit, in general
the same methods were used as in the present study.
The second large, previous study, which focused on
RB1 promoter methylation in glioblastoma, was performed by
Gonzalez-Gomez et al in 2003 in the University Hospital of
La Paz in Madrid (31). The study
included, among other tumor entities of the central nervous system,
42 glioblastomas. RB1 promoter methylation was identified in 9 of
the 42 tumors, corresponding to a rate of 21%. As reported by
Nakamura et al, a higher methylation rate was demonstrated
in secondary glioblastomas compared with primary glioblastomas. The
study included 32 primary and 10 secondary glioblastomas, with 15%
being of the primary (5/32 tumors) and 40% of the secondary
glioblastomas (4/10 tumors) being methylated. Gonzalez-Gomez et
al hypothesized that the hyper methylation of the RB1 promoter
represented a common epigenetic event that was associated with the
development of brain tumors (31).
This contradicted the results of our study where RB1-methylation
was identified in only 1.18% of cases. It must be noted again that
in the present study a lower rate of RB1-methylated tumors was to
be expected because the present study exclusively investigated
primary glioblastomas (according to the previous WHO system),
however, the percentage of RB1-methylated tumors (1.18%) was
markedly lower than that reported by Gonzalez-Gomez et al
(15%). Similar to Nakamura et al, Gonzalez-Gomez et
al used the older version of the WHO classification with a
markedly lower initial number of cases. The methods used by
Gonzalez-Gomez et al were similar to those used in the
present study.
In another study in 2002, Yin et al evaluated
the p14, p15, p16 and RB1 genes in patients with brain tumors. In
this study, Yin et al also used MS-PCR following bisulfite
conversion, which was performed using the CpGenome DNA Modification
kit (Intergen Health). None of the 30 glioblastomas examined
demonstrated RB1-methylation (32).
These results were similar to those of the present study, although
Yin et al (32) investigated
a smaller number of cases (n=30). Due to the low incidence of
methylation of RB1 and also p15INK4B and p16INK4A gene (4 and 7%),
they concluded that the methylation of these genes served a minor
role in the development of brain tumors in general (32). Based on the results of the present
study for RB1 methylation in 85 patients, the present study
supports the hypothesis that methylation of the RB1 gene does not
seem to serve a major role in the development of glioblastoma.
Yu et al (33) also evaluated the methylation status
of genes involved in the development of astrocytomas. Among others,
12 Glioblastomas were included in the tumors evaluated. In addition
to p14ARF and p15INK4b, no methylation was detected in the RB1-gene
in any of the glioblastomas. As in the previous studies, the same
methods were used as in the present study (33). Another study dealing with the
promoter methylation of cell cycle control genes was reported by
Ohta et al in 2006 on malignant astrocytomas (34). The study group included 31
glioblastomas and 23 anaplastic astrocytomas. In addition to RB1,
p14ARF, p15INK4b, p16INK4a, p21Waf1/Cip1, p27Kip1 and p73 were also
assessed. Methylation of the RB1 promoter was identified in three
of the tumors, which corresponded to ~6% of the tumors examined;
however, no differentiation was made between glioblastomas and
anaplastic astrocytomas. While ~50% of the tumors showed an
abnormal promoter methylation pattern for at least one of the genes
examined, none of the tumors had three or more genes affected.
Interestingly, there was no significant association between the
methylation status of the genes with clinicopathological
parameters. The methylation status of the genes had no effect on
PFS or OS, either individually or in combination. Ohta et al
thus concluded that promoter methylation served only a minor role
in relation to the development of malignant brain tumors. This was
consistent with the findings of the present study with respect to
RB1.
Genes of the RB1 signaling pathway and their
expression in astrocytomas were the focus of a study performed by
Ferreira et al in 2015 (35). From 58 analyzed samples, 23 were
glioblastomas. Methylation of the RB1 promoter was not detected in
any of the samples. Compared with the present study and the other
aforementioned studies, the methodology was different as Ferreira
et al used bisulfite sequencing, DNA sequencing following
bisulfite conversion (35,36). However, despite a different method,
the present study demonstrated a result which was in agreement with
the findings of Ferreira et al (35). A recent study, which performed gene
sequencing of 63 IDH-wild type glioblastomas (classified as
glioblastoma grade IV tumors), reported that focal homozygous RB1
deletion was present in only seven cases (11.10%) (37).
In conclusion, the present study demonstrated that
methylation of the RB1 promoter was not a relevant event in the
development and progression of glioblastoma. This was because it
was a very rare event in glioblastoma patients, with only one of 85
patients showing a methylation of the RB1 promoter. The same
patient was also the only survivor from the patients included in
the present study. However, this can only be reported as a case
report and could not be assessed statistically.
