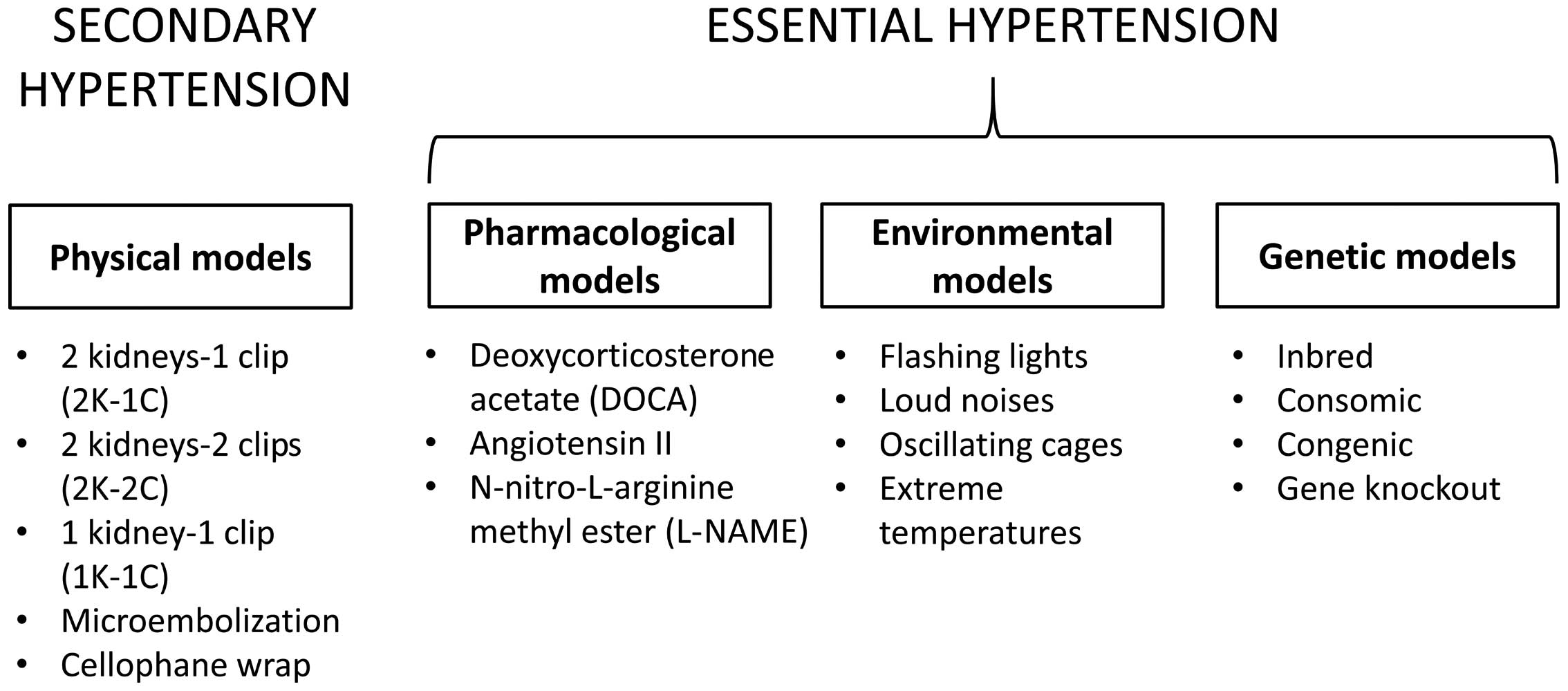
Hypertension is one of the most important risk
factors for the development of cardiovascular disease and is
responsible for >50% of the 17 million deaths per year worldwide
(1). It is a heterogeneous condition
with a number of etiologies and multiple, interacting genetic and
environmental factors (2). Its
incidence varies with age, plasma renin activity and sodium
sensitivity (3). The use of
pre-clinical animal models has significantly increased our
understanding of disease processes, as these permit the control of
the different contributing factors. However, no single system is
ideal, as there are species differences and other limitations of
these model systems (4,5). Species, including mice, have been popular
for the study of cardiometabolic disorders due to their amenability
to genetic and pharmacological modification (6–12). It is not
the aim of the present review to provide an exhaustive list of the
different models, but to discuss historically important model
systems whose use has significantly advanced our understanding of
hypertension.
Initial experiments for the investigation of
hypertension were performed in dogs. Such experiments included the
renovascular models developed by Goldblatt et al (15) in 1934. Subsequent models using rats,
rabbits, sheep and cats were developed (16). Pigs have also been used, in particular,
the Yucatan model for the study of DOCA-induced hypertension
(17).
Of the different species, rat has been a popular
model as a result of the availability of different inbred strains
and characteristics, including the SHR, Dahl salt-sensitive rats,
New Zealand and Milan strains (18).
Numerous justifications for using rats to model hypertension exist.
Firstly, its genome has been completely mapped, with a 99% sequence
homology to humans (19). Secondly,
the pathogenesis of hypertension in rats and humans are largely
similar in terms of arterial pressure development from childbirth,
response to environmental stressors, haemodynamic factors
(including vascular resistance), mechanisms regulating arteriolar
and venous constriction, neural modulation (including sympathetic
nerve activity) and renal vascular dynamics (including perfusion
parameters), as well as humoral influences by RAAS and NOS
(20). The advantages are that they
are low cost, with wide availability and easy to handle, maintain
and breed. However, these models also have their limitations.
