Increasing attention has been given to the role of
mitochondrial metabolism in cancer biology. The complex connection
between metabolism and tumorigenesis is a promising area of cancer
research. Mounting evidence has demonstrated that targeting
mitochondrial metabolism in cancer cells may present as a novel
strategy for anti-cancer therapy (7–11). As
summarized from current knowledge, the process of tumorigenesis and
mitochondrial biology inter-cross at multiple levels as follows: i)
Direct signals from mitochondria promote tumorigenesis; ii)
oncogenic signaling pathways alter mitochondrial functions; iii)
perturbation of mitochondrial functions have been shown to have a
major role in regulating metabolism and bioenergetics; iv)
mutations in mitochondrial DNA, proteins and enzymes result in
altered levels of metabolites, which support tumor development and
progression. The current review focuses on the importance of
various such classic alterations in cancer metabolism. Hence, by
understanding this aspect of metabolism, cancer biology may be
better understood and novel anti-cancer drugs may be developed.
All parts of the body require energy to work and
this energy is derived from consumption of food. Typically, all
food is broken down into smaller parts to generate the energy
source, ATP. ATP is a chemical energy generated via controlled
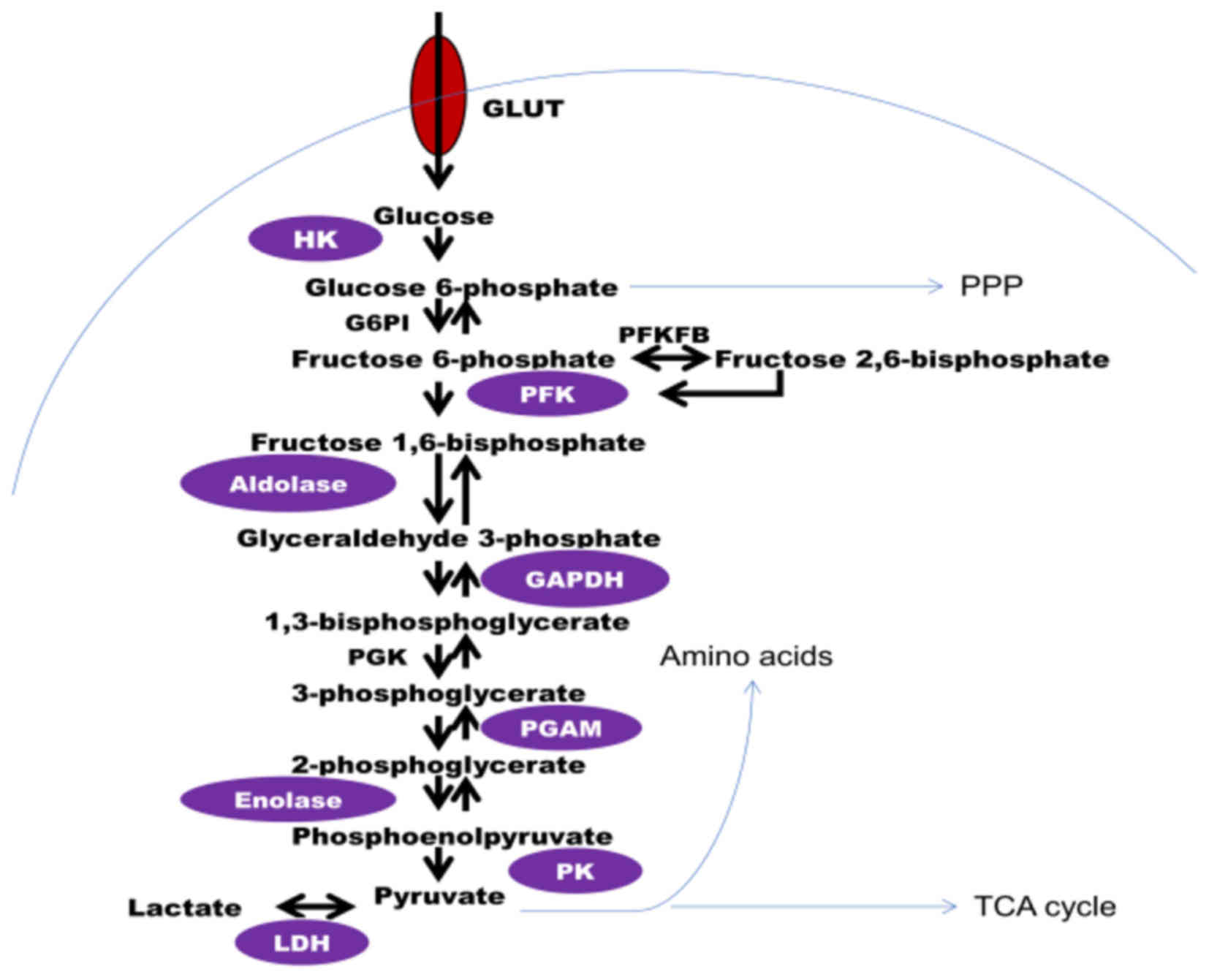
oxidation of glucose and other molecules. The process of the
breakdown of glucose, termed glycolysis, occurs in the cytoplasm of
mammalian cells. Glucose from food is taken up by specific glucose
transporters in the cell surface, and via a series of
enzyme-catalyzed reactions, broken down to pyruvate, the
end-product of glycolysis under aerobic conditions (Fig. 1). If there is a lack of oxygen supply,
pyruvate is converted to lactate (anaerobic glycolysis).
Theoretically, one molecule of glucose yields two molecules of
pyruvate and two molecules of ATP via glycolysis.
Since the early twentieth century, abnormalities of
glycolysis in cancer cells have been observed. Warburg (12), a German physiologist and a Nobel
laurate, observed that tumor cells depend solely on glycolysis for
energy production, even with an ample quantity of oxygen. This
phenomenon is since termed the Warburg effect of cancer cells. This
raises the question as to why cancer cells switch their metabolism
to aerobic glycolysis, unlike normal cells, which depend on
oxidative phosphorylation for energy production. While the exact
reasons remain unclear, the current explanations include: i)
Aerobic glycolysis, although less efficient than the classic
oxidative phosphorylation, provides rapid supply of ATP; ii)
glycolysis intermediates provide sufficient building blocks for
macromolecule synthesis required for the enhanced cell
proliferation. Due to this feature of cancer cells, studies have
been focused on novel strategies to selectively inhibit glucose
metabolism and/or glucose transport in cancer cells (13–16).
