Introduction
Abnormally high levels of iron have been reported in
the brain regions of patients with various neurodegenerative
diseases including Parkinson's disease (PD) and Alzheimer's disease
(AD) (1,2). In these diseases, progressive iron
accumulation has also been observed in activated microglia, as the
brain resident immune cells (3–5). Increased
cellular iron levels in activated microglia has been demonstrated
to enhance the release of cytokines and free radicals, which are
considered to participate in neuronal cell death (6). Therefore, increased understanding of the
interaction between microglia and candidate neuroprotective agents
under pathology-related, iron-rich conditions may aid the
development of more effective treatments for neurodegenerative
diseases.
A number of previous studies have demonstrated that
valproic acid (VPA), an anti-convulsant and mood-stabilizing drug,
was neuroprotective in cell culture and animal models of
neurodegenerative diseases (7–11). VPA has
been demonstrated to exert anti-inflammatory effects by decreasing
the expression of inflammatory and innate immune response genes in
human microglia and astrocytes (10,12,13).
Further research has indicated that VPA enhances microglial
phagocytosis of amyloid β1-42, the toxic protein fragment that
accumulates in the brain during AD (14). However, to the best of our knowledge,
it has not yet been investigated how VPA interacts with microglia
under pathology-related, iron-rich conditions. The present study
aimed to elucidate this interaction. In particular, it was
determined how VPA affects the production of nitric oxide (NO) and
interleukin 1β (IL-1β), as well as the transcription levels of
inducible NO synthase (iNOS) and IL-1β, using the established mouse
microglial cell line BV2. The effect of VPA on NO and IL-1β
production was the focus, as high concentrations of NO and IL-1β
released by activated microglia following a pathologic insult may
lead to neurotoxicity and has been reported in many
neurodegenerative diseases (15–19).
Furthermore, as it has been reported that the inducible nuclear
factor-κB (NF-κB) serves a critical role in regulating the
expression of inflammatory mediators and inflammatory cytokines
(20), including iNOS and IL-1β, the
effect of VPA on NF-κB nuclear translocation during microglial
activation under iron-rich conditions was also determined.
Materials and methods
Cell culture and treatment
Murine BV-2 microglial cells were obtained from
Professor James R. Connor (Pennsylvania State University, PA, USA).
The cells were maintained in Dulbecco's modified Eagle's medium
(DMEM) containing 5% fetal bovine serum (FBS), 2 mM L-glutamine,
100 µg/ml streptomycin and 100 U/ml penicillin (all from Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C under
humidified 95% O2 and 5% CO2. The cells were
seeded into 24-well plates at a density of 1×105/well or
in 96-well plates at a density of 5×104/well and
maintained at 37°C under humidified 95% O2 and 5%
CO2. After ~24 h, the medium was removed and replaced
with freshly prepared serum-free medium containing specified
concentrations of VPA (0, 0.8,1.6 and 3.2 mM) for a cell viability
assay. To detect intracellular iron, the medium was removed and
replaced with freshly prepared serum-free medium containing iron
(300 µg/ml) with or without lipopolysaccharide (LPS; 1 µg/ml). To
detect the gene expression of iNOS and IL-1β, as well as the
production of NO and IL-1β, the medium was removed and replaced
with freshly prepared serum-free medium with or without LPS (1
µg/ml) and/or iron (300 µg/ml) and/or a non-toxic dose of VPA.
Detection of intracellular iron
The form of iron used to treat the cells in the
present study was ferric citrate ammonium (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). Levels of intracellular iron were
measured using calcein acetoxymethyl ester (calcein-AM) (21,22). This
reagent contains AM ester derivatives of fluorescent indicators,
and its lipophilic structure is able to permeate cell membranes.
Inside the cell, nonspecific cytosolic esterases cleave the
lipophilic structure rapidly (23).
The fluorescence intensity of the calcein may be quenched by iron
binding. Thus, decreased fluorescence intensity indicates increased
intracellular iron levels (23–26). BV-2
cells (1×104 cells per well, 96-well plates) were
treated with LPS with or without iron in serum-free medium for 4
and 24 h. The BV-2 cells were incubated with 1 µM calcein AM at
37°C for 30 min, after which, excess calcein was removed by washing
with phosphate-buffered saline (PBS). The fluorescence intensity of
calcein was measured at 485 nm excitation and 538 nm emission on a
Synergy 4 HT Multi-Mode Microplate Reader (BioTek Instruments,
Winooski, VT, USA). Meanwhile, total protein concentration was
determined using a Pierce Bicinchoninic Acid Protein Assay kit
(Pierce, Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol; the fluorescence data were presented as
fluorescent intensity units per unit protein.
