Introduction
Liver fibrosis is a complex pathophysiological
process of numerous chronic liver diseases, which are characterized
by the deposition of the extracellular matrix (ECM). Hepatic
stellate cells (HSCs), fibroblasts and myofibroblasts participate
in the process via different mechanisms. The inevitable consequence
of sustainable liver fibrosis is liver cirrhosis and hepatic
cancer, and therefore, preventing liver fibrosis is the primary
measure. At present, studies focus on the mechanisms that
potentially delay the process of liver fibrosis and even reverse
it. Accumulating evidence has shown that mesenchymal cells have an
important role in hepatic fibrogenesis. The epithelial-mesenchymal
transition (EMT) is suggested as one of the important origins of
mesenchymal cells.
During the EMT, epithelial cells lose their
epithelial characteristics and gradually obtain a mesenchymal
phenotype. The source of the mesenchymal cells participating in
tissue repair and regeneration remains to be elucidated. Bi et
al (1) reported that alendronate
sodium significantly arrested the progression of liver fibrosis.
Deng et al (2) observed that
biliary epithelial cells (BECs) undergoing EMT may contribute to
fibrogenesis in biliary atresia by detecting the antigen for
cytokeratin-7 (CK-7), heat-shock protein 1 (HSP1), HSP47 and
α-smooth muscle actin (α-SMA) in liver sections from patients with
biliary atresia. This progress involves the switch of cadherin from
E-cadherin to N-cadherin, the dissolution of intracellular
connections, the upregulation of matrix remodeling factors and the
rearrangement of the cellular cytoskeleton.
Different cell types in liver fibrosis
Liver fibrogenic cells
Liver fibrogenic cells are a heterogenous cell
group, which includes the α-SMA+ myofibroblasts (MFs).
Liver fibrogenic cells may have a major role in liver fibrosis
according to recent studies, and the origin of these cells remains
to be elucidated. HSCs are considered the major source as they are
the main ECM-producing cells in the injured liver. Hepatic MFs may
also originate from bone marrow-derived mesenchymal cells and cells
from EMT and endothelial-mesenchymal transition (EnMT).
HSCs
Activation of HSCs is a central event in liver
fibrosis. Recently, a number of studies have demonstrated that HSCs
are derived from mesodermal-derived multipotent mesenchymal
progenitor cells. HSCs are significant in producing the ECM,
particularly collagen type 1, which is regulated by complex stimuli
and pathways. Transforming growth factor-β (TGF-β) is prominent
among these stimuli. TGF-β has 3 major isoforms: TGF-β1, TGF-β2 and
TGF-β3. Generally, TGF-β1 is stored in an inactivated state, and
once activated, it will enhance the transcription of the target
gene via its receptors to Smad proteins. As it responds to Smad,
the further matrix production in HSCs differs between acute and
chronic injury (3). In addition to
TGF-β, there are numerous other factors that exhibit profibrogenic
effects on HSCs, such as retinoids and angiotensin II (4–6). During
liver fibrosis, parenchymal injury and sustained inflammation
generate a large panel of signals that induce the activation of
quiescent HSCs. HSC activation is associated with the activation of
nuclear factor κB (NF-κB) and activator protein 1, which are
activated following the stimulation of intracellular signaling
cascades. Platelet-derived growth factor has been shown to activate
mitogen-activated protein kinase (MAPK) signaling, specifically
c-Jun N-terminal protein kinase, extracellular signal-regulated
kinase (ERK) and p38, and finally, regulate HSCs proliferation.
Following the activation of HSCs, a variety of changes in gene
transcription occur. The target genes include, but are not limited
to, the following: α-SMA, type 1 collagen, MMP-2, TGF-β1, TGF-β
receptors, TIMPs 1 and 2 (7,8). Persistent activation leads to changes in
HSC behavior, such as proliferation, chemotaxis, fibrogenesis and
cytokine release, and all these changes are discrete (9). Liver MFs originating from activated HSCs
exhibit high proliferative capacity, upregulate the expression of
typical mesenchymal cell markers, such as α-SMA, type 1 collagen,
fibronectin, desmin and vimentin intermediate filaments (10).
Portal fibroblasts (PFs)
The PFs are spindle shaped and exhibit biological
similarities with activated HSCs; however, they have different
genetic profiles and signaling responses (11,12). They
are of mesenchymal origins that undergo myofibroblastic
differentiation. PFs do not express α-SMA, glial fibrillary acidic
protein filaments and desmin, cluster of differentiation 146
(CD146) and cellular retinol-binding protein-1 proteins (13,14), nor
store retinoids, which is different from HSCs. In response to
tissue injury in liver fibrosis, PFs undergo myofibroblastic
activation. Proliferation of the MFs originated from PFs primarily
occurs in disease associated with ductular reaction and/or
cholestasis, in which the initial injury site is the portal area
(15,16).
Fibrocytes
Fibrocytes originated from hematopoietic stem cells
are capable to differentiate into MFs. Once tissue is damaged,
fibrocytes are recruited to the injured organ and secrete growth
factors. The migration of fibrocytes is regulated by C-C chemokine
receptor type 2 (CCR2) and CCR1. Studies have shown that the extent
of differentiation into MFs depends on different organs and the
type of injury (17,18).