Marked progress has been made in understanding the
molecular mechanisms leading to constitutive upregulation of
glycolysis in tumor cells. Various glycolytic enzymes are
multi-functional proteins whose expression levels are often
increased in cancer cells. For example, hexokinase (HK), the enzyme
that converts glucose to glucose 6-phosphate (G6P), the first step
of glycolysis, is involved in transcription regulation, and its
expression is often upregulated in tumor cells (17,18). The
majority of malignant cells display enhanced expression levels of
type II isoform (HK-II), which may contribute to the elevated
glycolysis (19,20). Phosphofructokinase (PFK), the enzyme
that catalyzes the rate limiting step of glycolysis, has been
identified to be upregulated in types of breast cancer (21,22).
Another critical regulator of glycolysis is the enzyme
6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase (Pfkfb), a
family of bifunctional enzymes that control the levels of fructose
2,6-bisphosphate, which in turn is a powerful allosteric activator
of PFK1. Two Pfkfb isoforms, type 2 and 3, are associated with
cancers (23–26). Subsequently, the enzyme aldolase that
catalyzes the reversible conversion of fructose-1,6-bisphosphate to
glyceraldehyde-3-phosphate and dihydroxyacetone phosphate, has been
demonstrated to be overexpressed in squamous cell lung carcinoma
(27). The well-known classic
glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH;
the housekeeping gene) is also implicated in cancer. Overexpression
of GAPDH is considered an important feature of numerous types of
cancer (28–30). GAPDH has been proposed as a promising
target for the treatment of carcinomas (31). Pyruvate kinase (PK), the enzyme that
catalyzes the irreversible phosphoryl group transfer from
phosphoenolpyruvate to pyruvate, yielding pyruvate and ATP, appears
to be involved in cancer; previous studies and our findings have
demonstrated that tumor cells overexpress the type M2 isoform, PKM2
(32–35). As the majority of cancer cells are
dependent on aerobic glycolysis for ATP production, the enzyme,
lactate dehydrogenase (LDH), which catalyzes the conversion of
pyruvate to lactate, is the key to determining the glycolytic
phenotype of cancer cells. Thus, LDH is a promising target for
anti-cancer therapy. The inhibition of LDH suppresses tumor
progression of lymphomas and pancreatic cancer xenografts (36). These results indicate that selectively
targeting glycolysis and/or glycolytic enzymes in tumor cells may
present as an effective approach for the treatment of different
types of cancer.
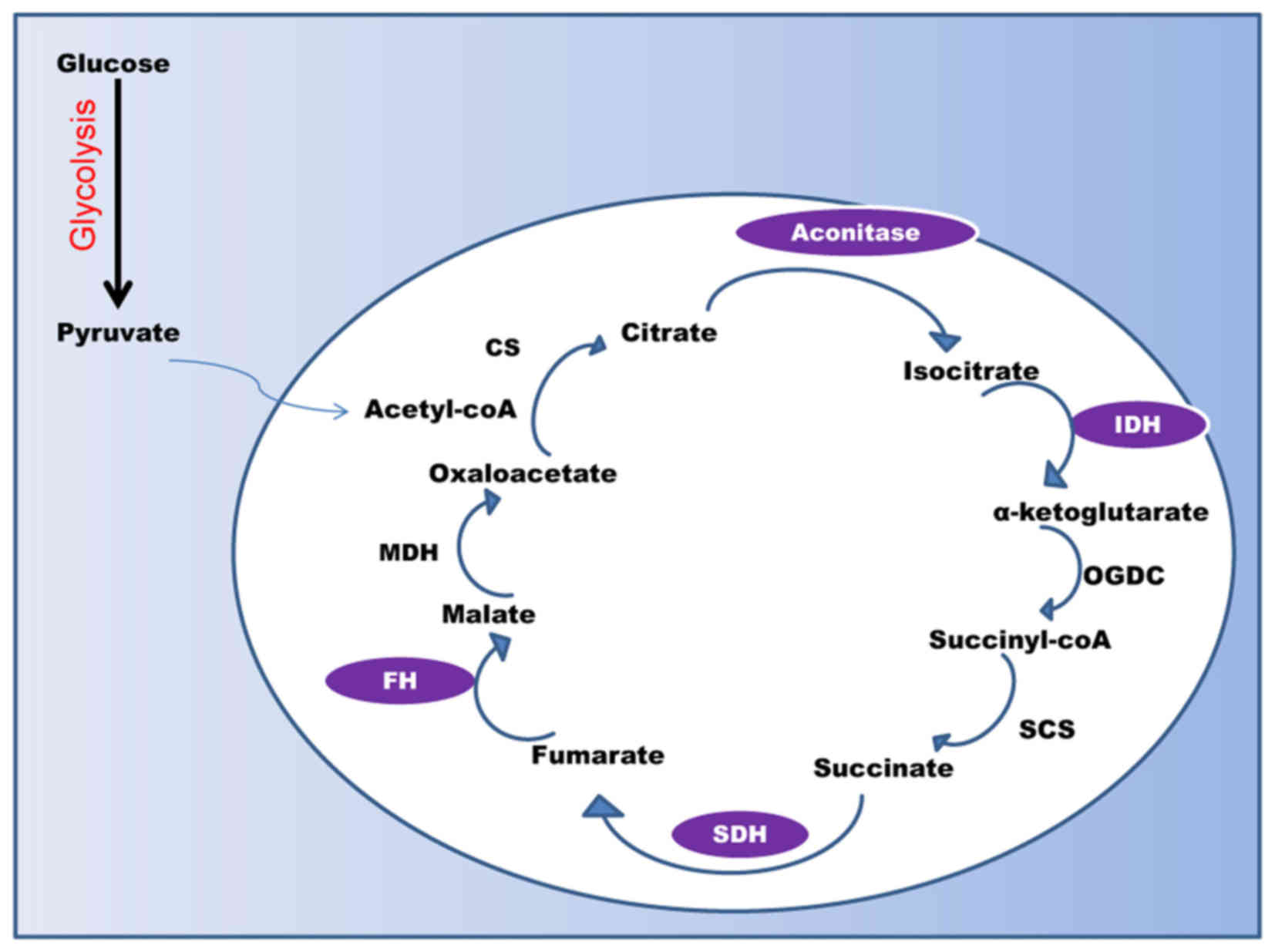
The Krebs cycle (the citric acid cycle or the TCA)
is a series of chemical reactions that generate energy via the
oxidation of pyruvate (Fig. 2). TCA
cycles occur in all aerobic living organisms. It provides
precursors for biosynthesis of compounds (such as amino acids), and
nicotinamide adenine dinucleotide (NADH), which is later used by
the electron transport chain to generate energy by converting NADH
to NAD+. The TCA cycle is the central metabolic hub of the cell
that occurs primarily in the mitochondria in contrast to
glycolysis, which occurs in the cytosol. Even a minor alteration in
these processes markedly influences mitochondrial energy
production. Although mutations in mitochondrial DNA have been
evaluated for over two decades (37–39), much
attention has been focused on the identification of mutations in
various TCA cycle enzymes (40,41). The
cycle consists of eight steps catalyzed by eight different enzymes.