Firstly, the identical genotype may not induce the same phenotype
in all animals (21) due to
contributions from numerous genes and the additional environmental
influences (22,23). Secondly, the same gene mutations and
deletion observed in rats may not induce to the identical
phenotypic effects in humans (24).
Larger animals have closer anatomical, physiological
and haemodynamic properties to humans when compared with small
animals, including rats and mice, making them particularly suitable
for the study of flow characteristics (25,26).
However, the major disadvantage is the high costs required for
their maintenance. Additionally, the domestication of dogs has led
to their decreasing use for research (27).
The kidney-clip models mimicking renal arterial
stenosis were first performed in dogs (15), which have been gradually replaced by
smaller animals. In the 2K-1C model, one of the two renal arteries
is constricted by a clip (28).
Initially, decreased renal arterial pressure in the clipped kidney
leads to increased plasma renin activity (PRA) with higher
circulating levels of renin and aldosterone (29). This is followed by the return of the
PRA to a near normal level, and finally by chronically elevated PRA
(30). Patients with renovascular
hypertension exhibit similar patterns of PRA (31). The underlying mechanism involves RAAS
activation, increased renin production and subsequent angiotensin
(Ang)-I release and conversion by Ang converting enzyme (ACE) to
Ang-II. The net effects are further vasoconstriction and increased
production of aldosterone level, which together lead to water and
salt retention, and an increased blood pressure. In addition, the
model also reveals increased sympathetic nerve activity that
further drives renin production (32).
The 2K-2C model, where both renal arteries are constricted,
resemble bilateral renal arterial stenosis in humans and the
mechanism involved is similar to the 2K-1C model, but with a more
severe phenotype (33).
In the 1K-1C model, unilateral nephrectomy is
performed with a constricting clip on the renal artery of remaining
kidney (34). This resembles patients
who suffer from RAS of the solitary kidney (35). Similar to the previous renal models,
initial elevation of blood pressure is due to RAAS activation.
However, because of the absence of a functional kidney, no
compensatory rise in sodium and water excretion is observed.
Consequently, more fluid is retained inside the body. In other
words, this is more volume- rather than RAAS-dependent. This is
consistent with the experimental findings that ACE inhibition was
unable to prevent chronic hypertension in renal artery stenosis of
a solitary kidney, however, was successful in doing so where a
normal functioning kidney is present (36).
Renal parenchymal hypertension is the commonest
cause of secondary hypertension and is responsible for up to 5% of
all cases (37). Subtotal nephrectomy
ablation, in which up to 5/6 of the kidney is removed, has been
performed to induce chronic renal disease (38). This model demonstrates glomerular,
tubular and interstitial injury, loss of nephrons and the
development of hypertension. It can be combined with the
introduction of excess salt into the diet to increase the severity
and speed of onset of this hypertension. The mechanism is dependent
on the RAAS and the hypertension can be reduced by ACE inhibition.
Renal ischemia has been produced by microembolisation, which led to
the development of nephrosclerosis and hypertension (39). Perinephric fibrosis has been induced by
wrapping the kidney in cellophane, mimicking fibrosis that occurs
after kidney transplantation (40).
Mineralocorticoids or their synthetic derivatives,
including DOCA, are used with sodium chloride in unilateral
nephrectomised rats to induce hypertension (41,42). Renin
is suppressed and fluid reabsorption is increased, thereby
producing a low renin-volume overload model of hypertension
(43). Using this model, key
sodium-independent mechanisms for mediating hypertension, including
upregulation of Ang-II receptors in the central nervous system
(44), elevated vasopressin (45), increased oxidative stress (46) and endothelin (47), have been identified. Aside from
elucidating the molecular mechanisms underlying renal hypertension,
it provides a useful platform for investigating the natural history
of disease, including any complications, such as
glomerulosclerosis, proteinuria, impaired endothelium-dependent
relaxation of the vasculature and cardiac hypertrophy can be
investigated (42). In the
DOCA-hypertensive Yucatan miniature swine model, excess dietary
salt is not required for sustaining hypertension due to enhanced
SNS activity at baseline, as evidenced by the increased plasma
norepinephrine level (48).
Glucocorticoids can also be used to induce hypertension (49). Although hypertension is produced via
RAAS activation, this approach is less effective than the DOCA-salt
method. An alternative is chronic infusion of RAAS components,
including Ang-II (50).
Nitric oxide (NO), a potent vasodilator derived from
the intact endothelium, is produced by NOS. This production is
triggered by vasoactive messengers, including acetylcholine
(51). A NO-deficient model was
produced by chronic infusion of N-nitro-L-arginine methyl ester
(L-NAME), a NOS inhibitor (52). A low
dose produced a volume-dependent increase in blood pressure
predominantly due to renal vasoconstriction and decreased
glomerular filtration (53). A high
dose led to both salt- and volume-independent hypertension since
the mechanism is renal and systemic vasoconstriction (54).
Environmental stress, including separate or
simultaneous introduction of flashing lights, loud noises and
oscillating cages (55–57), or long-term exposure to high salt, fat
or sugar in the diet, can be used to induce hypertension (58). Extremes of temperature, particularly
coldness, also induces a hypertensive phenotype, as observed in
rats exposed to 5°C for 3 weeks (59).