Cell viability assay
The cytotoxicity of VPA and iron were evaluated with
an MTT assay. BV2 cells were seeded in 96-well plates and cultured
as described above. After 24 h, the cells were exposed to VPA at
the concentrations between 0–3.2 mM or iron at the concentrations
0, 50, 100 and 300 µg/ml. After 24 h of treatment, the medium was
removed and replaced with 0.1 ml MTT reagent (0.5 mg/ml;
Sigma-Aldrich; Merck KGaA) in serum-free DMEM. The cells were then
incubated for 2 h at 37°C and 5% CO2. Following
incubation, the supernatant was removed, the formazan dye was
dissolved with dimethyl sulfoxide, and absorption at 570 nm was
measured using the Synergy 4 HT Multi-Mode Microplate Reader.
Reverse transcription polymerase chain
reaction (RT-PCR) analysis
BV-2 microglia (5×105 cells/well) were
cultured and treated as described above. After 6 h of treatment,
total RNA was extracted using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
Total RNA (1 µg) from each sample was added into a reaction mixture
containing 10× reaction buffer (2 µl), 25 mM MgCl2 (4
µl), dNTPs (2 µl), random primer (2 µl), ribonuclease inhibitor (1
µl), AMV reverse transcriptase (1 µl) and RNase free water (8 µl;
all from Promega Corporation, Madison, WI, USA). Each sample was
incubated at room temperature for 10 min, then at 42°C for 60 min,
followed by inactivation at 99°C for 5 min. To amplify the 311-bp
iNOS, 200-bp IL-1β and 300-bp GAPDH (reference) cDNA fragments, the
following primer sequences were used: For iNOS, forward,
5′-ATCCCGAAACGCTACACTTCC-3′ and reverse,
5′-GGCGAAGAACAATCCACAACTC-3′; for IL-1β, forward,
5′-GCTATGGCAACTGTCCCTGAAC-3′ and reverse,
5′-TGAGTGACACTGCCTTCCTCCTGAA-3′; and for GAPDH, forward,
5′-AAGCTCACTGGCATGGCCTTCC-3′ and reverse,
5′-TTGGAGGCCATGTAGGCCATGAG-3′. The cDNAs were amplified in a final
reaction volume of 25 µl containing 10× reaction buffer (2.5 µl),
25 mM MgCl2 (1.5 µl), dNTPs (0.5 µl), Taq DNA polymerase
(0.2 µl), forward and reverse primers (0.7 µl each), cDNA (5.0 µl)
and RNase free water (13.9 µl; all from Promega Corporation) under
the following conditions: Pre-denaturation at 95°C for 4 min
followed by 32 cycles of 95°C for 1 min, 60°C for 1 min, 72°C for 2
min, and a final extension at 72°C for 4 min. The PCR products were
visualized by electrophoresis on 1.5% agarose gel, followed by
staining with ethidium bromide. The amplification of specific genes
was verified by comparing their predicted size and their actual
size under ultraviolet (UV) light. Quantitative analysis of band
density was performed using Scion Image analysis software, version
4.02 (Scion Corporation, Frederick, MD, USA).
Nitrite assay
BV-2 microglia (5×105 cells/well) were
cultured and treated as described above. After 24 h of treatment,
the quantity of NO in the supernatant was estimated by measuring
accumulation of the stable NO metabolite nitrite
(NO2−) with a Griess Reagent kit (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. In brief, following treatment, 150 µl of cell culture
medium was collected and added to a 96-well plate, to which 20 µl
Griess reagent was added followed by 130 µl DMEM. The sample
reactions were incubated at room temperature for 30 min. Following
incubation, absorbance values were measured at 548 nm using the
Synergy 4 HT Multi-Mode Microplate Reader microplate reader. The
quantity of NO generated by the cells was calculated based on
concentrations of an NaNO2- standard curve.