Bone marrow-derived MFs
Certain hepatic MFs can also originate from the bone
marrow-derived mesenchymal stem cells (MSCs), which most likely
represent a population that is different from hematopoietic-derived
fibrocytes (9,19,20).
Other cells
Studies have shown that MFs may also be derived from
hepatocytes or cholangiocytes through EMT in the liver (21). Zeisberg et al (22) were the first to report the evidence for
hepatocyte EMT in vivo. They demonstrated that in the
transgenic mice challenged with CCL4, in which hepatocyte-derived
cells are permanently labeled by β-galactosidase (β-gal), 45% of
the cells expressing the fibroblast-specific protein 1 (FSP1) were
also positive for β-gal expression. Furthermore, the CCL4-induced
liver fibrosis can be limited by the inhibition of the TGF-β1
pathway. As a summary, the results demonstrated that hepatocyte EMT
was triggered by TGF-β1 and had a role in liver fibrosis.
Cholangiocytes symbolize a unique epithelial cell compartment in
the diseased liver. The biliary epithelial cells cannot be ruled
out of the assumption that liver epithelial cells undergo EMT in
liver fibrosis. Upon liver injury, cholangiocytes proliferate and
switch from a quiescent to a ‘reactive’ state. Reactive
cholangiocytes are known to express a variety of cytokines and
pro-fibrogenic growth factors. They are likely to contribute to
fibrosis and inflammation by promoting activation, proliferation
and collagen synthesis in the surrounding pro-fibrogenic cells
(23,24). However, Omenetti et al (25) showed a complete EMT in an immature
cholangiocyte cell line in vitro, suggesting the possibility
of direct contribution of cholangiocytes to fibrosis via EMT. In
biliary atresia, biliary epithelial cells expressed FSP-1 and
vimentin, while hepatocytes did not. Furthermore, the study showed
that the expression of mesenchymal markers in biliary epithelial
cells was observed in all liver disease with a ductular
proliferation component. In mice exposed to common bile duct
ligation (BDL), which is an experimental liver fibrosis model that
induces strong ductular reaction, biliary epithelial cells
underwent EMT, as shown by type I collagen and α-SMA expression
(26).
Basic concept of EMT
EMT allows the epithelial cells to lose their
polarity, to undergo complex biochemical changes and to assume
multiple mesenchymal cell phenotypes, which includes a
significantly increased production of ECM components, migratory
capacity, invasiveness and elevated resistance to apoptosis. The
progress was first described by Hay in 1995 in a chick model of
primitive streak formation (27). In
2003, it was agreed at the first meeting of The EMT International
Association, that epithelial-mesenchymal transformation and
epithelial-mesenchymal transdifferentiation would be termed EMT. In
March 2008, EMT was classified into three different subtypes at an
EMT meeting at Cold Spring Laboratory based on the different
biological contexts in which they occur (28,29). i) The
type 1 EMTs are associated with implantation, embryo formation and
organ development, neither cause organ fibrosis nor induce invasive
phenotype. ii) The type 2 EMTs, in contrast to type 1, are
connected to wound healing, tissue regeneration and organ fibrosis,
and involve secondary epithelial or endothelial cells transitioning
to resident tissue fibroblasts. As is observed during wound healing
and tissue regeneration, the type 2 EMTs are positively correlated
with inflammation and cease once inflammation is attenuated. iii)
The type 3 EMTs are part of the metastatic process, and occur in
neoplastic cells that have previously undergone genetic and
epigenetic changes.
Type 2 EMTs
Type 2 EMTs are associated with organ fibrosis and
regeneration occurring in the liver, lung, kidney and intestine.
FSP1, α-SMA and collagen 1 are the characterized markers of the
mesenchymal products generated by the EMTs during the development
of organ fibrosis (29–31). The aforementioned markers, along with
vimentin, desmin and discoidin domain receptor 2 (DDR2), have been
used to distinguish the epithelial cells that are undergoing EMTs
in response to ongoing inflammation. With the development of EMTs,
these cells continue to exhibit epithelial-specific morphology and
molecular markers, such as E-cadherin and cytokeratin, but showed
concomitant expression of FSP1 and α-SMA. When the epithelial cell
markers continue to be expressed, but the mesenchymal cells markers
have been already obtained, such cells possibly represent the
intermediate stage of EMT, or namely a partial EMT. Eventually
these cells ultimately shed all their epithelial markers (including
E-cadherin and zonula occludens-1) and acquire a fully fibroblastic
phenotype (31) (vimentin, α-SMA, FSP1
and β-catenin), and the cells have undergone complete EMT. In the
lineage studies, during the formation of fibroblasts in liver
tissues, renal and other organs including lung and heart, this
transition was strongly demonstrated (32–34). Studies
have demonstrated that endothelial cells can also be devoted to the
formation of mesenchymal cells via a process known as EnMT
(35). Li et al (36) studied mouse models with cell lineage
analysis and demonstrated that mesothelial cells (MCs) expressing
Wilms tumor 1 produce HSCs and MFs during liver fibrogenesis. The
results suggest that MCs participate in liver injury via
differentiation to HSCs and MFs and are able to undergo
mesothelial-mesenchymal transition.