In these animals, increases in plasma and urine catecholamines were
observed (60). These findings are
consistent with findings in humans, where those who work
chronically in cold areas develop hypertension (61) and higher values of blood pressure
recorded in winter compared with in the summer (62). Increased activity of the sympathetic
nervous system and RAAS activation appear to be the common
physiological mechanisms responsible for hypertension in the models
described above (60,63,64).
Genetic factors are estimated to influence up to 50%
of blood pressure variability in essential hypertension (65). The millennium genome project for
hypertension was initiated in 2000 to identify genetic variants
that predispose individuals to hypertension. This has involved a
combination of techniques, including a gene linkage approach using
single nucleotide polymorphisms, microsatellite markers and
systematic candidate gene analysis (66). In parallel with this has been the
development of genetic models using different animal species, which
have provided insights into the physiological mechanisms of
hypertension. These can be categorised into inbreeding, consomic,
congenic and subcongenic strains (18), which will be considered in turn.
The inbreeding method involves sibling mating of
hypertensive rats over several generations to produce strains with
genetic homogeneity when compared with the reference control
group.
SHRs have been used to determine the genes
responsible for hypertension, to evaluate complications of target
organs and the screening potential pharmacological agents for
treatment. In stroke-prone SHRs, it was shown that dietary
potassium supplementation decreases the risk of cerebrovascular
accidents, even when blood pressure was not lowered (72). At least three genetic loci have been
implicated in the early development of hypertension, with an
additional gene identified on chromosome 10 contributing to its
maintenance with aging (73). The New
Zealand hypertensive rats are similar to Japanese SHRs in
developing spontaneous hypertension (74), as do the Milan (20) and Lyon (75,76)
strains.
Dahl salt-sensitive (DS) rat strains are prone to
hypertension following administration of a low-salt diet (0.4%
NaCl), unlike Dahl salt-resistant (DR) rat strains, which remain
normotensive (77). DS strain rats fed
with a high-salt diet (8% NaCl) develop particularly severe
hypertension (78). The reason is that
the certain alleles at the genetic loci for ACE and guanylyl
cyclase A, causing DS rats to have increased ACE and decreased
atrial natriuretic factor (ANF, the ligand for guanylyl cyclase A)
(79). The Sabra strain also
demonstrates salt-sensitive hypertension (80).
The Fawn hooded hypertensive rats develop
hypertension due to glomerular sclerosis, and therefore serve as a
model for renal parenchymal disease (81). Sprague-Dawley rats, obese Zucker,
Wistar fatty rats have been used to assess the effects of
diet-induced obesity on the development of hypertension (82).
Transgenic technology can be used to investigate the
specific role of different genes in the regulation of blood
pressure (83). Broadly, the
approaches are generation of consomic and congenic strains, and
gene knockout.
A congenic strain refers to one in which a defined
chromosome segment is introduced to another by backcrossing with
appropriate selection (84). In the
case of a consomic strain, the entire chromosome is transferred
(85). For example, the mutant renin
gene from mouse was transferred to rats, producing elevated Ang-II
levels and hypertension (86), which
were prevented by ACE inhibition (87). Similarly, insertion of the human renin
gene into mice also consistently demonstrated activation of genes
involved in the RAAS (88,89).
Gene targeting permits targeted disruption,
including deletion, overexpression or subtle mutations, of a gene
product. Conditional knockout with tissue- and time-dependent
specificity is also possible, allowing investigation of the loss of
a particular gene at specific time points or in particular organs.
Gene knockout is often performed in mice because of the relative
ease in introducing genetic mutations, and this has led to an
increased understanding of different cardiovascular disorders with
potential for translational application (90–99).
Knockout of the angiotensinogen gene provided protection in
delaying the development of hypertension and increasing NO
availability compared with wild-type, thereby implicating the RAAS
as being critical in blood pressure regulation (100). However, each group demonstrated
similar extents of cardiac hypertrophy, suggesting RAAS-independent
mechanisms for this response. Knockout of genes encoding for
endothelial NOS (101) and atrial
natriuretic peptide develop hypertension, whereas Ang-II type 1a
receptor knockout rats demonstrate hypotension (102). The importance of the aldosterone
pathway was shown in a mineralocorticoid receptor mutation
conferring constitutive receptor activity led to early onset
hypertension (103). Liddle syndrome,
an autosomal dominant cause of pseudoaldosteronism, leading to
human hypertension, was shown to involve a mutated epithelial
sodium channel (104).
Certain limitations of genetic knockout models
require addressing. Firstly, the expression of certain gene
deletions may result in embryonic lethality so there is lack of
time to study the pathogenesis; secondly, redundancy among isoforms
causes phenotypic expression to be masked so sometimes double or
triple gene knockout is required. It is also important to note that
same gene deletion or overexpression in animals may lead to
different expression in phenotypes (24).
Different pharmacological, environmental and genetic
models using different animal species have provided useful and
valuable information on the aetiology, pathophysiology and
complications of human cardiovascular and metabolic disorders, and
a platform to examine the efficacy of pharmacotherapy (105–118).
However, a major limitation of these experimental models is the
anatomical differences between these animal species and humans
(119). Although the common mechanism
is RAAS activation across different species, species differences
must be carefully taken into consideration to ensure the safety of
newly developed pharmacological agents.
