Introduction
Intellectual disability (ID), defined as the
significant impairment of cognitive and adaptive behavior, affects
1.5–2% of the human population (1),
and therefore receives considerable attention from scientists
working in the fields of neuroscience and genetics. Studies have
shown that 10–15% of inherited severe ID is the result of genetic
aberrations on chromosome X, a condition termed X-Linked ID (XLID)
(2). XLID is subdivided into two major
categories, syndromic and non-syndromic, based on the presence or
absence of specific accompanying clinical, morphological,
neurological or metabolic abnormalities. The borders between
non-syndromic and syndromic are not always clearly defined,
particularly in small families where few individuals are affected.
Small families or individual cases are therefore usually
categorized as non-syndromic, contributing to the heterogeneity of
this category.
Through the intensive efforts of various research
groups and the utilization of powerful new genomic technologies,
>100 XLID genes have been reported so far, although it is
estimated that a substantial fraction of XLID genes remain to be
identified (3,4). The recently developed whole-exome
sequencing (WES) technology allows the rapid detection of variants
located within protein-coding regions of the genome that could not
be detected by technologies such as karyotyping and
array-comparative genomic hybridization (array-CGH). In the present
study, WES was used to screen two brothers with suspected XLID. The
analysis identified a novel variant in these patients within
HCFC1, a gene recently implicated in XLID.
Materials and methods
Clinical evaluation of patients
Patient 1 was the first of two male siblings born to
healthy non-consanguineous Greek-Cypriot parents and was initially
referred to the Clinical Genetics service (The Cyprus Institute of
Neurology and Genetics, Nicosia, Cyprus) at the age of 5 years and
2 months for evaluation regarding a history of developmental and
speech delay. The patient was born at 39 weeks gestation following
a normal vaginal delivery with a birth weight on the 9th
percentile, a birth length on the 25–50th percentile, and a birth
occipitofrontal circumference (ofc) on the 0.4th percentile. There
were no antenatal concerns, no history of a maternal intercurrent
infection or known exposure to teratogens. Developmentally the
patient sat without support at the age of 6 months and walked
independently at the age of 13 months. The patient presented with
moderate developmental motor and speech delay, as well as with
severe learning difficulties. Other characteristic findings at the
age of clinical evaluation were microcephaly (ofc <3rd
percentile) and hyperactivity. The weight and height of the patient
were between the 25–50th percentile. The patient had a beaked nose
with relatively large ears and synophrys. The hands and feet were
normal, including the palmar and plantar creases.
Patient 2 (the male sibling of patient 1) was
referred to the Department of Clinical Genetics at the age of 14
months for evaluation regarding developmental delay. The patient
was born prematurely at 35 weeks gestation following an emergency
C-section due to fetal distress and prolonged rupture of membranes
with a birth weight on the 25th percentile. At the age of 14
months, the patient was reported to be microcephalic and could only
walk with support. On examination at the age of 5 years, the
patient had severe developmental and speech delay. The patient was
microcephalic (ofc <3th percentile) with myopathic facies,
arched eyebrows, an open-mouthed appearance with a Cupid's bow
configuration of the upper lip and prognathism. The eyes of the
patient were prominent. The patient had a small and narrow alae
nasi, triangular nails, bilateral 2–3 toe syndactyly and bilateral
pes planus.
The endocrinology and ophthalmology evaluations
yielded normal results. Extensive metabolic investigations did not
reveal any abnormalities. In both siblings, karyotype, fragile X
syndrome, and high-resolution array-CGH analyses showed normal
results.