Mutations in genes that encode enzymes aconitase, isocitrate
dehydrogenase (IDH), succinate dehydrogenase (SDH), and fumarate
hydratase (FH) may lead to cancer. Aconitase catalyzes
isomerization of citrate to isocitrate via cis-aconitase. Altered
expression levels of aconitase are implicated in human prostate
cancer, wherein the normal citrate-producing glandular secretory
epithelial cells undergo a metabolic transformation to malignant
citrate-oxidizing cells, leading to abnormal citrate metabolism and
prostate malignancy (42). IDH
converts isocitrate to α-ketoglutarate (α-KG). Glioblastoma
multiforme, one of the most common and lethal types of brain
cancer, is characterized by IDH1 gene mutations (43). Similar studies discovered mutations in
IDH1 and IDH2 genes in the pathogenesis of malignant gliomas
(44). Mutations that occur in single
amino acid residue of IDH1 active sites not only result in the
novel ability for the mutant enzyme to convert α-KG to
2-hydroxyglutarate, which is proposed to contribute to the
formation and malignant progression of gliomas (45). FH is the enzyme that converts fumarate
to malate, and mutations in the FH gene are associated with
cutaneous, uterine and aggressive forms of renal cancer (46–48).
Cancer cells that harbor FH mutations produce up to 100-fold more
fumarate, and seven-fold more succinate, but decreased levels of
citrate and malate (49). FH
deficiency in tumor cells alters redox homeostasis to promote
tumorigenesis (48). Mutations in the
enzyme SDH, which catalyzes the oxidation of succinate to fumarate,
are implicated in pheochromocytoma, paraganglioma, renal cell
carcinoma and papillary thyroid cancers (50–52).
Reduced expression and loss of heterozygosity of the SDH gene are
observed in gastric and colon carcinoma (53). SDH downregulation results in succinate
accumulation leading to transmission of an oncogenic signal from
mitochondria to the cytosol (54).
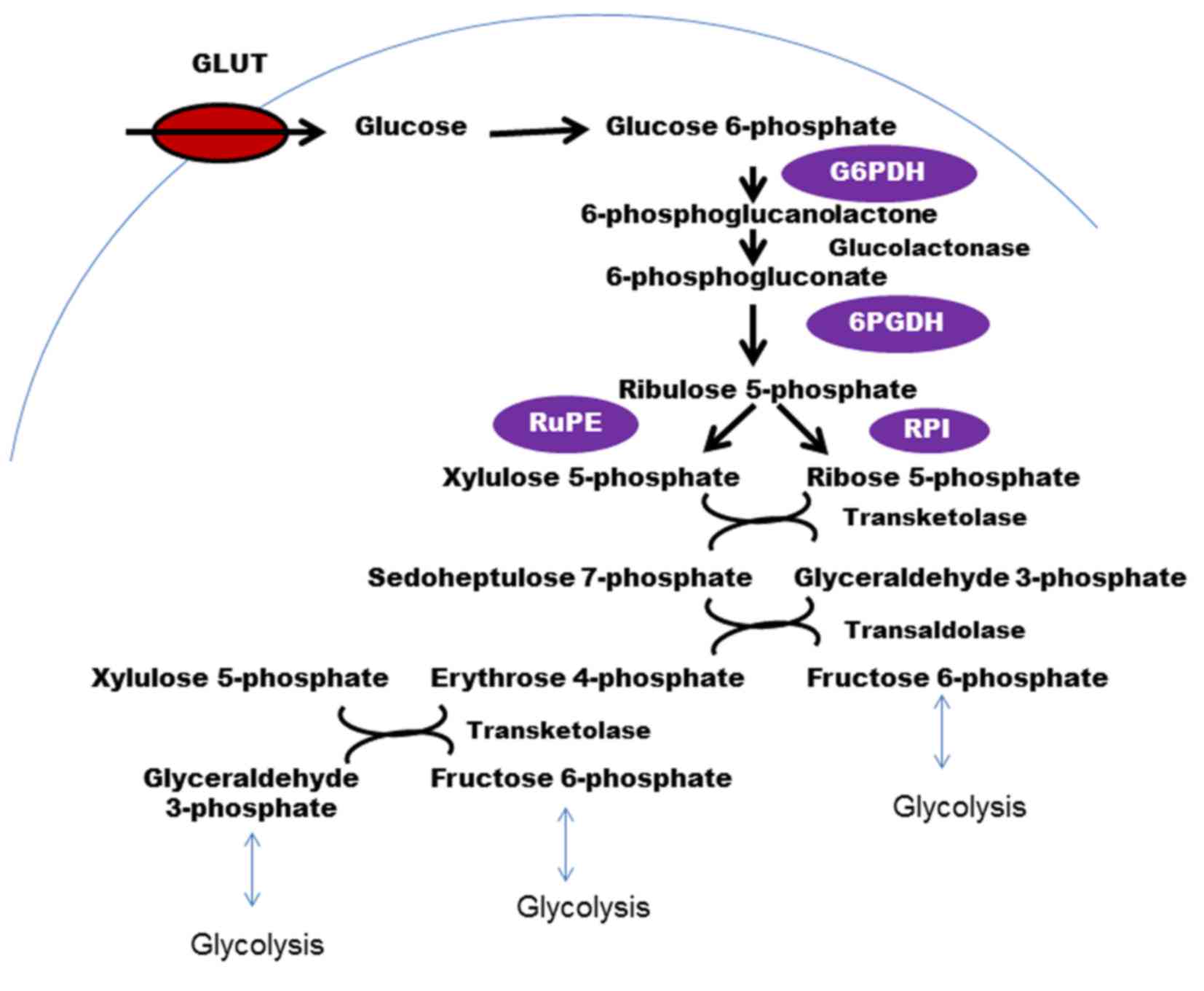
The PPP, which branches out from glycolysis at the
first committed step is the major catabolic pathway of glucose for
nucleotide synthesis in cancer cells (55–57). The
conversion of glucose to G6P, which is catalyzed by the enzyme HK,
is a common precursor for various metabolic glucose-consuming
routes (Fig. 3). Through this
pathway, cancer cells produce large quantities of ribose-5
phosphate (a precursor for nucleotide synthesis) and NAPDH (a
cofactor used in anabolic reactions). PPP runs parallel to
glycolysis and activation of these signaling pathways is a common
hallmark of tumor cells (58,59). As cancer cells are rapidly dividing,
the cells require a constant supply of nucleotides, and the
majority of the pentose phosphates are derived from the PPP. Thus,
PPP may influence the glycolytic flux. Various enzymes that execute
the PPP are implicated in different types of cancer. G6P
dehydrogenase (G6PD or G6PDH), the enzyme that catalyzes the
rate-limiting step in the PPP, and generates the first NADPH, is
highly overexpressed in certain tumors (60). Elevated levels of G6PD in association
with higher levels of PPP-derived metabolites are responsible for
clear-cell renal carcinoma-associated metabolic alterations
(61). Overexpression of G6PD in
human U2OS bone osteosarcoma epithelial cells enhances the
PPP-dependent production of NADPH (62). The same group also demonstrates that
simultaneous inhibition of glycolysis and PPP using
2-deoxy-d-glucose and 6-aminonicotinamide, respectively, induces
oxidative stress and sensitizes malignant human cancer cell lines
to radiotherapy, presumably via the induction of multiple cell
death modalities, including apoptosis, necrosis and mitotic
catastrophe (63). The next enzyme
that has a role in cancer is 6-phosphogluconate dehydrogenase
(6PGDH). 6PGDH catalyzes the oxidative decarboxylation of
6-phosphogluconate to ribulose 5-phosphate with a reduction of NADP
to NADPH. 6PGDH has been shown to be critical for lung
carcinogenesis and its inhibition may be a novel strategy to treat
glycolytic lung tumors (55).