The present review was funded by a Biotechnology and
Biological Sciences Research Council Doctoral Training Award at the
University of Cambridge awarded to Dr Gary Tse and the Economic and
Social Research Council grant awarded to Miss Yin Wah Fiona Chan at
the University of Cambridge.
|
1
|
Chen R, Dharmarajan K, Kulkarni VT,
Punnanithinont N, Gupta A, Bikdeli B, Mody PS and Ranasinghe I:
Most important outcomes research papers on hypertension. Circ
Cardiovasc Qual Outcomes. 6:e26–e35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galosy RA, Clarke LK, Vasko MR and
Crawford IL: Neurophysiology and neuropharmacology of
cardiovascular regulation and stress. Neurosci Biobehav Rev.
5:137–175. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinberger MH: Salt sensitivity of blood
pressure in humans. Hypertension. 27:481–490. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarikonda KV, Watson RE, Opara OC and
Dipette DJ: Experimental animal models of hypertension. J Am Soc
Hypertens. 3:158–165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lerman LO, Chade AR, Sica V and Napoli C:
Animal models of hypertension: An overview. J Lab Clin Med.
146:160–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tse G, Tse V and Yeo JM: Ventricular
anti-arrhythmic effects of heptanol in hypokalaemic,
Langendorff-perfused mouse hearts. Biomed Rep. 4:313–324.
2016.PubMed/NCBI
|
|
7
|
Tse G, Tse V, Yeo JM and Sun B: Atrial
anti arrhythmic effects of heptanol in Langendorff perfused mouse
hearts. PLoS One. 11:e01488582016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tse G, Wong ST, Tse V and Yeo JM:
Restitution analysis of alternans using dynamic pacing and its
comparison with S1S2 restitution in heptanol-treated, hypokalaemic
Langendorff-perfused mouse hearts. Biomed Rep. 4:673–680.
2016.PubMed/NCBI
|
|
9
|
Tse G, Sun B, Wong ST, Tse V and Yeo JM:
Ventricular anti arrhythmic effects of hypercalcaemia treatment in
hyperkalaemic, Langendorff perfused mouse hearts. Biomed Rep.
5:301–310. 2016.PubMed/NCBI
|
|
10
|
Tse G, Hothi SS, Grace AA and Huang CL:
Ventricular arrhythmogenesis following slowed conduction in
heptanol-treated, Langendorff-perfused mouse hearts. J Physiol Sci.
62:79–92. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tse G and Yeo JM: Conduction abnormalities
and ventricular arrhythmogenesis: The roles of sodium channels and
gap junctions. Int J Cardiol Heart Vasc. 9:75–82. 2015.PubMed/NCBI
|
|
12
|
Tse G, Yeo JM, Tse V, Kwan SK and Sun B:
Gap junction inhibition by heptanol increases ventricular
arrhythmogenicity by decreasing conduction velocity without
affecting repolarization properties or myocardial refractoriness in
Langendorff perfused mouse hearts. Mol Med Rep. (In press).
PubMed/NCBI
|
|
13
|
Leong XF, Ng CY and Jaarin K: Animal
models in cardiovascular research: Hypertension and
atherosclerosis. BioMed Res Int. 2015:5287572015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Doggrell SA and Brown L: Rat models of
hypertension, cardiac hypertrophy and failure. Cardiovasc Res.
39:89–105. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goldblatt H, Lynch J, Hanzal RF and
Summerville WW: Studies on experimental: I. the production of
persistent elevation of systolic blood pressure by means of renal
ischenmia. J Exp Med. 9:347–379. 1934. View Article : Google Scholar
|
|
16
|
Gross DR: Animal models of
hypertensionAnimal models in cardiovascular research. Springer;
Netherlands, Dordrecht: pp. 475–482. 1994, View Article : Google Scholar
|
|
17
|
Terris JM and Simmonds RC: The Yucatan
miniature swine: An improved pig model for the study of
desoxycorticosterone-acetate (DOCA) and aldosterone hypertension.
Proc Soc Exp Biol Med. 171:79–82. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yagil Y and Yagil C: Genetic models of
hypertension in experimental animals. Exp Nephrol. 9:1–9. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Twigger S, Lu J, Shimoyama M, Chen D,
Pasko D, Long H, Ginster J, Chen CF, Nigam R, Kwitek A, et al: Rat
Genome Database (RGD): Mapping disease onto the genome. Nucleic
Acids Res. 30:125–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Trippodo NC and Frohlich ED: Similarities
of genetic (spontaneous) hypertension. Man and rat. Circ Res.
48:309–319. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stoll M and Jacob HJ: Genetic rat models
of hypertension: Relationship to human hypertension. Curr Hypertens
Rep. 3:157–164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fuentes RM, Notkola IL, Shemeikka S,
Tuomilehto J and Nissinen A: Familial aggregation of blood
pressure: A population-based family study in eastern Finland. J Hum
Hypertens. 14:441–445. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perry IJ, Whincup PH and Shaper AG:
Environmental factors in the development of essential hypertension.