An EMT can be identified in rat fetal liver cells in
response to growth factors (epithelial growth factor and TGF-β) and
dimethyl sulfoxide (37,38). HSCs cultured in vitro were shown
to coexpress epithelial and mesenchymal markers, which provided
indirect evidence of EMT (39,40). Increasing evidence has shown that TGF-β
can induce an EMT in mice hepatocytes in vitro. The
mechanism demonstrated that TGF-β induced EMT via a MAPK-dependent
pathway and a Smad2/3-dependent pathway. Studies have shown that
hepatic growth factors can decrease the level of TGF-β, restore
E-cadherin, and decrease the amount of active matrix
metalloproteinase 9 (MMP-9) (41)
potentially. Other studies have demonstrated that the
E-cadherin/β-catenin signaling axis also has an important role for
EMT involving epithelial cells. Bone morphogenetic protein 7
(BMP-7) has been used in the mouse model of liver, kidney and lung
fibrosis, and the results demonstrated that BMP-7 functions as an
endogenous inhibitor of TGF-β induced EMT (31). TGF-β1 is recognized as a major cytokine
in organ fibrosis and is an inducer of collagen production and HSC
proliferation (42).
Biomarkers of EMT
To demonstrate the EMT, a variety of biomarkers have
been used. Among these markers, some are acquired and some are
attenuated during the process of transition. The following are a
few of the commonly used markers and mechanisms.
A change or switch of E-cadherin during the EMT in
tissue fibrosis, cancer and embryonic development is the
prototypical epithelial cell marker. During the transition, the
expression of E-cadherin is decreased, and in addition, EMT is
promoted by the loss of E-cadherin function (43,44). The
switches from E-cadherin to N/OB-cadherin have been increasingly
used in recent years to monitor the progress of EMT during
embryonic development, fibrosis and cancer progression. Integrins
are other EMT markers, which in general have limited utility, as
various integrins are expressed on mesenchymal and epithelial
cells. DDR2 upregulates MMP1 and cell motility upon binding to type
1 or type × collagen, and is associated with types 2 and 3 EMT.
FSP-1 (also known as S100A4 and MTS-1), is a member of the
Ca2+-binding S100 proteins. In tissue fibrosis, FSP-1 is
expressed by epithelial cells undergoing type 2 EMT transition to
mesenchymal cells, and it has been used as a prototypical marker
for detecting EMT in fibrosis and cancer. Vimentin, another marker
of EMT, is expressed in various cells including fibroblasts and
endothelial cells, and it should not be treated as a typical marker
of type 2 EMT as adult epithelial cells express vimentin in
response to different insults (45).
Fibronectin serves as a scaffold for the ECM, which has been used
as an indicator of type 1 EMT. The increased fibronectin expression
is associated with type 2 and type 3 EMT in vitro.
EMT in liver fibrosis: TGF-β/Smad and
non-Smad signaling pathway
TGF-β is believed to be a potent inducer of EMT and
a key mediator of wound healing, fibrosis (46) and cancer. TGF-β1 is a well-established
cytokine that induces the profibrogenic pathway and fibrosis in
liver (47). Furthermore, TGF-β1
expression is also associated with morphological alterations, such
as EMT in hepatocytes and changes in survival signaling pathways
(48). In the TGF-β signaling pathway,
active TGF-β1 ligands initiate signaling by binding to TGF-β
receptor type I (TβRI) and TβRII serine/threonine kinases. TβRI
phosphorylates Smad2 and Smad3, which form a complex with Smad4 and
translocate to the nucleus. Smad proteins convey signals from TGF-β
to nucleus. Once in the nucleus, the complex of Smads can regulate
the transcription of target genes. Activation of several Smad
independent pathways have been identified as crucial for EMT
induction by TGF-β, which includes phosphoinositide 3-kinase
(PI3K)-Akt (49), focal adhesion
kinase (50), p38 MAPK (51), and ERK (52). Recent studies have implicated
Krüppel-like factor-8 (53),
hyaluronan synthase 2 (54) and
microRNA miR-203 (55) as critical
regulators of EMT. Kim et al (56) demonstrated that the NF-κB decoy
oligodeoxynucleotide inhibited the EMT process in fibrotic liver
in vivo. The overexpression of TGF-β1 is associated with
liver fibrosis in diverse animal models and in patients with
chronic liver disease. TGF-β1 crucially controls the expression of
ECM network components, such as fibrillar collagens and
fibronectin, ECM-degrading protease inhibitors plasminogen
activator inhibitor-1 and TIMPs) and finally regulates ECM
deposition. The activity of TGF-β1 is strongly induced during
chronic liver injury with links between connective tissue growth
factor and TGF-β1 in the HSC activation process (57), which acquire myofibroblastic features
and produce ECM proteins in turn. TGF-β1 initiates and maintains
the EMT in a variety of biological systems by activating major
signaling pathways and transcriptional regulators integrated in
extensive cellular networks. It has been suggested that the loss of
E-cadherin expression in MDCKII cells exposed to TGF-β1 occurs
through a Smad-independent mechanism, which includes the MAPK and
PI3K pathways with expression of Snail. However, a complete
transition to the mesenchymal phenotype additionally requires Smad
signaling. A previous study reported that TGF-β1 participates in
the regulation of the Notch signaling pathway (58). A series of the previously mentioned
genes and others described to be involved in EMT (includes
Notch2 and Snail) were identified as TGF-β1 target
genes. Park et al (59)
reported that geniposide suppresses EMT, which leads to liver
fibrosis by inhibiting multiple TGF-β1-mediated molecular mediators
involved in hepatic injury. Lee et al (60) demonstrated that apamin suppressed the
TGF-β1-induced hepatocyte EMT in vitro and CCl4-injected
fibrosis in vivo.