WES and variant analysis
DNA was extracted from the examined individuals
using the Qiagen DNA extraction kit (Qiagen, Hilden, Germany)
according to the manufacturer's protocol. WES was performed on DNA
samples from the affected male siblings as a service by Oxford Gene
Technology (Kidlington, UK). Exome enrichment was performed using
the SureSelect exome enrichment kit v1.2 (Agilent Technologies,
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and the
concentration of each library was determined using the Agilent qPCR
NGS Library Quantification kit. Prior to sequencing, the samples
were pooled to a concentration of 10 nM per sample. Sequencing of
the enriched exome was performed on the Illumina HiSeq 2000 using
TruSeq v3 chemistry (Illumina, Inc., San Diego, CA, USA). For the
first patient there were 25, 241 and 930 paired reads, 67.48% of
bases were on target, and 92.69% of target bases were covered at
>20×. For the second patient there were 24, 926 and 649 paired
reads, 67.39% of bases were on target, and 92.60% of bases were
covered at >20×.
For the bionformatics analysis of the WES data the
reads were mapped to genome assembly hg19 using the Burrows-Wheeler
Aligner package, version 0.6.2 (5).
Local realignment of the mapped reads around potential
insertion/deletion (indel) sites was carried out with the Genome
Analysis Tool kit (GATK) version 1.6 (6). Duplicate reads were marked using Picard
version 1.83 (http://picard.sourceforge.net/). Base quality (Phred
scale) scores were recalibrated using the covariance recalibration
of GATK. SNP and indel variants were named using the GATK Unified
Genotyper for each sample. SNP novelty was determined against dbSNP
release 135 in February 2013 (7).
Further screening of variants was performed using the exome variant
server (EVS) database in May 2014 (http://evs.gs.washington.edu/EVS/). Pathogenicity
prediction for identified variants was performed using the Polyphen
(http://genetics.bwh.harvard.edu/pph/data/), Sift
(http://sift.jcvi.org/), and Condel (http://bg.upf.edu/fannsdb/) tools.
Sanger sequencing
Purification of polymerase chain reaction products
was performed using Exo-SapIT (Affymetrix, Santa Clara,
CA, USA). Sequencing reactions were performed using Applied
Biosystems BigDye terminator v1.1 and products were analyzed on the
ABI 3130 sequencer. The Primer sequences are available on
request.
Results
Variations
A total of 46,134 variations were observed for
patient 1 and 45,711 variations for patient 2. Of these, there were
45,784 overlapped genes for patient 1 and 45,369 for patient 2. For
variants not present in dbSNP 135 there were 3,462 for patient 1
and 3,483 for patient 2. Of these, 675 and 682, respectively, were
predicted to have serious consequences (missense, frameshift,
indel, essential splice-site, nonsense and nonstop) respectively.
Based on the suspected XLID inheritance of the disease, we
prioritized variants detected in the WES data that were: i) Located
on the X chromosome, ii) found in both affected patients, iii) rare
variants not present in the dsSNP database, and iv) predicted to
have serious phenotypic consequences by in silico tools (Sift,
Polyphen or Condel).
Only two variants fulfilled the criteria outlined
above: A non-synonymous variant in the HCFC1 gene
(ChrX:153,221,808 GRCh37; p.Ala897Val) and a non-synonymous variant
in the zinc finger protein, X-linked (ZFX) gene
(ChrX:24,197,706). Further screening of these two variants in the
EVS database found that the HCFC1 variant was not present in
any of the screened individuals, while the ZFX variant was
present in 8 out of 10,555 chromosomes. Additionally, mutations in
ZFX had not been previously associated with ID.
Consequently, the ZFX variant was less likely to be the
cause of the ID in these patients.