Br Med Bull. 50:246–259. 1994.PubMed/NCBI
|
|
24
|
Williams SM, Haines JL and Moore JH: The
use of animal models in the study of complex disease: All else is
never equal or why do so many human studies fail to replicate
animal findings? BioEssays. 26:170–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fuster V, Lie JT, Badimon L, Rosemark JA,
Badimon JJ and Bowie EJ: Spontaneous and diet-induced coronary
atherosclerosis in normal swine and swine with von Willebrand
disease. Arteriosclerosis. 5:67–73. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fekete A, Rényi Vámos A and Szitás A:
Experimental model of renovascular hypertension in dogs. Int Urol
Nephrol. 2:391–400. 1970. View Article : Google Scholar
|
|
27
|
Hasiwa N, Bailey J, Clausing P, Daneshian
M, Eileraas M, Farkas S, Gyertyán I, Hubrecht R, Kobel W,
Krummenacher G, et al: Critical evaluation of the use of dogs in
biomedical research and testing in Europe. ALTEX. 28:326–340. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wiesel P, Mazzolai L, Nussberger J and
Pedrazzini T: Two-kidney, one clip and one-kidney, one clip
hypertension in mice. Hypertension. 29:1025–1030. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Okamura T, Miyazaki M, Inagami T and Toda
N: Vascular renin-angiotensin system in two-kidney, one clip
hypertensive rats. Hypertension. 8:560–565. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamasaki S: Divided renal and caval vein
plasma renin activity in two-kidney two-clip hypertension in
rabbits and variations of blood pressure, plasma volume and renal
function following unilateral nephrectomy. J Urol. 138:1457–1460.
1987.PubMed/NCBI
|
|
31
|
Sato K, Abe K, Seino M, Yasujima M, Imai
Y, Sato M, Hiwatari M, Omata K, Tanno M, Kohzuki M, et al: Renal
vein plasma renin activity in patients with unilateral renovascular
hypertension. Jpn Circ J. 52:431–436. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sawamura T and Nakada T: Role of dopamine
in the striatum, renin-angiotensin system and renal sympathetic
nerve on the development of two-kidney, one-clip Goldblatt
hypertension. J Urol. 155:1108–1111. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeng J, Zhang Y, Mo J, Su Z and Huang R:
Two kidney, two clip renovascular hypertensive rats can be used as
stroke prone rats. Stroke. 29:1708–1713. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murphy WR, Coleman TG, Smith TL and Stanek
KA: Effects of graded renal artery constriction on blood pressure,
renal artery pressure, and plasma renin activity in Goldblatt
hypertension. Hypertension. 6:68–74. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feldman L, Beberashvili I, Averbukh Z and
Weissgarten J: Renal artery stenosis of solitary kidney: The
dilemma. Ren Fail. 26:525–529. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brunner HR, Kirshman JD, Sealey JE and
Laragh JH: Hypertension of renal origin: Evidence for two different
mechanisms. Science. 174:1344–1346. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Preston RA and Epstein M: Renal
parenchymal disease and hypertension. Semin Nephrol. 15:138–151.
1995.PubMed/NCBI
|
|
38
|
van Koppen A, Verhaar MC, Bongartz LG and
Joles JA: 5/6th nephrectomy in combination with high salt diet and
nitric oxide synthase inhibition to induce chronic kidney disease
in the Lewis rat. J Vis Exp. 77:e503982013.
|
|
39
|
Moore S and Mersereau WA: Microembolic
renal ischemia, hypertension, and nephrosclerosis. Arch Pathol.
85:623–630. 1968.PubMed/NCBI
|
|
40
|
Page IH: A method for producing persistent
hypertension by cellophane. Science. 89:273–274. 1939. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Selye H, Hall CE and Rowley EM: Malignant
hypertension produced by treatment with desoxycorticosterone
acetate and sodium chloride. Can Med Assoc J. 49:88–92.
1943.PubMed/NCBI
|
|
42
|
Iyer A, Chan V and Brown L: The DOCA-salt
hypertensive rat as a model of cardiovascular oxidative and
inflammatory stress. Curr Cardiol Rev. 6:291–297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Williams SK and Ogedegbe G: Unraveling the
mechanism of renin-angiotensin- aldosterone system activation and
target organ damage in hypertensive blacks. Hypertension. 59:10–11.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wilson KM, Sumners C, Hathaway S and
Fregly MJ: Mineralocorticoids modulate central angiotensin II
receptors in rats. Brain Res. 382:87–96. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Möhring J, Möhring B, Petri M and Haack D:
Vasopressor role of ADH in the pathogenesis of malignant DOC
hypertension. Am J Physiol. 232:F260–F269. 1977.PubMed/NCBI
|
|
46
|
Li L, Chu Y, Fink GD, Engelhardt JF,
Heistad DD and Chen AF: Endothelin-1 stimulates arterial VCAM-1
expression via NADPH oxidase-derived superoxide in
mineralocorticoid hypertension. Hypertension. 42:997–1003. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Matsumura Y, Hashimoto N, Taira S, Kuro T,
Kitano R, Ohkita M, Opgenorth TJ and Takaoka M: Different
contributions of endothelin-A and endothelin-B receptors in the
pathogenesis of deoxycorticosterone acetate-salt-induced
hypertension in rats. Hypertension. 33:759–765. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zambraski EJ, Ciccone CD and Izzo JL Jr:
The role of the sympathetic nervous system in 2-kidney
DOCA-hypertensive Yucatan miniature swine. Clin Exp Hypertens A.