Enzyme-linked immunosorbent assay
(ELISA)
BV-2 microglia (5×105 cells/well) were
treated as described above. After 24 h of treatment, the cell
culture medium was centrifuged for 10 min at 3,000 × g and the
supernatant was collected for quantification. The concentration of
IL-1β in the culture supernatants was measured using an ELISA kits
from R&D Systems, Inc. (Minneapolis, MN, USA; cat. no. MLB00C)
according to the manufacturer's protocol. The results were
expressed in pg/ml.
Measurement of NF-κB p65 nuclear
translocation
Immunofluorescence assay
BV-2 microglia were seeded on 6-chamber slides at a
density of 2×104 cells/chamber and allowed to grow for
24 h in growth medium at 37°C under humidified 95% O2
and 5% CO2. The medium was removed and replaced with
freshly prepared serum-free medium with or without LPS (1 μg/ml)
and/or iron (300 µg/ml) and/or a non-toxic dose of VPA. After 4 h
of treatment, the medium was removed and the cells were fixed with
4% paraformaldehyde at 4°C for 5 min, washed with PBS and
permeabilized with 100% methanol for 5 min at −20°C. To stain the
p65 subunit of NF-κB, the cells were washed with PBS and incubated
in PBS containing 1% bovine serum albumin (BSA; Sigma-Aldrich;
Merck KGaA) and rabbit anti-NF-κB p65 antibody (cat. no.
STCSC-33020; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at
1:100 dilution for 1 h at room temperature. Following a PBS wash,
the cells were incubated at room temperature for an additional 1 h
with Alexa-Fluor 488-conjugated goat anti-rabbit secondary antibody
(cat. no. A27034; Invitrogen; Thermo Fisher Scientific, Inc.) at
1:1,000 dilution in PBS containing 1% BSA. Following another PBS
wash, the cell nuclei were stained with DAPI for 10 min at room
temperature. Finally, the cover slips were mounted with 50%
glycerol and examined with a Leica confocal microscope (Leica
Microsystems, Inc., Buffalo Grove, IL, USA).
NF-κB ELISA assay
BV-2 microglia were seeded in 6-well plates at a
density of 5×105cells/well. The cells were cultured and
treatment as described above. After 4 h of treatment, the medium
was removed and the cells were washed twice with ice-cold PBS. The
cells were scraped off with a rubber policeman cell scraper and
centrifuged at 200 × g for 10 min at 4°C. To prepare nuclear
extracts, cells were resuspended in a buffer containing 10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.9),
1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol and 0.2
mM phenylmethylsulfonyl fluoride (PMSF), which was followed by
vortexing for 15 sec prior to standing at 4°C for 12 min. The
samples were then centrifuged at 200 × g for 3 min at 4°C. The
pelleted nuclei were resuspended in 30 µl buffer containing 20 mM
HEPES (pH 7.9), 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2,
0.2 mM EDTA, 0.5 mM dithiothreitol and 0.2 mM PMSF and incubated
for 20 min on ice, prior to centrifugation of the nuclear lysates
at 10,000 × g for 3 min at 4°C. Supernatants containing the
solubilized nuclear proteins were collected, and the quantity of
NF-κB in the nuclear fractions was measured with a NF-κB p65 ELISA
kit, (cat. no. ab176647; Abcam, Cambridge, UK) according the
manufacturer's instructions.
Statistical analysis
All results were expressed as the mean ± standard
error of the mean from at least three independent experiments
performed in triplicate. Multiple comparisons of data were
performed using by one-way analysis of variance followed by
Bonferroni post-hoc tests. The analyses were conducted using
SigmaStat software version 3.5 (Systat Software, Inc., San Jose,
CA, USA). P<0.05 was considered to indicate statistical
significance.