Ribulose-5 phosphate isomerase, another critical enzyme in the PPP,
which catalyzes the conversion of ribulose-5 phosphate to ribose-5
phosphate and xylulose-5 phosphate (Xu5P), is also associated with
cancer (55). Ribose-5 phosphate is
important as it is a precursor for de novo nucleotide
synthesis in rapidly proliferating cancer cells. Xu5P increases the
levels of PFKFB, which activates PFK1 and increases glycolytic flux
(64). Thus, all of these studies
implicate that the regulation of PPP is vital for cancer cell
survival and proliferation. Furthermore, increased glycolytic flux
in cancer cells may be regulated directly or indirectly by PPP, and
hence, this may represent a promising strategy for treatment of
cancer cells.
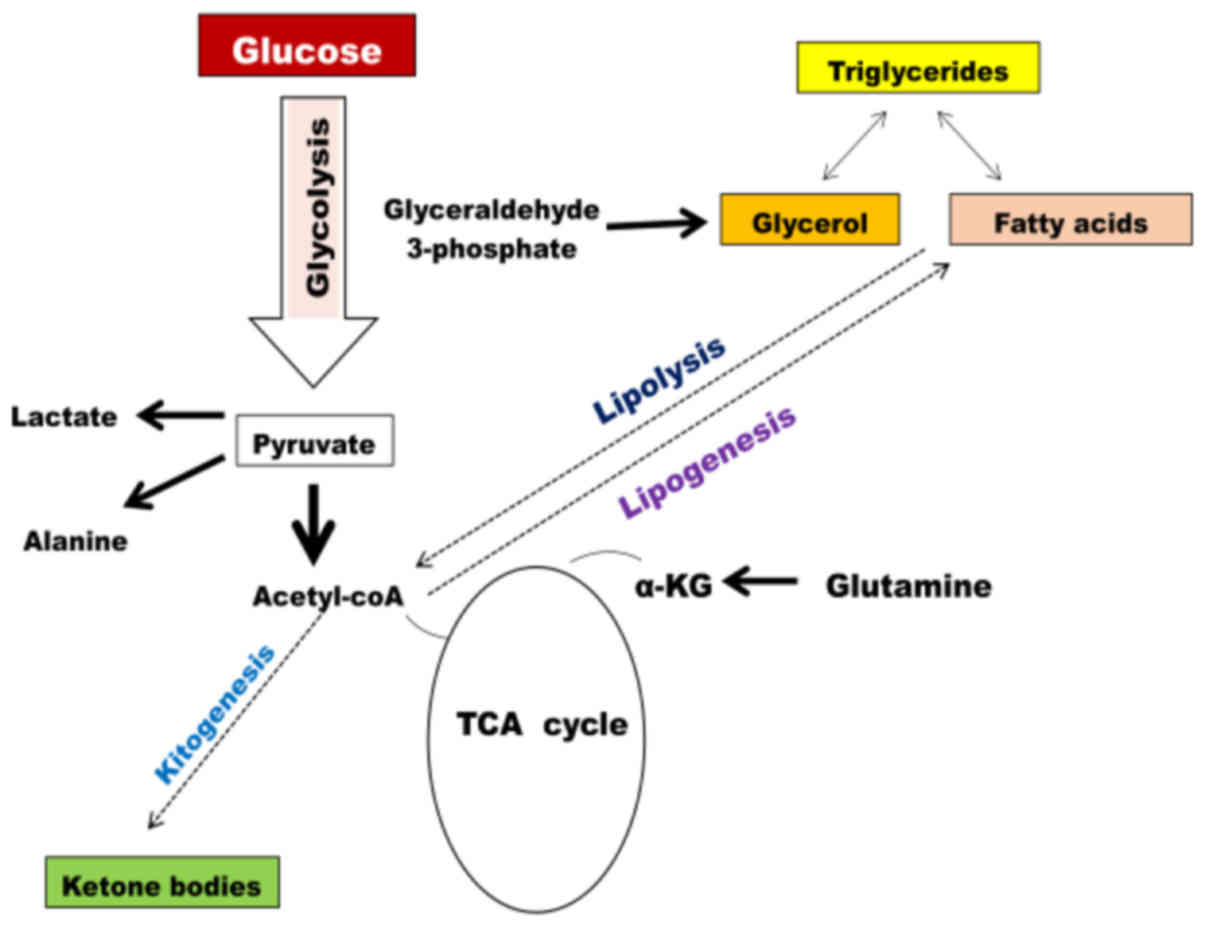
The role of lipid metabolism in cancer cells has
long been disregarded; over the past decade, the increased rate of
lipid metabolism in cancer cells is being recognized as the
prominent hallmark of transformed cells (83–85).
Lipids are a diverse group of molecules composed of fat,
triglycerides, phospholipid, cholesterols and cholesterol esters
(Fig. 5). Lipids form the major
component of cell membranes (phospholipid bilayer), hormones
(steroid hormones, such as cholesterol) and certain lipid-soluble
vitamins. Hence, lipids perform various roles in the body, from
providing energy to muscles to producing hormones (86). In rapidly proliferating cancer cells,
there is an overwhelming requirement for macromolecule synthesis.