8:411–424. 1986.PubMed/NCBI
|
|
49
|
Dahl LK, Knudsen KD, Heine M and Leitl G:
Effects of chronic excess salt ingestion. Genetic influence on the
development of salt hypertension in parabiotic rats: Evidence for a
humoral factor. J Exp Med. 126:687–699. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
McCubbin JW, DeMoura RS, Page IH and
Olmsted F: Arterial hypertension elicited by subpressor amounts of
angiotensin. Science. 149:1394–1395. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moncada S and Higgs A: The
L-arginine-nitric oxide pathway. N Engl J Med. 329:2002–2012. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ribeiro MO, Antunes E, de Nucci G,
Lovisolo SM and Zatz R: Chronic inhibition of nitric oxide
synthesis. A new model of arterial hypertension. Hypertension.
20:298–303. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lahera V, Salazar J, Salom MG and Romero
JC: Deficient production of nitric oxide induces volume-dependent
hypertension. J Hypertens Suppl. 10:(Suppl 7). S173–S177. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zatz R and Baylis C: Chronic nitric oxide
inhibition model six years on. Hypertension. 32:958–964. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Buñag RD, Takeda K and Riley E:
Spontaneous remission of hypertension in awake rats chronically
exposed to shaker stress. Hypertension. 2:311–318. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Smookler HH, Goebel KH, Siegel MI and
Clarke DE: Hypertensive effects of prolonged auditory, visual, and
motion stimulation. Fed Proc. 32:2105–2110. 1973.PubMed/NCBI
|
|
57
|
McCann SM, Rothballer AB, Yeakel EH and
Shenkin HA: Adrenalectomy and blood pressure of rats subjected to
auditory stimulation. Am J Physiol. 155:128–131. 1948.PubMed/NCBI
|
|
58
|
Kaufman LN, Peterson MM and Smith SM:
Hypertension and sympathetic hyperactivity induced in rats by
high-fat or glucose diets. Am J Physiol. 260:E95–E100.
1991.PubMed/NCBI
|
|
59
|
Fregly MJ: Effects of Extremes of
temperature on hypertensive rats. Am J Physiol. 176:275–281.
1954.PubMed/NCBI
|
|
60
|
Papanek PE, Wood CE and Fregly MJ: Role of
the sympathetic nervous system in cold-induced hypertension in
rats. J Appl Physiol (1985). 71:300–306. 1991.PubMed/NCBI
|
|
61
|
Donaldson GC, Robinson D and Allaway SL:
An analysis of arterial disease mortality and BUPA health screening
data in men, in relation to outdoor temperature. Clin Sci (Lond).
92:261–268. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Brennan PJ, Greenberg G, Miall WE and
Thompson SG: Seasonal variation in arterial blood pressure. Br Med
J (Clin Res Ed). 285:919–923. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Coste SC, Qi Y, Brooks VL, McCarron DA and
Hatton DC: Captopril and stress-induced hypertension in the
borderline hypertensive rat. J Hypertens. 13:1391–1398. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hwang IS, Ho H, Hoffman BB and Reaven GM:
Fructose-induced insulin resistance and hypertension in rats.
Hypertension. 10:512–516. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Garcia EA, Newhouse S, Caulfield MJ and
Munroe PB: Genes and hypertension. Curr Pharm Des. 9:1679–1689.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tabara Y, Kohara K and Miki T: Millennium
Genome Project for Hypertension: Hunting for genes for
hypertension: The millennium genome project for hypertension.
Hypertens Res. 35:567–573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mann JF, Phillips MI, Dietz R, Haebara H
and Ganten D: Effects of central and peripheral angiotensin
blockade in hypertensive rats. Am J Physiol. 234:H629–H637.
1978.PubMed/NCBI
|
|
68
|
Yan L, Zhang JD, Wang B, Lv YJ, Jiang H,
Liu GL, Qiao Y, Ren M and Guo XF: Quercetin inhibits left
ventricular hypertrophy in spontaneously hypertensive rats and
inhibits angiotensin II-induced H9C2 cells hypertrophy by enhancing
PPAR-γ expression and suppressing AP-1 activity. PLoS One.
8:e725482013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bagby SP, McDonald WJ and Mass RD: Serial
renin-angiotensin studies in spontaneously hypertensive and
Wistar-Kyoto normotensive rats. Transition from normal- to
high-renin status during the established phase of spontaneous
hypertension. Hypertension. 1:347–354. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Okamoto K and Aoki K: Development of a
strain of spontaneously hypertensive rats. Jpn Circ J. 27:282–293.
1963. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kurtz TW and Morris RC Jr: Biological
variability in Wistar-Kyoto rats. Implications for research with
the spontaneously hypertensive rat. Hypertension. 10:127–131. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tobian L, Lange JM, Ulm KM, Wold LJ and
Iwai J: Potassium prevents death from strokes in hypertensive rats
without lowering blood pressure. J Hypertens Suppl. 2:S363–S366.