Sanger sequencing
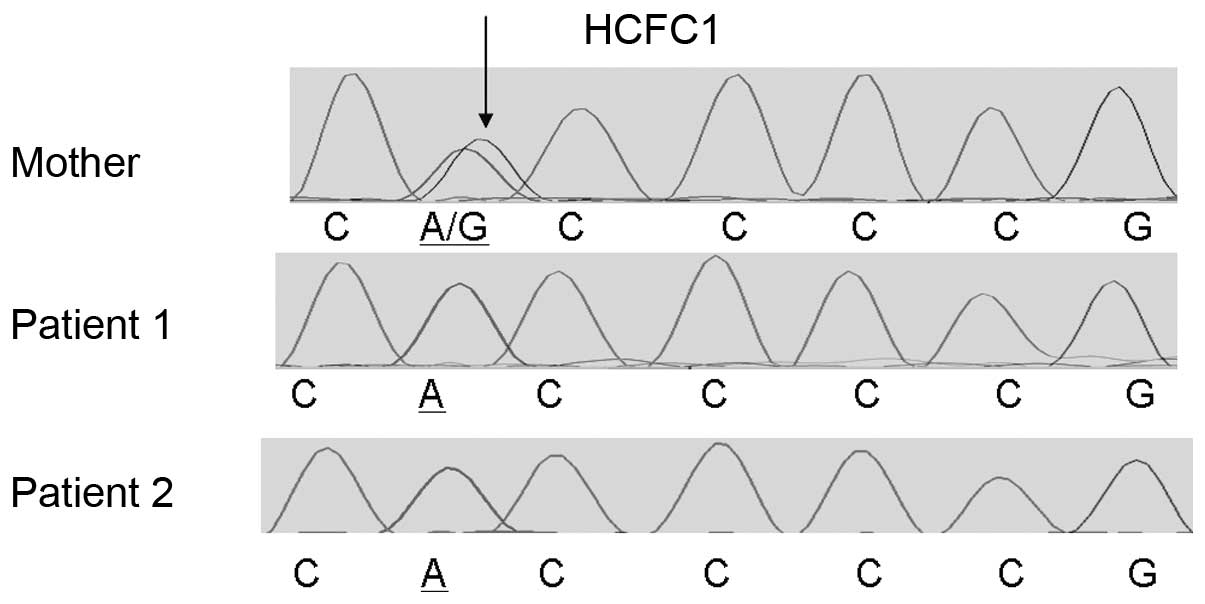
The identified HCFC1 variant was present in the two
male siblings as confirmed by Sanger sequencing using custom
primers and the Big Dye terminator v1.1, while their mother was a
heterozygous carrier (Fig. 1).
Additionally, Sanger sequencing of the HCFC1 variant showed that
this was not present in 100 neurotypical individuals from the
Cypriot population. This rare variant affects an evolutionary
conserved amino acid (Fig. 2),
supporting its functional importance.
Discussion
HCFC1 encodes a large protein of >2,000
amino acids made up of kelch repeats, HCF-proteolysis repeats,
protein interacting domains, basic domain, acidic region, and
fibronectin domains (Fig. 3). In
recent years studies have reported an association of mutations
affecting HCFC1 with ID, metabolic disorders and dysmorphias.
Mutations affecting conserved amino acids within the HCFC1 kelch
domains have been associated with defective cobalamin metabolism
and neurological symptoms by affecting transcription of
MMAHC (8). Gérard et al
(9) recently reported a p.Tyr103His
mutation in two male siblings with ID and cobalamin disorder. These
male siblings also exhibited dysmorphic features and microcephaly.
Experimental evidence from the examination of patient-derived cell
lines and in vitro assays suggest that mutations within
kelch domains result in almost complete loss of HCFC1 function and
result in cobalamin disorder, while HCFC1 mutations in other
regions result in much milder effects on protein function and
cobalamin metabolism (8,10). Loss-of-function of the zebrafish
homolog of HCFC1 has also been associated with craniofacial
abnormalities through dysregulation of MMAHC (11). It has been suggested that mutations in
non-kelch domains of HCFC1, which result in partial
loss-of-function, cause neurological disorders in the absence of
cobalamin deficiency due to HCFC1 having other gene targets that
are important for neurological development (8,10).