The hedgehog (Hh) pathway has been identified as an
essential morphogene for tissue remodeling in adult tissue. Hh
ligands, Sonic Hh (Shh), Indian Hh (Ihh), Desert Hh (Dhh), bind to
the patch (Ptc), releasing smoothened (Smo) into the cytosol
(61). The aforementioned released Smo
promotes the translocation of the cytoplasmic glioblastoma family
(Glis: Gli1, Gli2 and Gli3) into the nucleus, which acts as a
transcriptional factor, activating Hh signaling (62–64).
Evidence has shown that Hh signaling is activated in damaged liver,
where it regulates tissue reconstruction. The level of Hh
expression was suggested to be parallel to the degree of fibrosis
(65). Furthermore, Hh signaling has
been demonstrated to activate quiescent hepatic stellate cells into
MF-HSCs (66) (Fig. 1).
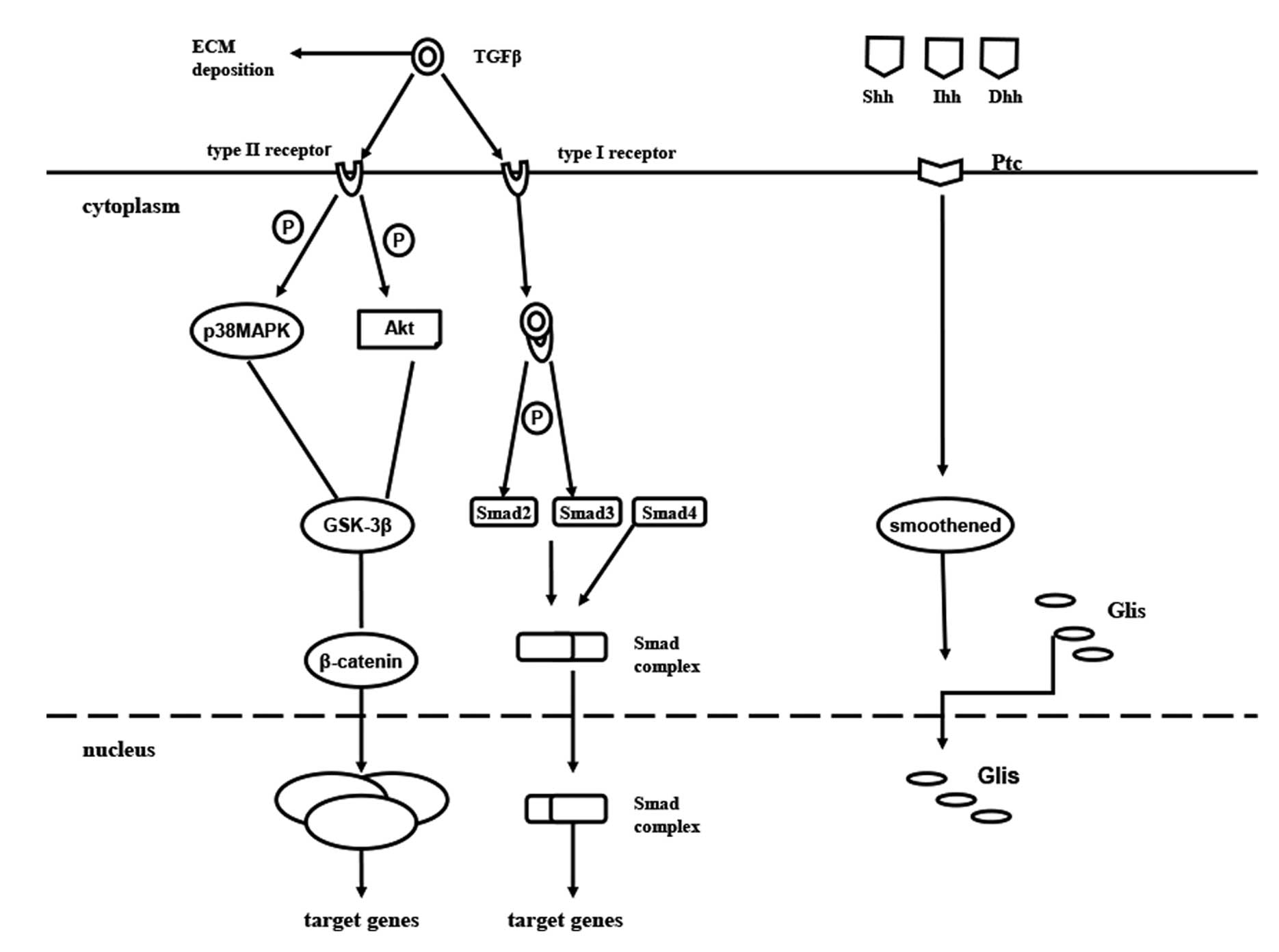 | Figure 1.Cellular signaling pathways of EMT in
liver fibrosis. Active TGFβ1 ligands initiate signaling by binding
to TβRI and TβRII serine/threonine kinases. TβRI phosphorylates
Smad2 and Smad3, which form a complex with Smad4 and translocate to
the nucleus. Smad proteins convey signals from TGF-β to the
nucleus. Once in the nucleus, the complex of Smad can regulate the
transcription of target genes. EMT, epithelial-mesenchymal
transition; TGF, transforming growth factor; TβRI, TGFβ receptor
type I; P, phosphorylate; MAPK, mitogen-activated protein kinase;
GSK-3β, glycogen synthase kinase-3β; ILK, integrin-linked kinase;
TCF/LEF-1 complex, T-cell factor/lymphoid enhancer-binding factor-1
complex; Hh, hedgehog; Shh, Sonic Hh; Ihh, Indian Hh; Dhh, Desert
Hh; Ptc, patch; ECM, extracellular matrix; Glis, glioblastoma
family. |
Controversy
Hepatocyte EMT
EMT was first demonstrated to occur in the fibrosis
tissue in the kidney, in vitro (30). Following this, a mice model of renal
fibrosis induced by unilateral ureteral obstruction lost the
epithelial marker E-cadherin and gained mesenchymal cells markers
(such as α-SMA). As the origin of fibroblastic cells remains under
debate, it is appealing that the liver epithelial cells may have
the possibility participate to fibrosis via EMT. Hepatocyte EMT was
observed when cells were incubated with TGF-β1 (67), which was characterized by a decrease in
epithelial marker E-cadherin expression and concomitant acquisition
of mesenchymal markers (type I collagen and vimentin). While
substantial experimental evidence supports that EMT makes a
contribution to embryonic development and tumor metastasis, and
renal fibrosis, the role of EMT in liver fibrosis remains under
debate. Taura et al (68) bred
the triple transgenic mice expressing ROSA26 stop β-gal, Albumin
Cre and collagen α1 (1) green
fluorescent protein and induced fibrosis by CCl4 injections. The
study examined the expression of four different mesenchymal