1984.PubMed/NCBI
|
|
73
|
Mashimo T, Nabika T, Matsumoto C, Tamada
T, Ueno K, Sawamura M, Ikeda K, Kato N, Nara Y and Yamori Y: Aging
and salt-loading modulate blood pressure QTLs in rats. Am J
Hypertens. 12:1098–1104. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Smirk FH and Hall WH: Inherited
hypertension in rats. Nature. 182:727–728. 1958. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vincent M, Bornet H, Berthezene F, Dupont
J and Sassard J: Thyroid function and blood pressure in two new
strains of spontaneously hypertensive and normotensive rats. Clin
Sci Mol Med. 54:391–395. 1978.PubMed/NCBI
|
|
76
|
Vincent M, Gomez-Sanchez CE, Bataillard A
and Sassard J: Steroids during development of genetic hypertension
in rats of Lyon strain. Am J Physiol. 257:H506–H510.
1989.PubMed/NCBI
|
|
77
|
Rapp JP and Dene H: Development and
characteristics of inbred strains of Dahl salt-sensitive and
salt-resistant rats. Hypertension. 7:340–349. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dahl LK, Heine M and Tassinari L: Effects
of chronic excess salt ingestion. Further demonstration that
genetic factors influence the development of hypertension: Evidence
from experimental hypertension due to cortisone and to adrenal
regeneration. J Exp Med. 122:533–545. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Deng Y and Rapp JP: Cosegregation of blood
pressure with angiotensin converting enzyme and atrial natriuretic
peptide receptor genes using Dahl salt-sensitive rats. Nat Genet.
1:267–272. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zamir N, Gutman Y and Ben-Ishay D:
Hypertension and brain catecholamine distribution in the Hebrew
University Sabra, H and N rats. Clin Sci Mol Med Suppl.
4:105s–107s. 1978.PubMed/NCBI
|
|
81
|
Kuijpers MH and Gruys E: Spontaneous
hypertension and hypertensive renal disease in the fawn-hooded rat.
Br J Exp Pathol. 65:181–190. 1984.PubMed/NCBI
|
|
82
|
Dobrian AD, Davies MJ, Prewitt RL and
Lauterio TJ: Development of hypertension in a rat model of
diet-induced obesity. Hypertension. 35:1009–1015. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Mullins LJ, Bailey MA and Mullins JJ:
Hypertension, kidney, and transgenics: A fresh perspective. Physiol
Rev. 86:709–746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lagrange D and Fournié GJ: Generation of
congenic and consomic rat strains. Methods Mol Biol. 597:243–266.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Cowley AW Jr, Liang M, Roman RJ, Greene AS
and Jacob HJ: Consomic rat model systems for physiological
genomics. Acta Physiol Scand. 181:585–592. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mullins JJ, Peters J and Ganten D:
Fulminant hypertension in transgenic rats harbouring the mouse
Ren-2 gene. Nature. 344:541–544. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Montgomery HE, Kiernan LA, Whitworth CE,
Fleming S, Unger T, Gohlke P, Mullins JJ and McEwan JR: Inhibition
of tissue angiotensin converting enzyme activity prevents malignant
hypertension in TGR(mREN2)27. J Hypertens. 16:635–643. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Sigmund CD, Jones CA, Kane CM, Wu C, Lang
JA and Gross KW: Regulated tissue- and cell-specific expression of
the human renin gene in transgenic mice. Circ Res. 70:1070–1079.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chung O, Schips T, Rohmeiss P, Gretz N,
Strauch M and Unger T: Protein excretion and renal adaptation of
transgenic mRen2 rats to changing oral sodium loads. J Hypertens
Suppl. 11:S188–S189. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tse G: Novel conduction repolarization
indices for the stratification of arrhythmic risk. J Geriatr
Cardiol. (In press). PubMed/NCBI
|
|
91
|
Tse G: (Tpeak Tend)/QRS and (Tpeak
Tend)/(QT x QRS): Novel markers for predicting arrhythmic risk in
the Brugada syndrome. Europace. (In press).
|
|
92
|
Tse G and Yan BP: Electrophysiological
mechanisms of long and short QT syndromes: Insights from mouse
models. IJC Heart & Vasculature. (In press).
|
|
93
|
Tse G and Yan BP: Novel arrhythmic risk
markers incorporating QRS dispersion: QRSd × (Tpeak - Tend)/QRS and
QRSd × (Tpeak - Tend)/(QT × QRS). Ann Noninvasive Electrocardiol.
Aug 18–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Tse G, Lai ET, Lee AP, Yan BP and Wong SH:
Electrophysiological mechanisms of gastrointestinal
arrhythmogenesis: Lessons from the heart. Front Physiol. 7:2302016.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tse G, Wong ST, Tse V and Yeo JM:
Variability in local action potential durations, dispersion of
repolarization and wavelength restitution in aged wild type and
Scn5a/− mouse hearts modelling human Brugada syndrome. J Geriatr
Cardiol. (In press). PubMed/NCBI
|
|
96
|
Tse G: Mechanisms of cardiac arrhythmias.
J Arrhythm. 32:75–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tse G, Yeo JM, Chan YW, Lai ET and Yan BP:
What is the arrhythmic substrate in viral myocarditis? Insights
from clinical and animal studies. Front Physiol. 7:3082016.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Tse G and Yan BP: Traditional and novel
electrocardiographic conduction and repolarization markers of
sudden cardiac death. Europace. (In press).
|
|
99
|
Sun B, Chen Z, Gu J, et al: The roles of
tight and gap junctions in the pathogenesis of high fat diet
induced atherosclerosis. Int J Clin Exp Pathol. (In press).
|
|
100
|
Sun Z, Cade R, Zhang Z, Alouidor J and Van
H: Angiotensinogen gene knockout delays and attenuates cold-induced
hypertension. Hypertension. 41:322–327. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Shesely EG, Maeda N, Kim H-S, Desai KM,
Krege JH, Laubach VE, Sherman PA, Sessa WC and Smithies O: Elevated
blood pressures in mice lacking endothelial nitric oxide synthase.