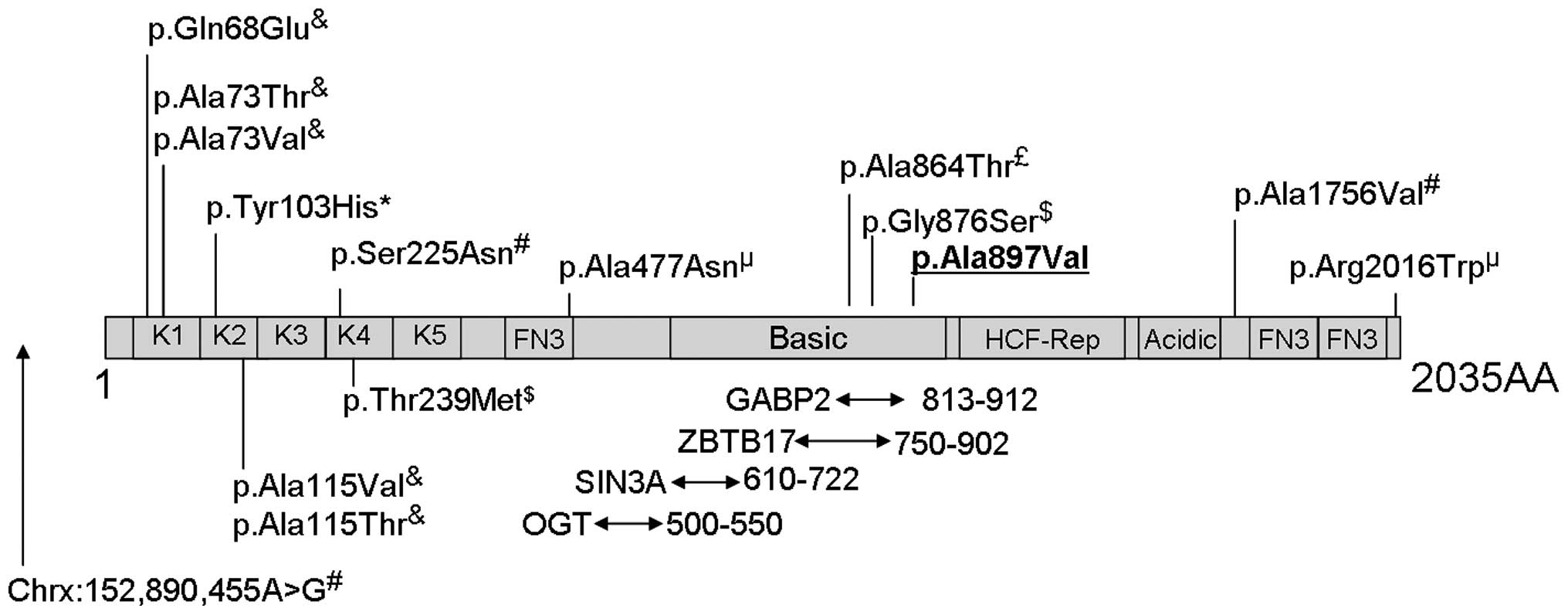 | Figure 3.Schematic representation of the host
cell factor C1 (HCFC1) protein and its domains based on information
from Uniprot (www.uniprot.org; entry P51610) and
from published studies. The diagram is not drawn to scale. Kelch
domains (K1-K5), fibronectin-type 3 (FN3), basic domain,
HCF-proteolysis repeats (HCF-Rep) and acidic domains are shown.
Also shown are the protein interaction regions and variants
reported in the literature to be associated with intellectual
disabilities. Areas of interaction between HCFC1 and the SIN3A,
ZBTB17, OGT and GABP2 proteins are indicated by arrows underneath
the diagram, with the numbers next to the arrows reporting the
location of the interacting domains. The p.Ala897Val variant
identified in the present study is underlined in bold. Superscripts
above the variants in the diagram indicate the reference reporting
this variant: £(3),&(8), *(9),
µ(10),
$(13),
#(14). |
Examination of patient 1 revealed normal levels of
cobalamin, homocysteine and folate in the blood [436 pg/ml
cobalamin (normal range, 208–964 pg/ml), 9.1 mmol/l homocysteine
(normal range, 5–15 mmol/l) and 5.4 ng/ml folate (normal range,
3–15 ng/ml)]. Consequently, no supporting evidence that the
pathogenic phenotype observed in this case is associated with
defective cobalamin metabolism was identified.