Hence, cancer cells also demonstrate a high dependence on lipids
(83). One of the enzymes involved in
the synthesis of de novo fatty acids is ATP citrate lyase
(ACLY). ACLY catalyzes the conversion of mitochondrial-derived
citrate to oxaloacetate and cytosolic acetyl-CoA. Thus, ACLY links
de novo lipogenesis to gluconeogenesis and the Krebs cycle
(87). Studies have demonstrated that
higher expression levels of ACLY correlated with advanced stages of
cancer and lymph node metastasis in tissue samples from gastric
adenocarcinoma patients (88).
However, targeting ACLY by microRNA-22 (miR-22) suppresses cancer
cell proliferation and invasion in osteosarcoma, prostate, cervical
and lung cancer cells (89). Another
study demonstrates that ACLY is required for low molecular weight
isoform of cyclin E mediated transformation, migration, and
invasion of breast cancer cells in vitro along with tumor
growth in vivo (90).
Acetyl-CoA carboxylase (ACC) is the rate-limiting enzyme in fatty
acid synthesis. ACC carboxylates acetyl-CoA to form malonyl-CoA. In
patients with squamous cell carcinoma of the head and neck, there
is an association between phosphorylated AMP-activated protein
kinase and ACC expression, and the therapeutic outcome is that high
phosphorylated-ACC expression is associated with a worse overall
survival rate in the patients (91).
Similarly, ACC1 expression is upregulated in patients with
hepatocellular carcinoma (HCC), and upregulation of ACC1 is also
significantly correlated with the poorer overall survival of, and
disease recurrence in HCC patients (92). Fatty acid synthase (FASN), which
catalyzes the final step in fatty acid synthesis, is often
overexpressed in human cancers (93,94).
Inhibition of FASN suppresses invasion and migration of HCC cells
(95). In contrast to enhanced fatty
acid synthesis, certain types of cancer rely on the mitochondrial
fatty acid oxidation (FAO) for ATP production (96). Although the mechanism that upregulates
FAO in cancer remains unclear, it is proposed that FAO may confer
benefits beyond ATP production (96).
The FAO contributes to maintenance of redox homeostasis, and cell
survival in hematopoietic stem cells and leukemia cells (97). Carnitine palmitoyltransferase (CPT1),
the enzyme that catalyzes the initial step of FAO, is implicated in
various types of cancer (96,98,99). CPT1
upregulation increases FAO, ATP production and endows resistance to
metabolic stress.
Increased glycose consumption, lactate production,
PPP, lipid metabolism, and amino acid synthesis are commonly
observed metabolic profile in almost all types of cancer cell. This
type of metabolic profiling of tumor cells has been proposed to
support their rapid cell growth (100). High rates of glycolysis leading to
lactate production (aerobic glycolysis or the Warburg effect)
distinguish cancer cells from normal cells (12,13).
Glucose is a remarkable fuel for cancer cell, and a precursor for
the supply of various metabolic intermediates, which are utilized
for lipid, amino acid and nucleotide synthesis. Glutamine serves as
another important source of fuel in cancer cells (65). Glutamine enters the mitochondria to
replenish the Krebs cycle intermediates (66–69).
Glutamine enters the Krebs cycle to produce α-KG, succinate,
fumarate and malate. Highly proliferative cancer cells have a high
demand for the rapid synthesis of lipids, amino acids and
nucleotides (83–87). Tumor cells also divert carbon from
glycolysis into the PPP (58), by
which cancer cells synthesize macromolecules, such as nucleic
acids. In addition, citrate and acetyl-coA are key intermediates
for lipid synthesis (88–90). Since these metabolic pathways are
interconnected, understanding the mechanism(s) leading to this
metabolic switch in cancer cells is of utmost importance.
Metabolic reprogramming of cancer cells is
recognized as one of the hallmarks of cancer. In this review
article, the core dysregulated metabolic pathways and enzymes
contributing to cancer cell proliferation, differentiation and
metastasis, as well as the central role of mitochondria in
orchestrating metabolic reprogramming were summarized. The close
connection between these metabolic pathways, the role of
mitochondria and redox regulation of tumor cells represents a
promising strategy to target cancer growth. Thus, targeting these
important metabolic enzymes and/or mitochondrial metabolic pathways
may offer a valid and novel anti-cancer therapeutic strategy.
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dang CV: Links between metabolism and
cancer. Genes Dev. 26:877–890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Newmeyer DD and Ferguson-Miller S:
Mitochondria: Releasing power for life and unleashing the
machineries of death. Cell. 112:481–490. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
5
|
Detmer SA and Chan DC: Functions and
dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol.
8:870–879. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: More than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wallace DC: Mitochondria and cancer. Nat
Rev Cancer. 12:685–698. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weinberg SE and Chandel NS: Targeting
mitochondria metabolism for cancer therapy. Nat Chem Biol. 11:9–15.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wen S, Zhu D and Huang P: Targeting cancer
cell mitochondria as a therapeutic approach. Future Med Chem.
5:53–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang F, Ogasawara MA and Huang P: Small
mitochondria-targeting molecules as anti-cancer agents. Mol Aspects
Med. 31:75–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carew JS and Huang P: Mitochondrial
defects in cancer. Mol Cancer. 1:92002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeBerardinis RJ: Is cancer a disease of
abnormal cellular metabolism? New angles on an old idea. Genet Med.
10:767–777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Seyfried TN and Shelton LM: Cancer as a
metabolic disease. Nutr Metab (Lond). 7:72010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pelicano H, Martin DS, Xu RH and Huang P:
Glycolysis inhibition for anticancer treatment. Oncogene.
25:4633–4646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Niederacher D and Entian KD:
Characterization of Hex2 protein, a negative regulatory element
necessary for glucose repression in yeast. FEBS J. 200:311–319.
1991.
|
|
18
|
Herrero P, Galíndez J, Ruiz N,
Martínez-Campa C and Moreno F: Transcriptional regulation of the
Saccharomyces cerevisiae HXK1, HXK2 and GLK1 genes. Yeast.
11:137–144. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rempel A, Mathupala SP, Griffin CA,
Hawkins AL and Pedersen PL: Glucose catabolism in cancer cells:
Amplification of the gene encoding type II hexokinase. Cancer Res.
56:2468–2471. 1996.PubMed/NCBI
|
|
20
|
Bustamante E and Pedersen PL: High aerobic
glycolysis of rat hepatoma cells in culture: Role of mitochondrial
hexokinase. Proc Natl Acad Sci USA. 74:pp. 3735–3739. 1977;
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
El-Bacha T, de Freitas MS and Sola-Penna
M: Cellular distribution of phosphofructokinase activity and
implications to metabolic regulation in human breast cancer. Mol
Genet Metab. 79:294–299. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zancan P, Sola-Penna M, Furtado CM and Da
Silva D: Differential expression of phosphofructokinase-1 isoforms
correlates with the glycolytic efficiency of breast cancer cells.