Supplementary Material
Supporting Data
Acknowledgements
The authors wish to thank Professor Klaus-Dieter
Zang, previous Head of the Human genetic department for his
academic support. The authors wish to thank Sigrid Welsch, our
technician in the neurosurgery laboratory, for her technical
assistance. The authors wish to thank Dr Kettern Julia and Dr
Jürgens-Wemheuer Wiebke from the neuropathology department for the
MGMT gel image.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
The study was conceptualized by SU. SU and GB onfirm
the authenticity of all the raw data. The methodology was
established by CS. Statistical evaluation was performed by CS.
Acquisition and validation of clinical data was performed by RK.
Formal analysis of the data were performed by GB and WSS.
Methodical proceedings, GB. Resources, JO. Data curation was
performed by GB. The original draft of the manuscript was written
by MH and review and editing was performed by SU. Supervision was
provided by RK, JO and SU. Project administration was performed by
RK. All authors have read and agreed to the published version of
the manuscript.
Ethics approval and consent to
participate
The present study was performed according to the
guidelines of the Declaration of Helsinki and approved by the
Ethics Committee of the General Medical Council of the State
Saarland (approval no. 43/99). Informed consent for participation
was obtained from all subjects involved in the study.
Patient consent for publication
Informed consent for publication was obtained from
all subjects involved in the study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
WHO
|
World Health Organization
|
|
IDH
|
isocitrate dehydrogenase
|
|
DNMT1
|
DNA methyltransferase 1
|
|
5caC
|
5-carboxylcytosine
|
|
RB1
|
retinoblastoma gene
|
|
pRB
|
retinoblastoma protein
|
|
LOH
|
loss of heterozygosity
|
|
CDKN2A
|
cyclin dependent kinase inhibitor
2A
|
|
MS-PCR
|
methylation-specific PCR
|
|
MGMT
|
O-6-methylguanine-DNA methyl
transferase
|
|
PFS
|
progression-free survival
|
|
OS
|
overall survival
|
|
CTCF
|
CCCTC-binding factor
|
|
cMRI
|
cerebral Magnetic Resonance
Imaging
|
References
|
1
|
Sturm D, Witt H, Hovestadt V, Khuong-Quang
DA, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et
al: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic
and biological subgroups of glioblastoma. Cancer Cell. 22:425–437.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Strichman-Almashanu LZ, Lee RS, Onyango
PO, Perlman E, Flam F, Frieman MB and Feinberg AP: A genome-wide
screen for normally methylated human CpG islands that can identify
novel imprinted genes. Genome Res. 12:543–554. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gill RM, Hamel PA, Zhe J, Zacksenhaus E,
Gallie BL and Phillips RA: Characterization of the human RB1
promoter and of elements involved in transcriptional regulation.
Cell Growth Differ. 5:467–474. 1994.PubMed/NCBI
|
|
4
|
Wu H and Zhang Y: Reversing DNA
methylation: Mechanisms, genomics, and biological functions. Cell.
156:45–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sparkes RS, Murphree AL, Lingua RW,
Sparkes MC, Field LL, Funderburk SJ and Benedict WF: Gene for
hereditary retinoblastoma assigned to human chromosome 13 by
linkage to esterase D. Science. 219:971–973. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sellers WR and Kaelin WG Jr: Role of the
retinoblastoma protein in the pathogenesis of human cancer. J Clin
Oncol. 15:3301–3312. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Buchkovich K, Duffy LA and Harlow E: The
retinoblastoma protein is phosphorylated during specific phases of
the cell cycle. Cell. 58:1097–1105. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khani P, Nasri F, Khani Chamani F, Saeidi
F, Sadri Nahand J, Tabibkhooei A and Mirzaei H: Genetic and
epigenetic contribution to astrocytic gliomas pathogenesis. J
Neurochem. 148:188–203. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Henson JW, Schnitker BL, Correa KM, von
Deimling A, Fassbender F, Xu HJ, Benedict WF, Yandell DW and Louis
DN: The retinoblastoma gene is involved in malignant progression of
astrocytomas. Ann Neurol. 36:714–721. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruas M and Peters G: The p16INK4a/CDKN2A
tumor suppressor and its relatives. Biochim Biophys Acta.
1378:F115–F177. 1998.PubMed/NCBI
|
|
12
|
Ueki K, Ono Y, Henson JW, Efird JT, von
Deimling A and Louis DN: CDKN2/p16 or RB alterations occur in the
majority of glioblastomas and are inversely correlated. Cancer Res.
56:150–153. 1996.PubMed/NCBI
|
|
13
|
Ichimura K, Schmidt EE, Goike HM and
Collins VP: Human glioblastomas with no alterations of the CDKN2A
(p16INK4A, MTS1) and CDK4 genes have frequent mutations of the
retinoblastoma gene. Oncogene. 13:1065–1072. 1996.PubMed/NCBI
|
|
14
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Müllenbach R, Lagoda PJ and Welter C: An
efficient salt-chloroform extraction of DNA from blood and tissues.