The variant identified in the present study
(p.Ala897Val) is located within the GA-binding protein (GABP)
interaction domain. Interaction of GABP with HCFC1 with
transcription factors, including GABP, is believed to be essential
for the ability of HCFC1 to regulate the transcription of a large
number of genes (12). Within the same
region, two variants have been identified in an autistic patient
(p.Gly876Ser) (13) and in a patient
with ID (p.Ala864Thr) (3). The
findings of the present study are consistent with mutations within
this region of HCFC1 being associated with ID or ASD and with
dysmorphic features, without clinical manifestation of cobalamin
deficiency.
References
|
1
|
Leonard H and Wen X: The epidemiology of
mental retardation: Challenges and opportunities in the new
millennium. Ment Retard Dev Disabil Res Rev. 8:117–134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De Brouwer AP, Yntema HG, Kleefstra T,
Lugtenberg D, Oudakker AR, de Vries BB, van Bokhoven H, Van Esch H,
Frints SG, Froyen G, et al: Mutation frequencies of X-linked mental
retardation genes in families from the EuroMRX consortium. Hum
Mutat. 28:207–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tarpey PS, Smith R, Pleasance E, Whibley
A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, et
al: A systematic, large-scale resequencing screen of X-chromosome
coding exons in mental retardation. Nat Genet. 41:535–543. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bassani S, Zapata J, Gerosa L, Moretto E,
Murru L and Passafaro M: The neurobiology of X-linked intellectual
disability. Neuroscientist. 19:541–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler Transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wheeler DL, Barrett T, Benson DA, et al:
Database resources of the National Center for Biotechnology
Information. Nucleic Acids Res. 35(Database issue): D5–D12. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu HC, Sloan JL, Scharer G, Brebner A,
Quintana AM, Achilly NP, Manoli I, Coughlin CR II, Geiger EA,
Schneck U, et al: An X-linked cobalamin disorder caused by
mutations in transcriptional coregulator HCFC1. Am J Hum Genet.
93:506–514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gérard M, Morin G, Bourillon A, Colson C,
Mathieu S, Rabier D, de Villemeur Billette T, de Baulny Ogier H and
Benoist JF: Multiple congenital anomalies in two boys with mutation
in HCFC1 and cobalamin disorder. Eur J Med Genet. 58:148–153. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jolly LA, Nguyen LS, Domingo D, Sun Y,
Barry S, Hancarova M, Plevova P, Vlckova M, Havlovicova M,
Kalscheuer VM, et al: HCFC1 loss-of-function mutations disrupt
neuronal and neural progenitor cells of the developing brain. Hum
Mol Genet. 24:3335–3347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quintana AM, Geiger EA, Achilly N,
Rosenblatt DS, Maclean KN, Stabler SP, Artinger KB, Appel B and
Shaikh TH: Hcfc1b, a zebrafish ortholog of HCFC1, regulates
craniofacial development by modulating mmachc expression. Dev Biol.
396:94–106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Michaud J, Praz V, Faresse James N,
Jnbaptiste CK, Tyagi S, Schütz F and Herr W: HCFC1 is a common
component of active human CpG-island promoters and coincides with
ZNF143, THAP11, YY1 and GABP transcription factor occupancy. Genome
Res. 23:907–916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Piton A, Gauthier J, Hamdan FF, Lafrenière
RG, Yang Y, Henrion E, Laurent S, Noreau A, Thibodeau P, Karemera
L, et al: Systematic resequencing of X-chromosome synaptic genes in
autism spectrum disorder and schizophrenia. Mol Psychiatry.
16:867–880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang L, Jolly LA, Willis-Owen S, Gardner
A, Kumar R, Douglas E, Shoubridge C, Wieczorek D, Tzschach A, Cohen
M, Hackett A, Field M, Froyen G, Hu H, Haas SA, Ropers HH,
Kalscheuer VM, Corbett MA and Gecz J: A noncoding, regulatory
mutation implicates HCFC1 in nonsyndromic intellectual disability.
Am J Hum Genet. 91:694–702. 2012. View Article : Google Scholar : PubMed/NCBI
|