Mol Genet Metab. 100:372–378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Clem BF, O'Neal J, Tapolsky G, Clem AL,
Imbert-Fernandez Y, Kerr DA II, Klarer AC, Redman R, Miller DM,
Trent JO, et al: Targeting 6-phosphofructo-2-kinase (PFKFB3) as a
therapeutic strategy against cancer. Mol Cancer Ther. 12:1461–1470.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Atsumi T, Chesney J, Metz C, Leng L,
Donnelly S, Makita Z, Mitchell R and Bucala R: High expression of
inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
(iPFK-2; PFKFB3) in human cancers. Cancer Res. 62:5881–5887.
2002.PubMed/NCBI
|
|
25
|
Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ,
Kim JW, Park SW and Kim KS: Androgen stimulates glycolysis for de
novo lipid synthesis by increasing the activities of hexokinase 2
and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in
prostate cancer cells. Biochem J. 433:225–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Okar DA, Manzano A, Navarro-Sabatè A,
Riera L, Bartrons R and Lange AJ: PFK-2/FBPase-2: Maker and breaker
of the essential biofactor fructose-2,6-bisphosphate. Trends
Biochem Sci. 26:30–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li C, Xiao Z, Chen Z, Zhang X, Li J, Wu X,
Li X, Yi H, Li M, Zhu G, et al: Proteome analysis of human lung
squamous carcinoma. Proteomics. 6:547–558. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tokunaga K, Nakamura Y, Sakata K, Fujimori
K, Ohkubo M, Sawada K and Sakiyama S: Enhanced expression of a
glyceraldehyde-3-phosphate dehydrogenase gene in human lung
cancers. Cancer Res. 47:5616–5619. 1987.PubMed/NCBI
|
|
29
|
Schek N, Hall BL and Finn OJ: Increased
glyceraldehyde-3-phosphate dehydrogenase gene expression in human
pancreatic adenocarcinoma. Cancer Res. 48:6354–6359.
1988.PubMed/NCBI
|
|
30
|
Epner DE, Partin AW, Schalken JA, Isaacs
JT and Coffey DS: Association of glyceraldehyde-3-phosphate
dehydrogenase expression with cell motility and metastatic
potential of rat prostatic adenocarcinoma. Cancer Res.
53:1995–1997. 1993.PubMed/NCBI
|
|
31
|
Krasnov GS, Dmitriev AA, Snezhkina AV and
Kudryavtseva AV: Deregulation of glycolysis in cancer:
Glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target.
Expert Opin Ther Targets. 17:681–693. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng C, Gao Y, Wang C, Yu X, Zhang W, Guan
H, Shan Z and Teng W: Aberrant overexpression of pyruvate kinase M2
is associated with aggressive tumor features and the BRAF mutation
in papillary thyroid cancer. J Clin Endocrinol Metab.
98:E1524–E1533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Azoitei N, Becher A, Steinestel K, Rouhi
A, Diepold K, Genze F, Simmet T and Seufferlein T: PKM2 promotes
tumor angiogenesis by regulating HIF-1α through NF-κB activation.
Mol Cancer. 15:32016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu W, Cao Y, Zhang Y, Li S, Gao J, Wang
XA, Mu J, Hu YP, Jiang L, Dong P, et al: Up-regulation of PKM2
promote malignancy and related to adverse prognostic risk factor in
human gallbladder cancer. Sci Rep. 6:263512016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wittwer JA, Robbins D, Wang F, Codarin S,
Shen X, Kevil CG, Huang TT, Van Remmen H, Richardson A and Zhao Y:
Enhancing mitochondrial respiration suppresses tumor promoter
TPA-induced PKM2 expression and cell transformation in skin
epidermal JB6 cells. Cancer Prev Res (Phila). 4:1476–1484. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Le A, Cooper CR, Gouw AM, Dinavahi R,
Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL and Dang
CV: Inhibition of lactate dehydrogenase A induces oxidative stress
and inhibits tumor progression. Proc Natl Acad Sci USA. 107:pp.
2037–2042. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Linnane AW, Marzuki S, Ozawa T and Tanaka
M: Mitochondrial DNA mutations as an important contributor to
ageing and degenerative diseases. Lancet. 1:642–645. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Taylor RW and Turnbull DM: Mitochondrial
DNA mutations in human disease. Nat Rev Genet. 6:389–402. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fliss MS, Usadel H, Caballero OL, Wu L,
Buta MR, Eleff SM, Jen J and Sidransky D: Facile detection of
mitochondrial DNA mutations in tumors and bodily fluids. Science.
287:2017–2019. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cardaci S and Ciriolo MR: TCA cycle
defects and cancer: When metabolism tunes redox state. Int J Cell
Biol. 2012:1618372012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rustin P, Bourgeron T, Parfait B, Chretien
D, Munnich A and Rötig A: Inborn errors of the Krebs cycle: A group
of unusual mitochondrial diseases in human. Biochim Biophys Acta.
1361:185–197. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Singh KK, Desouki MM, Franklin RB and
Costello LC: Mitochondrial aconitase and citrate metabolism in
malignant and nonmalignant human prostate tissues. Mol Cancer.
5:142006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1 and IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dang L, White DW, Gross S, Bennett BD,
Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et
al: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature. 462:739–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Toro JR, Nickerson ML, Wei MH, Warren MB,
Glenn GM, Turner ML, Stewart L, Duray P, Tourre O, Sharma N, et al:
Mutations in the fumarate hydratase gene cause hereditary
leiomyomatosis and renal cell cancer in families in North America.
Am J Hum Genet. 73:95–106. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen YB, Brannon AR, Toubaji A, Dudas ME,
Won HH, Al-Ahmadie HA, Fine SW, Gopalan A, Frizzell N, Voss MH, et
al: Hereditary leiomyomatosis and renal cell carcinoma
syndrome-associated renal cancer: Recognition of the syndrome by
pathologic features and the utility of detecting aberrant
succination by immunohistochemistry. Am J Surg Pathol. 38:627–637.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Frezza C, Zheng L, Folger O, Rajagopalan
KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, Hedley
A, et al: Haem oxygenase is synthetically lethal with the tumour
suppressor fumarate hydratase. Nature. 477:225–228. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gaude E and Frezza C: Defects in
mitochondrial metabolism and cancer. Cancer Metab. 2:102014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Neumann HP, Pawlu C, Pęczkowska M, Bausch
B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA,
et al: European-American Paraganglioma Study Group: Distinct
clinical features of paraganglioma syndromes associated with SDHB
and SDHD gene mutations. JAMA. 292:943–951. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pollard PJ, Wortham NC and Tomlinson IP:
The TCA cycle and tumorigenesis: The examples of fumarate hydratase
and succinate dehydrogenase. Ann Med. 35:632–639. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pollard PJ, Brière JJ, Alam NA, Barwell J,
Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, et al:
Accumulation of Krebs cycle intermediates and over-expression of
HIF1α in tumours which result from germline FH and SDH mutations.