Results
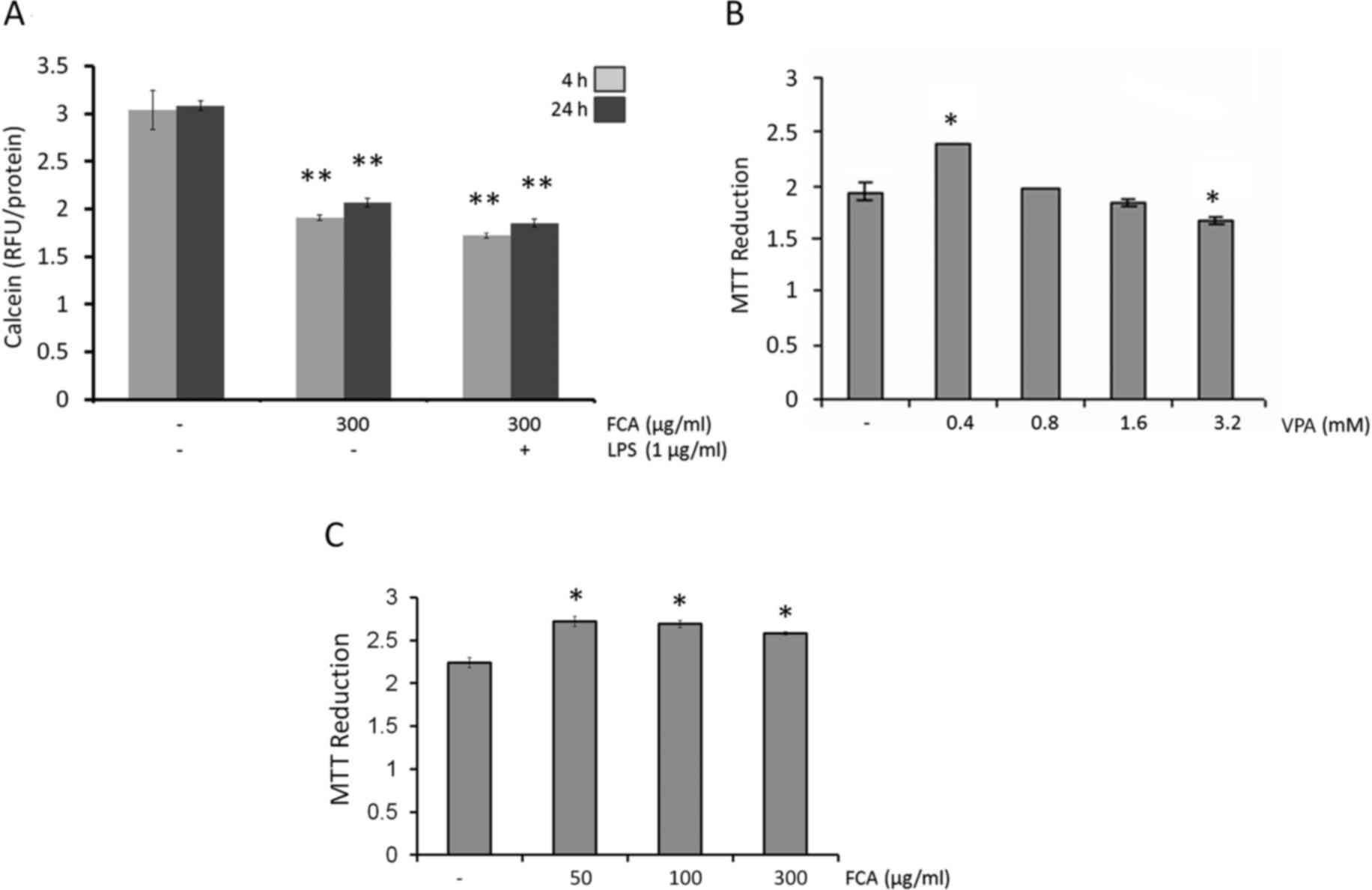
Iron-rich microglial cells
The present study first aimed to mimic the
intracellular iron loading of activated microglial cells observed
in neurodegenerative diseases. The levels of intracellular iron
were measured using calcein AM, taking decreased fluorescence
intensity as indication of increased intracellular iron levels. As
depicted in Fig. 1A, intracellular
iron was detected in untreated cells at 4 and 24 h. These
intracellular iron levels were increased at 4 h and 24 h on
treatment with iron (300 µg/ml; both P<0.05). If the cells were
supplemented with iron during LPS treatment, intracellular iron
levels increased marginally above the levels observed for iron
treatment alone. These results demonstrated that the addition of
iron to the microglial medium is sufficient to cause intracellular
iron accumulation in microglial cells.
Effects of VPA and iron on the
viability of BV2 cells
To determine the potential cytotoxic effect of VPA
and iron on BV2 microglial cells, the cells were treated with
various concentrations (0–3.2 mM) of VPA or iron (0–300 µg/ml).
After 24 h of treatment, cell viability was evaluated by MTT assay.