Proc Natl Acad Sci USA. 93:13176–13181. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sugaya T, Nishimatsu S, Tanimoto K,
Takimoto E, Yamagishi T, Imamura K, Goto S, Imaizumi K, Hisada Y,
Otsuka A, et al: Angiotensin II type 1a receptor-deficient mice
with hypotension and hyperreninemia. J Biol Chem. 270:18719–18722.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Geller DS, Farhi A, Pinkerton N, Fradley
M, Moritz M, Spitzer A, Meinke G, Tsai FT, Sigler PB and Lifton RP:
Activating mineralocorticoid receptor mutation in hypertension
exacerbated by pregnancy. Science. 289:119–123. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Shimkets RA, Warnock DG, Bositis CM,
Nelson-Williams C, Hansson JH, Schambelan M, Gill JR Jr, Ulick S,
Milora RV, Findling JW, et al: Liddle's syndrome: Heritable human
hypertension caused by mutations in the beta subunit of the
epithelial sodium channel. Cell. 79:407–414. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Tse G, Wong ST, Tse V, Lee YT, Lin HY and
Yeo JM: Cardiac dynamics: Alternans and arrhythmogenesis. J
Arrhythm. (In press).
|
|
106
|
Tse G, Wong ST, Tse V and Yeo JM:
Monophasic action potential recordings: Which is the recording
electrode? J Basic Clin Physiol Pharmacol. Apr 30–2016.(Epub ahead
of print). View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Tse G, Lai ET, Yeo JM, Tse V and Wong SH:
Mechanisms of electrical activation and conduction in the
gastrointestinal system: Lessons from cardiac electrophysiology.
Front Physiol. 7:1822016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Tse G, Wong ST, Tse V and Yeo JM:
Depolarization vs. repolarization: What is the mechanism of
ventricular arrhythmogenesis underlying sodium channel
haploinsufficiency in mouse hearts? Acta Physiol (Oxf). (In
press).
|
|
109
|
Chen Z, Sun B, Tse G, Jiang J and Xu W:
Reversibility of both sinus node dysfunction and reduced HCN4 mRNA
expression level in an atrial tachycardia pacing model of
tachycardia bradycardia syndrome in rabbit hearts. Int J Clin Exp
Pathol. (In press).
|
|
110
|
Tse G, Lai ET, Tse V and Yeo JM: Molecular
and electrophysiological mechanisms underlying cardiac
arrhythmogenesis in diabetes mellitus. J Diabetes Res. 2016:1–8.
2016. View Article : Google Scholar
|
|
111
|
Tse G: Both transmural dispersion of
repolarization and transmural dispersion of refractoriness are poor
predictors of arrhythmogenicity: A role for the index of Cardiac
Electrophysiological Balance (QT/QRS)? J Geriatr Cardiol. (In
press). PubMed/NCBI
|
|
112
|
Tse G, Wong ST, Tse V and Yeo JM:
Determination of action potential wavelength restitution in Scn5a/-
mouse hearts modelling human Brugada syndrome. J Physiol. (In
press).
|
|
113
|
Choy L, Yeo JM, Tse V, Chan SP and Tse G:
Cardiac disease and arrhythmogenesis: Mechanistic insights from
mouse models. Int J Cardiol Heart Vasc. 12:1–10. 2016.PubMed/NCBI
|
|
114
|
Hu Z, Chen Z, Wang Y, et al: Effects of
granulocyte colony stimulating factor on rabbit carotid and swine
heart models of chronic obliterative arterial disease. Mol Med Rep.
(In press).
|
|
115
|
Tse G, Yan BP, Chan YW, Tian XY and Huang
Y: Reactive oxygen species, endoplasmic reticulum stress and
mitochondrial dysfunction: The link with cardiac arrhythmogenesis.
Front Physiol. 7:3132016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Tse G, Ali A, Prasad SK, Vassiliou V and
Raphael CE: Atypical case of post partum cardiomyopathy: an overlap
syndrome with arrhythmogenic right ventricular cardiomyopathy?
BJR|case reports. 1:20150182. 2015.
|
|
117
|
Tse G, Ali A, Alpendurada F, Prasad S,
Raphael CE and Vassiliou V: Tuberculous constrictive pericarditis.
Res Cardiovasc Med. 4:e296142015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Vassiliou V, Chin C, Perperoglou A, Tse G,
Ali A, Raphael C, Jabbour A, Newby D, Pennell D, Dweck M, et al: 93
ejection fraction by cardiovascular magnetic resonance predicts
adverse outcomes post aortic valve replacement. Heart. 100:(Suppl
3). A53–A54. 2014. View Article : Google Scholar
|
|
119
|
Hasenfuss G: Animal models of human
cardiovascular disease, heart failure and hypertrophy. Cardiovasc
Res. 39:60–76. 1998. View Article : Google Scholar : PubMed/NCBI
|