markers, which were FSP-1, α-SMA, vimentin and desmin. In these
studies, the lack of expression of yellow fluorescent protein (YFP)
supports the conclusion that EMT does not contribute to fibrosis in
these models. Furthermore, the complete absence of its
colocalization with YFP suggests that liver epithelial cells do not
transition to either mesenchymal cells or MFs via EMT in the mouse
models examined (69).
Cholangiocyte EMT
Cholangiocyte EMT was recently challenged with
lineage-tracing methodology. Scholten et al (70) studied several strains expressing Cre
under cholangiocyte-, HSC- or FSP-1-specific promoters in two liver
fibrosis models (chronic CCl4 intoxication and common BDL) with
Cre-Lox technology for lineage tracing. Following permanent genetic
Cre-mediated labeling of cholangiocytes, the fundamental experiment
traced the fate of cells expressing K19 in this case. The study
concluded that EMT of cholangiocytes identified by genetic labeling
does not contribute to liver fibrosis in mice.
Conclusion
There have been considerable advances in the
understanding of the mechanisms of the EMT. The possibility that
EMT makes a contribution to liver fibrogenesis reinforced that not
only HSCs, but bone marrow-derived cells and circulating
fibrocytes, could contribute to this process. The research of EMT
in the next few years holds a significant potential as a viable
therapeutic target. Future research probes into the molecular
similarities and differences among the EMT programs. Furthermore,
the identification of the signaling pathways that lead to
activation of EMT programs during liver fibrosis is providing novel
insights into the plasticity of cellular phenotypes and possible
therapeutic interventions.
References
|
1
|
Bi WR, Jin CX, Xu GT and Yang CQ: Effect
of alendronate sodium on the expression of mesenchymal-epithelial
transition markers in mice with liver fibrosis. Exp Ther Med.
5:247–252. 2013.PubMed/NCBI
|
|
2
|
Deng YH, Pu CL, Li YC, Zhu J, Xiang C,
Zhang MM and Guo CB: Analysis of biliary epithelial-mesenchymal
transition in portal tract fibrogenesis in biliary atresia. Dig Dis
Sci. 56:731–740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yoshida K and Matsuzaki K: Differential
regulation of TGF-β/Smad signaling in hepatic stellate cells
between acute and chronic liver injuries. Front Physiol. 3:532012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moreno-Alvarez P, Sosa-Garrocho M,
Briones-Orta MA, González-Espinosa C, Medina-Tamayo J, Molina-Jijón
E, Pedraza-Chaverri J and Macías-Silva M: Angiotensin II increases
mRNA levels of all TGF-beta isoforms in quiescent and activated rat
hepatic stellate cells. Cell Biol Int. 34:969–978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee YS and Jeong WI: Retinoic acids and
hepatic stellate cells in liver disease. J Gastroenterol Hepatol.