Trends Genet. 5:3911989.PubMed/NCBI
|
|
16
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: A novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Tilborg AA, Morolli B, Giphart-Gassler
M, de Vries A, van Geenen DA, Lurkin I, Kros JM and Zwarthoff EC:
Lack of genetic and epigenetic changes in meningiomas without NF2
loss. J Pathol. 208:564–573. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Simpson DJ, Hibberts NA, McNicol AM,
Clayton RN and Farrell WE: Loss of pRb expression in pituitary
adenomas is associated with methylation of the RB1 CpG island.
Cancer Res. 60:1211–1216. 2000.PubMed/NCBI
|
|
19
|
Esteller M, Garcia-Foncillas J, Andion E,
Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB and Herman JG:
Inactivation of the DNA-repair gene MGMT and the clinical response
of gliomas to alkylating agents. N Engl J Med. 343:1350–1354. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karnofsky Performance Status-an overview |
ScienceDirect Topics [Internet]. [zitiert 8. August 2022].
Available from. https://www.sciencedirect.com/topics/nursing–and–
health–professions/karnofsky–performance-status
|
|
22
|
Schiff D, Lee EQ, Nayak L, Norden AD,
Reardon DA and Wen PY: Medical management of brain tumors and the
sequelae of treatment. Neuro Oncol. 17:488–504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
J Dabrowski M and Wojtas B: Global DNA
methylation patterns in human gliomas and their interplay with
other epigenetic modifications. Int J Mol Sci. 20:34782019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taylor KH, Kramer RS, Davis JW, Guo J,
Duff DJ, Xu D, Caldwell CW and Shi H: Ultradeep bisulfite
sequencing analysis of DNA methylation patterns in multiple gene
promoters by 454 sequencing. Cancer Res. 67:8511–8518. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Raineri S and Mellor J: IDH1: Linking
metabolism and epigenetics. Front Genet. 9:4932018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chinnam M and Goodrich DW: RB1,
development, and cancer. Curr Top Dev Biol. 94:129–169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Indovina P, Pentimalli F, Casini N, Vocca
I and Giordano A: RB1 dual role in proliferation and apoptosis:
Cell fate control and implications for cancer therapy. Oncotarget.
6:17873–17890. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aubrey BJ, Kelly GL, Janic A, Herold MJ
and Strasser A: How does p53 induce apoptosis and how does this
relate to p53-mediated tumour suppression? Cell Death Differ.
25:104–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakamura M, Yonekawa Y, Kleihues P and
Ohgaki H: Promoter hypermethylation of the RB1 gene in
glioblastomas. Lab Invest. 81:77–82. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gonzalez-Gomez P, Bello MJ, Alonso ME,
Arjona D, Lomas J, de Campos JM, Isla A and Rey JA: CpG island
methylation status and mutation analysis of the RB1 gene essential
promoter region and protein-binding pocket domain in nervous system
tumours. Br J Cancer. 88:109–114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin D, Xie D, Hofmann WK, Miller CW, Black
KL and Koeffler HP: Methylation, expression, and mutation analysis
of the cell cycle control genes in human brain tumors. Oncogene.
21:8372–8378. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu J, Zhang H, Gu J, Lin S, Li J, Lu W,
Wang Y and Zhu J: Methylation profiles of thirty four promoter-CpG
islands and concordant methylation behaviours of sixteen genes that
may contribute to carcinogenesis of astrocytoma. BMC Cancer.
4:652004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ohta T, Watanabe T, Katayama Y, Yoshino A,
Yachi K, Ogino A, Komine C and Fukushima T: Aberrant promoter
hypermethylation profile of cell cycle regulatory genes in
malignant astrocytomas. Oncol Rep. 16:957–963. 2006.PubMed/NCBI
|
|
35
|
Ferreira WAS, Araújo MD, Anselmo NP, de
Oliveira EHC, Brito JRN, Burbano RR, Harada ML and Borges Bdo N:
Expression analysis of genes involved in the RB/E2F pathway in
astrocytic tumors. PLoS One. 10:e01372592015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y and Tollefsbol TO: DNA methylation
detection: Bisulfite genomic sequencing analysis. Methods Mol Biol.
791:11–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Suwala AK, Stichel D, Schrimpf D, Maas
SLN, Sill M, Dohmen H, Banan R, Reinhardt A, Sievers P, Hinz F, et
al: Glioblastomas with primitive neuronal component harbor a
distinct methylation and copy-number profile with inactivation of
TP53, PTEN, and RB1. Acta Neuropathol. 142:179–189. 2021.
View Article : Google Scholar : PubMed/NCBI
|