The results indicted that VPA at concentrations up to 1.6 mM
(Fig. 1B) and iron at concentrations
up to 300 µg/ml (Fig. 1C) were
non-toxic against BV2 cells with respect to the untreated controls.
Therefore, the highest non-toxic concentration of VPA (1.6 mM) and
300 µg/ml iron were used in subsequent assays.
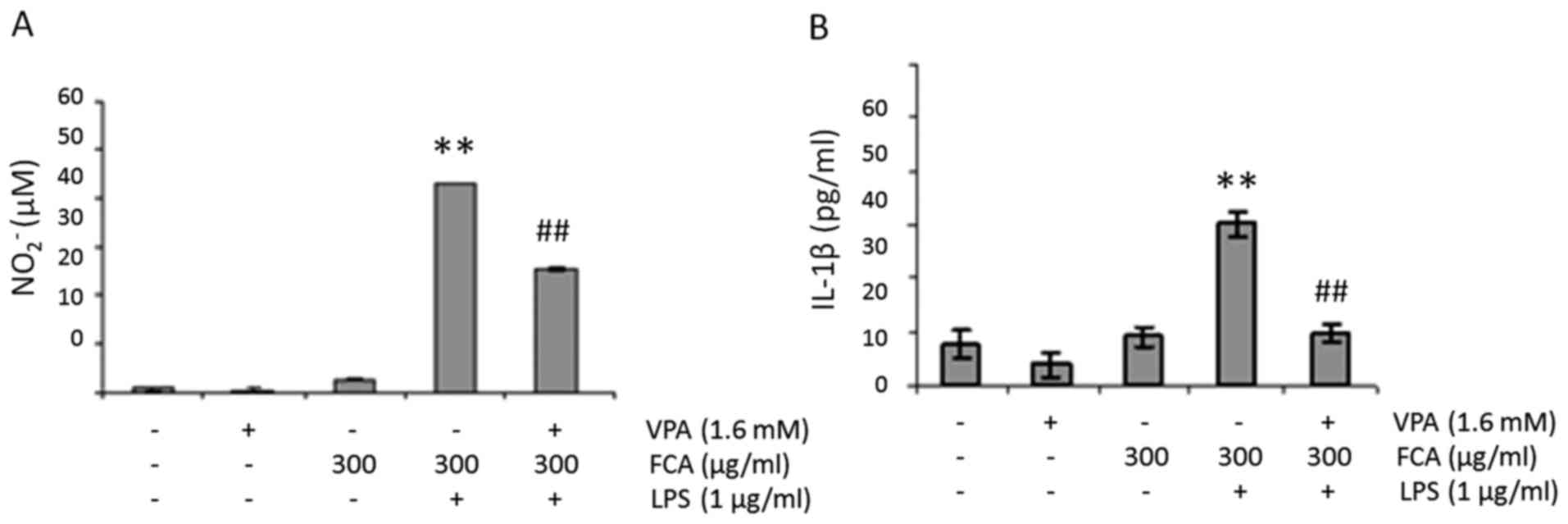
VPA attenuates NO and IL-1β production
in iron-rich activated microglia
To determine the effect of VPA on NO production in
iron-rich activated microglia, BV2 cells were challenged with 1
µg/ml LPS in the presence or absence of 300 µg/ml iron and/or 1.6
mM VPA for 24 h. NO2− levels in the cell
culture media were subsequently determined. As presented in
Fig. 2A, the addition of iron or VPA
had no effect on baseline NO2− level.
However, when cells were treated with LPS in the presence of iron,
NO2− production was significantly increased
when compared with that of the untreated controls (P<0.01). More
notably, when the cells were treated with LPS and iron in the
presence of VPA, NO2− levels were
significantly reduced when compared with LPS + iron treatment alone
(P<0.01).
Similar to NO production, when cells were treated
with LPS in the presence of iron, IL-1β levels significantly
increased compared with that in the untreated controls (P<0.01);
IL-1β levels also significantly decreased when the cells were
treated with LPS and iron in the presence of VPA when compared with
LPS + iron treatment alone (P<0.01; Fig. 2B).