Hum Mol Genet. 14:2231–2239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Habano W, Sugai T, Nakamura S, Uesugi N,
Higuchi T, Terashima M and Horiuchi S: Reduced expression and loss
of heterozygosity of the SDHD gene in colorectal and gastric
cancer. Oncol Rep. 10:1375–1380. 2003.PubMed/NCBI
|
|
54
|
Selak MA, Armour SM, MacKenzie ED,
Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB
and Gottlieb E: Succinate links TCA cycle dysfunction to
oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell.
7:77–85. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Deberardinis RJ, Sayed N, Ditsworth D and
Thompson CB: Brick by brick: Metabolism and tumor cell growth. Curr
Opin Genet Dev. 18:54–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Riganti C, Gazzano E, Polimeni M, Aldieri
E and Ghigo D: The pentose phosphate pathway: An antioxidant
defense and a crossroad in tumor cell fate. Free Radic Biol Med.
53:421–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jiang P, Du W and Wu M: Regulation of the
pentose phosphate pathway in cancer. Protein Cell. 5:592–602. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jonas SK, Benedetto C, Flatman A, Hammond
RH, Micheletti L, Riley C, Riley PA, Spargo DJ, Zonca M and Slater
TF: Increased activity of 6-phosphogluconate dehydrogenase and
glucose-6-phosphate dehydrogenase in purified cell suspensions and
single cells from the uterine cervix in cervical intraepithelial
neoplasia. Br J Cancer. 66:185–191. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lucarelli G, Galleggiante V, Rutigliano M,
Sanguedolce F, Cagiano S, Bufo P, Lastilla G, Maiorano E, Ribatti
D, Giglio A, et al: Metabolomic profile of glycolysis and the
pentose phosphate pathway identifies the central role of
glucose-6-phosphate dehydrogenase in clear cell-renal cell
carcinoma. Oncotarget. 6:13371–13386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
D'Alessandro A, Amelio I, Berkers CR,
Antonov A, Vousden KH, Melino G and Zolla L: Metabolic effect of
TAp63α: Enhanced glycolysis and pentose phosphate pathway,
resulting in increased antioxidant defense. Oncotarget.
5:7722–7733. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sukhatme VP and Chan B: Glycolytic cancer
cells lacking 6-phosphogluconate dehydrogenase metabolize glucose
to induce senescence. FEBS Lett. 586:2389–2395. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Nishimura M and Uyeda K: Purification and
characterization of a novel xylulose 5-phosphate-activated protein
phosphatase catalyzing dephosphorylation of
fructose-6-phosphate,2-kinase:fructose-2,6-bisphosphatase. J Biol
Chem. 270:26341–26346. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wise DR and Thompson CB: Glutamine
addiction: A new therapeutic target in cancer. Trends Biochem Sci.
35:427–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
DeBerardinis RJ and Cheng T: Q's next: The
diverse functions of glutamine in metabolism, cell biology and
cancer. Oncogene. 29:313–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dang CV: Glutaminolysis: Supplying carbon
or nitrogen or both for cancer cells? Cell Cycle. 9:3884–3886.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:619–634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hensley CT, Wasti AT and DeBerardinis RJ:
Glutamine and cancer: Cell biology, physiology, and clinical
opportunities. J Clin Invest. 123:3678–3684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB, et al: Myc regulates a transcriptional program that stimulates
mitochondrial glutaminolysis and leads to glutamine addiction. Proc
Natl Acad Sci USA. 105:pp. 18782–18787. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Stepulak A, Luksch H, Gebhardt C,
Uckermann O, Marzahn J, Sifringer M, Rzeski W, Staufner C, Brocke
KS, Turski L, et al: Expression of glutamate receptor subunits in
human cancers. Histochem Cell Biol. 132:435–445. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Durán RV, Oppliger W, Robitaille AM,
Heiserich L, Skendaj R, Gottlieb E and Hall MN: Glutaminolysis
activates Rag-mTORC1 signaling. Mol Cell. 47:349–358. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jain M, Nilsson R, Sharma S, Madhusudhan
N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB and Mootha
VK: Metabolite profiling identifies a key role for glycine in rapid
cancer cell proliferation. Science. 336:1040–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Amelio I, Cutruzzolá F, Antonov A,
Agostini M and Melino G: Serine and glycine metabolism in cancer.
Trends Biochem Sci. 39:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hasegawa S, Ichiyama T, Sonaka I, Ohsaki
A, Okada S, Wakiguchi H, Kudo K, Kittaka S, Hara M and Furukawa S:
Cysteine, histidine and glycine exhibit anti-inflammatory effects
in human coronary arterial endothelial cells. Clin Exp Immunol.
167:269–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Alarcon-Aguilar FJ, Almanza-Perez J,
Blancas G, Angeles S, Garcia-Macedo R, Roman R and Cruz M: Glycine
regulates the production of pro-inflammatory cytokines in lean and
monosodium glutamate-obese mice. Eur J Pharmacol. 599:152–158.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cruz M, Maldonado-Bernal C,
Mondragón-Gonzalez R, Sanchez-Barrera R, Wacher NH,
Carvajal-Sandoval G and Kumate J: Glycine treatment decreases
proinflammatory cytokines and increases interferon-γ in patients
with type 2 diabetes. J Endocrinol Invest. 31:694–699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhang WC, Shyh-Chang N, Yang H, Rai A,
Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, et al: Glycine
decarboxylase activity drives non-small cell lung cancer
tumor-initiating cells and tumorigenesis. Cell. 148:259–272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Locasale JW: Serine, glycine and
one-carbon units: Cancer metabolism in full circle. Nat Rev Cancer.
13:572–583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Possemato R, Marks KM, Shaul YD, Pacold
ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et
al: Functional genomics reveal that the serine synthesis pathway is
essential in breast cancer. Nature. 476:346–350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Locasale JW, Grassian AR, Melman T,
Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen
T, Sharfi H, et al: Phosphoglycerate dehydrogenase diverts
glycolytic flux and contributes to oncogenesis. Nat Genet.
43:869–874. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mattaini KR, Sullivan MR and Vander Heiden
MG: The importance of serine metabolism in cancer. J Cell Biol.