27(Suppl 2): S75–S79. 2012. View Article : Google Scholar
|
|
6
|
Rippe RA and Brenner DA: From quiescence
to activation: Gene regulation in hepatic stellate cells.
Gastroenterology. 127:1260–1262. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mann J, Oakley F, Akiboye F, Elsharkawy A,
Thorne AW and Mann DA: Regulation of myofibroblast
transdifferentiation by DNA methylation and MeCP2: Implications for
wound healing and fibrogenesis. Cell Death Differ. 14:275–285.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsukamoto H, She H, Hazra S, Cheng J and
Miyahara T: Anti-adipogenic regulation underlies hepatic stellate
cell transdifferentiation. J Gastroenterol Hepatol. 21(Suppl 3):
S102–S105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elpek GÖ: Cellular and molecular
mechanisms in the pathogenesis of liver fibrosis: An update. World
J Gastroenterol. 20:7260–7276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fausther M, Lavoie EG and Dranoff JA:
Contribution of Myofibroblasts of Different Origins to Liver
Fibrosis. Curr Pathobiol Rep. 1:225–230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bosselut N, Housset C, Marcelo P, Rey C,
Burmester T, Vinh J, Vaubourdolle M, Cadoret A and Baudin B:
Distinct proteomic features of two fibrogenic liver cell
populations: Hepatic stellate cells and portal myofibroblasts.
Proteomics. 10:1017–1028. 2010.PubMed/NCBI
|
|
12
|
Dranoff JA and Wells RG: Portal
fibroblasts: Underappreciated mediators of biliary fibrosis.
Hepatology. 51:1438–1444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Uchio K, Tuchweber B, Manabe N, Gabbiani
G, Rosenbaum J and Desmoulière A: Cellular retinol-binding
protein-1 expression and modulation during in vivo and in vitro
myofibroblastic differentiation of rat hepatic stellate cells and
portal fibroblasts. Lab Invest. 82:619–628. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iwaisako K, Brenner DA and Kisseleva T:
What's new in liver fibrosis? The origin of myofibroblasts in liver
fibrosis. J Gastroenterol Hepatol. 27(Suppl 2): S65–S68. 2012.
View Article : Google Scholar
|
|
15
|
Tuchweber B, Desmoulière A,
Bochaton-Piallat ML, Rubbia-Brandt L and Gabbiani G: Proliferation
and phenotypic modulation of portal fibroblasts in the early stages
of cholestatic fibrosis in the rat. Lab Invest. 74:265–278.
1996.PubMed/NCBI
|
|
16
|
Kinnman N, Francoz C, Barbu V, Wendum D,
Rey C, Hultcrantz R, Poupon R and Housset C: The myofibroblastic
conversion of peribiliary fibrogenic cells distinct from hepatic
stellate cells is stimulated by platelet-derived growth factor
during liver fibrogenesis. Lab Invest. 83:163–173. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quan TE, Cowper S, Wu SP, Bockenstedt LK
and Bucala R: Circulating fibrocytes: Collagen-secreting cells of
the peripheral blood. Int J Biochem Cell Biol. 36:598–606. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Strieter RM, Keeley EC, Burdick MD and
Mehrad B: The role of circulating mesenchymal progenitor cells,
fibrocytes, in promoting pulmonary fibrosis. Trans Am Clin Climatol
Assoc. 120:49–59. 2009.PubMed/NCBI
|
|
19
|
Kisseleva T, Uchinami H, Feirt N,
Quintana-Bustamante O, Segovia JC, Schwabe RF and Brenner DA: Bone
marrow-derived fibrocytes participate in pathogenesis of liver
fibrosis. J Hepatol. 45:429–438. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kisseleva T and Brenner DA: The phenotypic
fate and functional role for bone marrow-derived stem cells in
liver fibrosis. J Hepatol. 56:965–972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Forbes SJ and Parola M: Liver fibrogenic
cells. Best Pract Res Clin Gastroenterol. 25:207–217. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeisberg M, Yang C, Martino M, Duncan MB,
Rieder F, Tanjore H and Kalluri R: Fibroblasts derive from
hepatocytes in liver fibrosis via epithelial to mesenchymal
transition. J Biol Chem. 282:23337–23347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Milani S, Herbst H, Schuppan D, Stein H
and Surrenti C: Transforming growth factors beta 1 and beta 2 are
differentially expressed in fibrotic liver disease. Am J Pathol.