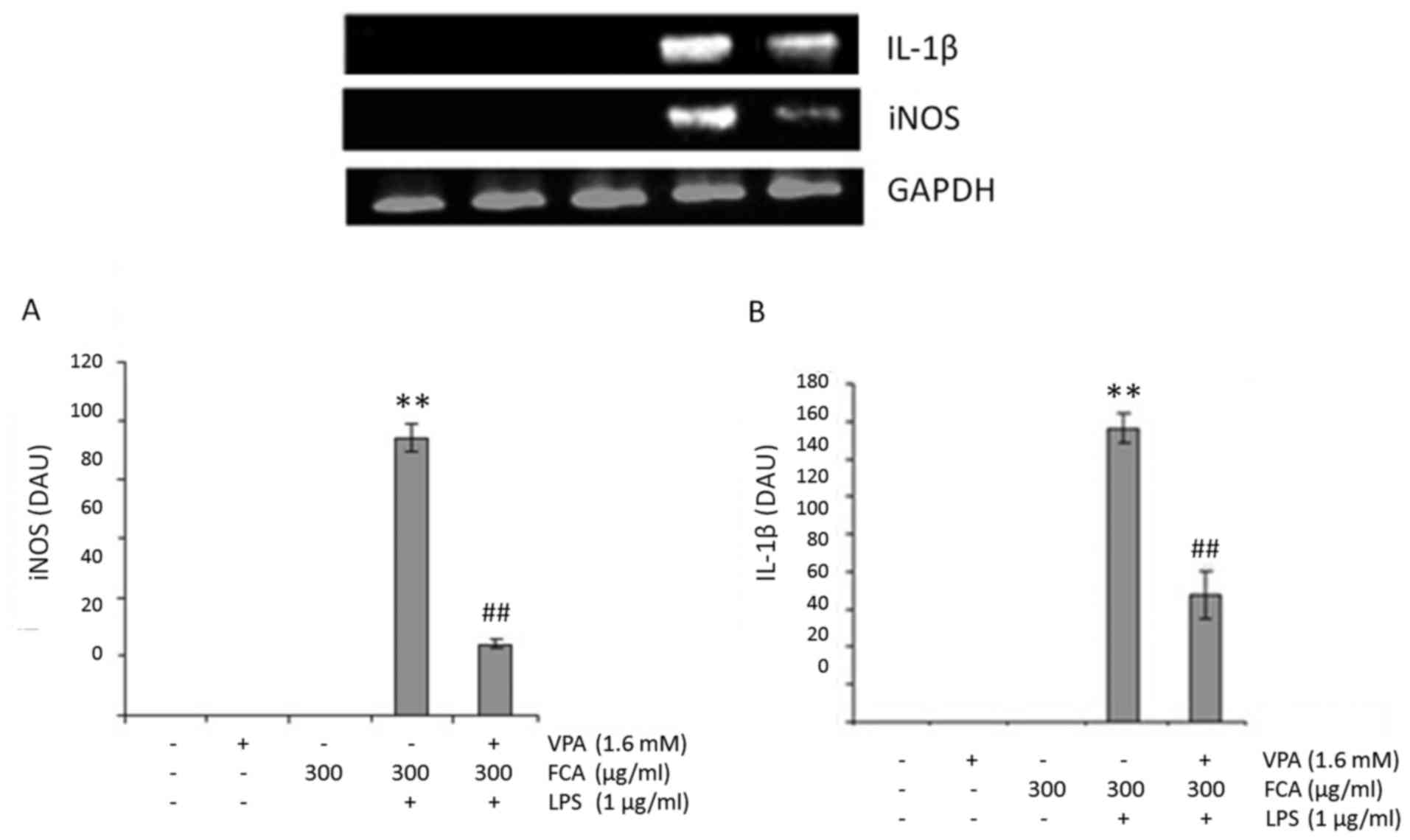
VPA attenuates gene expression of iNOS
and IL-1β in iron-rich activated microglia
To determine if VPA affects microglial NO and IL-1β
production at the transcription level, iNOS and IL-1β mRNA
expression in BV2 cells was assessed at 6 h under the conditions
indicated in Fig. 3A and B,
respectively. Treatment with VPA was identified to significantly
decrease the mRNA expression of iNOS and IL-1β in LPS-treated cells
in the presence of iron, with respect to LPS + iron treatment alone
(both P<0.01). These results suggest that VPA inhibits
NO2− and IL-1β production by downregulating
iNOS and IL-1β mRNA expression respectively.