214:249–257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Baenke F, Peck B, Miess H and Schulze A:
Hooked on fat: The role of lipid synthesis in cancer metabolism and
tumour development. Dis Model Mech. 6:1353–1363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Santos CR and Schulze A: Lipid metabolism
in cancer. FEBS J. 279:2610–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Currie E, Schulze A, Zechner R, Walther TC
and Farese RV Jr: Cellular fatty acid metabolism and cancer. Cell
Metab. 18:153–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Vance JE and Vance DE: Biochemistry of
lipids, lipoproteins and membranes. Elsevier; Amsterdam: 2002,
View Article : Google Scholar
|
|
87
|
Bauer DE, Hatzivassiliou G, Zhao F,
Andreadis C and Thompson CB: ATP citrate lyase is an important
component of cell growth and transformation. Oncogene.
24:6314–6322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Qian X, Hu J, Zhao J and Chen H: ATP
citrate lyase expression is associated with advanced stage and
prognosis in gastric adenocarcinoma. Int J Clin Exp Med.
8:7855–7860. 2015.PubMed/NCBI
|
|
89
|
Xin M, Qiao Z, Li J, Liu J, Song S, Zhao
X, Miao P, Tang T, Wang L, Liu W, et al: miR-22 inhibits tumor
growth and metastasis by targeting ATP citrate lyase: Evidence in
osteosarcoma, prostate cancer, cervical cancer and lung cancer.
Oncotarget. 7:44252–44265. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lucenay KS, Doostan I, Karakas C, Bui T,
Ding Z, Mills GB, Hunt KK and Keyomarsi K: Cyclin E associates with
the lipogenic enzyme ATP-citrate lyase to enable malignant growth
of breast cancer cells. Cancer Res. 76:2406–2418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Su YW, Lin YH, Pai MH, Lo AC, Lee YC, Fang
IC, Lin J, Hsieh RK, Chang YF and Chen CL: Association between
phosphorylated AMP-activated protein kinase and acetyl-CoA
carboxylase expression and outcome in patients with squamous cell
carcinoma of the head and neck. PLoS One. 9:e961832014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang MD, Wu H, Fu GB, Zhang HL, Zhou X,
Tang L, Dong LW, Qin CJ, Huang S, Zhao LH, et al: Acetyl-coenzyme A
carboxylase alpha promotion of glucose-mediated fatty acid
synthesis enhances survival of hepatocellular carcinoma in mice and
patients. Hepatology. 63:1272–1286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bauerschlag DO, Maass N, Leonhardt P,
Verburg FA, Pecks U, Zeppernick F, Morgenroth A, Mottaghy FM, Tolba
R, Meinhold-Heerlein I, et al: Fatty acid synthase overexpression:
Target for therapy and reversal of chemoresistance in ovarian
cancer. J Transl Med. 13:1462015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ogino S, Kawasaki T, Ogawa A, Kirkner GJ,
Loda M and Fuchs CS: Fatty acid synthase overexpression in
colorectal cancer is associated with microsatellite instability,
independent of CpG island methylator phenotype. Hum Pathol.
38:842–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gong J, Shen S, Yang Y, Qin S, Huang L,
Zhang H, Chen L, Chen Y, Li S, She S, et al: Inhibition of FASN
suppresses migration, invasion and growth in hepatoma carcinoma
cells by deregulating the HIF-1α/IGFBP1 pathway. Int J Oncol.
50:883–892. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Carracedo A, Cantley LC and Pandolfi PP:
Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev
Cancer. 13:227–232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ito K and Suda T: Metabolic requirements
for the maintenance of self-renewing stem cells. Nat Rev Mol Cell
Biol. 15:243–256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zaugg K, Yao Y, Reilly PT, Kannan K,
Kiarash R, Mason J, Huang P, Sawyer SK, Fuerth B, Faubert B, et al:
Carnitine palmitoyltransferase 1C promotes cell survival and tumor
growth under conditions of metabolic stress. Genes Dev.
25:1041–1051. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
McGarry JD and Brown NF: The mitochondrial
carnitine palmitoyltransferase system. From concept to molecular
analysis. Eur J Biochem. 244:1–14. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Coller HA: Is cancer a metabolic disease?
Am J Pathol. 184:4–17. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Tan DJ, Bai RK and Wong LJ: Comprehensive
scanning of somatic mitochondrial DNA mutations in breast cancer.
Cancer Res. 62:972–976. 2002.PubMed/NCBI
|
|
102
|
Liu VW, Shi HH, Cheung AN, Chiu PM, Leung
TW, Nagley P, Wong LC and Ngan HY: High incidence of somatic
mitochondrial DNA mutations in human ovarian carcinomas. Cancer
Res. 61:5998–6001. 2001.PubMed/NCBI
|
|
103
|
Richard SM, Bailliet G, Páez GL, Bianchi
MS, Peltomäki P and Bianchi NO: Nuclear and mitochondrial genome
instability in human breast cancer. Cancer Res. 60:4231–4237.
2000.PubMed/NCBI
|
|
104
|
Ishikawa K, Takenaga K, Akimoto M,
Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y and Hayashi
J: ROS-generating mitochondrial DNA mutations can regulate tumor
cell metastasis. Science. 320:661–664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Swalwell H, Kirby DM, Blakely EL, Mitchell
A, Salemi R, Sugiana C, Compton AG, Tucker EJ, Ke BX, Lamont PJ, et
al: Respiratory chain complex I deficiency caused by mitochondrial
DNA mutations. Eur J Hum Genet. 19:769–775. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kwong JQ, Henning MS, Starkov AA and
Manfredi G: The mitochondrial respiratory chain is a modulator of
apoptosis. J Cell Biol. 179:1163–1177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Osellame LD, Blacker TS and Duchen MR:
Cellular and molecular mechanisms of mitochondrial function. Best
Pract Res Clin Endocrinol Metab. 26:711–723. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Shen YH, Wang XL and Wilcken DE: Nitric
oxide induces and inhibits apoptosis through different pathways.
FEBS Lett. 433:125–131. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Seiler N and Raul F: Polyamines and
apoptosis. J Cell Mol Med. 9:623–642. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Agostinelli E, Tempera G, Molinari A,
Salvi M, Battaglia V, Toninello A and Arancia G: The physiological
role of biogenic amines redox reactions in mitochondria. New
perspectives in cancer therapy. Amino Acids. 33:175–187. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Grancara S, Ohkubo S, Artico M,
Ciccariello M, Manente S, Bragadin M, Toninello A and Agostinelli
E: Milestones and recent discoveries on cell death mediated by
mitochondria and their interactions with biologically active
amines. Amino Acids. 48:2313–2326. 2016. View Article : Google Scholar : PubMed/NCBI
|