139:1221–1229. 1991.PubMed/NCBI
|
|
24
|
Pinzani M, Milani S, Herbst H, DeFranco R,
Grappone C, Gentilini A, Caligiuri A, Pellegrini G, Ngo DV,
Romanelli RG and Gentilini P: Expression of platelet-derived growth
factor and its receptors in normal human liver and during active
hepatic fibrogenesis. Am J Pathol. 148:785–800. 1996.PubMed/NCBI
|
|
25
|
Omenetti A, Porrello A, Jung Y, Yang L,
Popov Y, Choi SS, Witek RP, Alpini G, Venter J and Vandongen HM:
Hedgehog signaling regulates epithelial-mesenchymal transition
during biliary fibrosis in rodents and humans. J Clin Invest.
118:3331–3342. 2008.PubMed/NCBI
|
|
26
|
Xia JL, Dai C, Michalopoulos GK and Liu Y:
Hepatocyte growth factor attenuates liver fibrosis induced by bile
duct ligation. Am J Pathol. 168:1500–1512. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Strutz F, Okada H, Lo CW, Danoff T, Carone
RL, Tomaszewski JE and Neilson EG: Identification and
characterization of a fibroblast marker: FSP1. J Cell Biol.
130:393–405. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Okada H, Danoff TM, Kalluri R and Neilson
EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am
J Physiol. 273:F563–F574. 1997.PubMed/NCBI
|
|
31
|
Zeisberg M, Hanai J, Sugimoto H, Mammoto
T, Charytan D, Strutz F and Kalluri R: BMP-7 counteracts
TGF-beta1-induced epithelial-to-mesenchymal transition and reverses
chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee HY, Jeon HS, Song EK, Han MK, Park SI,
Lee SI, Yun HJ, Kim JR, Kim JS, Lee YC, et al: CD40 ligation of
rheumatoid synovial fibroblasts regulates RANKL-mediated
osteoclastogenesis: Evidence of NF-kappaB-dependent, CD40-mediated
bone destruction in rheumatoid arthritis. Arthritis Rheum.
54:1747–1758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iwano M, Plieth D, Danoff TM, Xue C, Okada
H and Neilson EG: Evidence that fibroblasts derive from epithelium
during tissue fibrosis. J Clin Invest. 110:341–350. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Potenta S, Zeisberg E and Kalluri R: The
role of endothelial-to-mesenchymal transition in cancer
progression. Br J Cancer. 99:1375–1379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y, Wang J and Asahina K: Mesothelial
cells give rise to hepatic stellate cells and myofibroblasts via
mesothelial-mesenchymal transition in liver injury. Proc Natl Acad
Sci USA. 110:2324–2329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pagan R, Martín I, Llobera M and Vilaró S:
Epithelial-mesenchymal transition of cultured rat neonatal
hepatocytes is differentially regulated in response to epidermal
growth factor and dimethyl sulfoxide. Hepatology. 25:598–606. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Valdés F, Alvarez AM, Locascio A, Vega S,
Herrera B, Fernández M, Benito M, Nieto MA and Fabregat I: The
epithelial mesenchymal transition confers resistance to the
apoptotic effects of transforming growth factor beta in fetal rat
hepatocytes. Mol Cancer Res. 1:68–78. 2002.PubMed/NCBI
|
|
39
|
Sicklick JK, Choi SS, Bustamante M, McCall
SJ, Pérez EH, Huang J, Li YX, Rojkind M and Diehl AM: Evidence for
epithelial-mesenchymal transitions in adult liver cells. Am J
Physiol Gastrointest Liver Physiol. 291:G575–G583. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xue ZF, Wu XM and Liu M: Hepatic
regeneration and the epithelial to mesenchymal transition. World J
Gastroenterol. 19:1380–1386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang J and Liu Y: Blockage of tubular
epithelial to myofibroblast transition by hepatocyte growth factor
prevents renal interstitial fibrosis. J Am Soc Nephrol. 13:96–107.
2002.PubMed/NCBI
|
|
42
|
Eghbali-Fatourechi G, Sieck GC, Prakash
YS, Maercklein P, Gores GJ and Fitzpatrick LA: Type I procollagen
production and cell proliferation is mediated by transforming
growth factor-beta in a model of hepatic fibrosis. Endocrinology.
137:1894–1903. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hay ED and Zuk A: Transformations between
epithelium and mesenchyme: Normal, pathological and experimentally
induced. Am J Kidney Dis. 26:678–690. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Witzgall R, Brown D, Schwarz C and
Bonventre JV: Localization of proliferating cell nuclear antigen,
vimentin, c-Fos and clusterin in the postischemic kidney. Evidence
for a heterogenous genetic response among nephron segments and a
large pool of mitotically active and dedifferentiated cells. J Clin
Invest. 93:2175–2188. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Klass BR, Grobbelaar AO and Rolfe KJ:
Transforming growth factor beta1 signalling, wound healing and
repair: A multifunctional cytokine with clinical implications for
wound repair, a delicate balance. Postgrad Med J. 85:9–14. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Martin M, Lefaix J and Delanian S:
TGF-beta1 and radiation fibrosis: A master switch and a specific
therapeutic target? Int J Radiat Oncol Biol Phys. 47:277–290. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Del Castillo G, Murillo MM,
Alvarez-Barrientos A, Bertran E, Fernández M, Sánchez A and
Fabregat I: Autocrine production of TGF-beta confers resistance to
apoptosis after an epithelial-mesenchymal transition process in
hepatocytes: Role of EGF receptor ligands. Exp Cell Res.
312:2860–2871. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor beta-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cicchini C, Laudadio I, Citarella F,
Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L and
Tripodi M: TGFbeta-induced EMT requires focal adhesion kinase (FAK)
signaling. Exp Cell Res. 314:143–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bhowmick NA, Zent R, Ghiassi M, McDonnell
M and Moses HL: Integrin beta 1 signaling is necessary for
transforming growth factor-beta activation of p38MAPK and
epithelial plasticity. J Biol Chem. 276:46707–46713. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang H, Liu L, Wang Y, Zhao G, Xie R, Liu
C, Xiao X, Wu K, Nie Y, Zhang H and Fan D: KLF8 involves in
TGF-beta-induced EMT and promotes invasion and migration in gastric
cancer cells. J Cancer Res Clin Oncol. 139:1033–1042. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Porsch H, Bernert B, Mehić M, Theocharis
AD, Heldin CH and Heldin P: Efficient TGFβ-induced
epithelial-mesenchymal transition depends on hyaluronan synthase
HAS2. Oncogene. 32:4355–4365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ding X, Park SI, McCauley LK and Wang CY:
Signaling between transforming growth factor β (TGF-β) and
transcription factor SNAI2 represses expression of microRNA miR-203
to promote epithelial-mesenchymal transition and tumor metastasis.
J Biol Chem. 288:10241–10253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim KH, Lee WR, Kang YN, Chang YC and Park
KW: Inhibitory effect of nuclear factor-κB decoy
oligodeoxynucleotide on liver fibrosis through regulation of the
epithelial-mesenchymal transition. Hum Gene Ther. 25:721–729. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Leask A and Abraham DJ: TGF-beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zavadil J, Cermak L, Soto-Nieves N and
Böttinger EP: Integration of TGF-beta/Smad and Jagged1/Notch
signalling in epithelial-to-mesenchymal transition. EMBO J.
23:1155–1165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Park JH, Yoon J, Lee KY and Park B:
Effects of geniposide on hepatocytes undergoing
epithelial-mesenchymal transition in hepatic fibrosis by targeting
TGFβ/Smad and ERK-MAPK signaling pathways. Biochimie. 113:26–34.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee WR, Kim KH, An HJ, Kim JY, Lee SJ, Han
SM, Pak SC and Park KK: Apamin inhibits hepatic fibrosis through
suppression of transforming growth factor β1-induced hepatocyte
epithelial-mesenchymal transition. Biochem Biophys Res Commun.
450:195–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang S, Lee Y, Kim J, Hyun J, Lee K, Kim Y
and Jung Y: Potential role of Hedgehog pathway in liver response to
radiation. PLoS One. 8:e741412013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ingham PW and McMahon AP: Hedgehog
signaling in animal development: Paradigms and principles. Genes
Dev. 15:3059–3087. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
van den Brink GR: Hedgehog signaling in
development and homeostasis of the gastrointestinal tract. Physiol
Rev. 87:1343–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Varjosalo M and Taipale J: Hedgehog:
Functions and mechanisms. Genes Dev. 22:2454–2472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kahila Bar-Gal G, Kim MJ, Klein A, Shin
DH, Oh CS, Kim JW, Kim TH, Kim SB, Grant PR, Pappo O, et al:
Tracing hepatitis B virus to the 16th century in a Korean mummy.
Hepatology. 56:1671–1680. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Choi SS, Omenetti A, Witek RP, Moylan CA,
Syn WK, Jung Y, Yang L, Sudan DL, Sicklick JK, Michelotti GA, et
al: Hedgehog pathway activation and epithelial-to-mesenchymal
transitions during myofibroblastic transformation of rat hepatic
cells in culture and cirrhosis. Am J Physiol Gastrointest Liver
Physiol. 297:G1093–G1106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kaimori A, Potter J, Kaimori JY, Wang C,
Mezey E and Koteish A: Transforming growth factor-beta1 induces an
epithelial-to-mesenchymal transition state in mouse hepatocytes in
vitro. J Biol Chem. 282:22089–22101. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Taura K, Miura K, Iwaisako K, Osterreicher
CH, Kodama Y, Penz-Osterreicher M and Brenner DA: Hepatocytes do
not undergo epithelial-mesenchymal transition in liver fibrosis in
mice. Hepatology. 51:1027–1036. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lee SJ, Kim KH and Park KK: Mechanisms of
fibrogenesis in liver cirrhosis: The molecular aspects of
epithelial-mesenchymal transition. World J Hepatol. 6:207–216.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Scholten D, Osterreicher CH, Scholten A,
Iwaisako K, Gu G, Brenner DA and Kisseleva T: Genetic labeling does
not detect epithelial-to-mesenchymal transition of cholangiocytes
in liver fibrosis in mice. Gastroenterology. 139:987–998. 2010.
View Article : Google Scholar : PubMed/NCBI
|