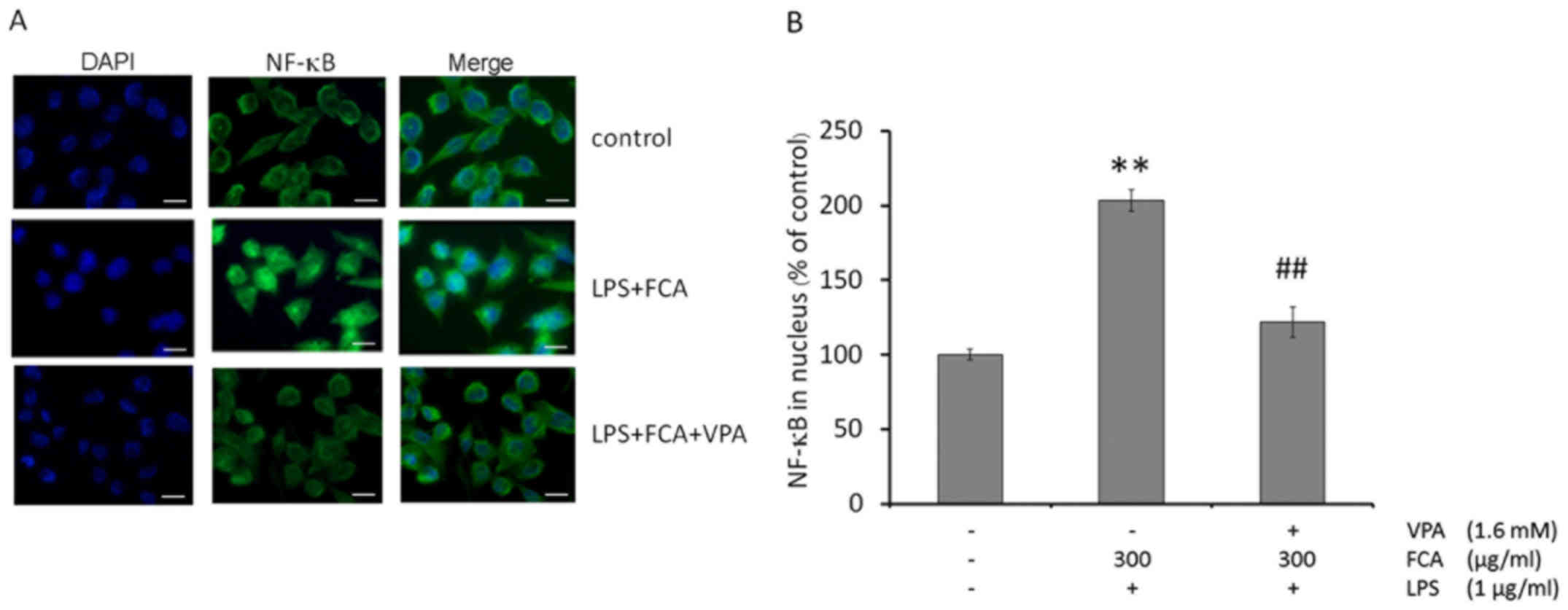
Effects of VPA on NF-κB nuclear
translocation in iron-rich activated microglia
To determine the effect of VPA on NF-κB nuclear
translocation in iron-rich activated microglia, levels of the NF-κB
p65 subunit in the nuclei of activated BV-2 microglia were assessed
by immunofluorescence microscopy. As depicted in Fig. 4A, NF-κB p65 was mainly localized to
the cytoplasm of untreated cells. Exposing BV-2 microglia to LPS
with iron increased the nuclear localization of NF-κB p65; while
treatment with VPA blocked NF-κB nuclear translocation in the
iron-rich activated microglia.
Consistent with the results from the
immunofluorescence assay, quantification of nuclear NF-κB p65 by
ELISA demonstrated that the treatment of BV-2 microglia with LPS
and iron significantly increased the nuclear localization of NF-κB
p65, compared with that in untreated cells (P<0.01); however,
VPA treatment significantly reduced NF-κB nuclear localization in
the iron-rich activated microglia (P<0.01; Fig. 4B).
Discussion
Dysregulation of iron homeostasis leads to the
production of neurotoxic substances and reactive oxygen species,
resulting in iron-induced oxidative stress (4). Elevated levels of iron is a pathological
hallmark of AD, a disease in which iron accumulates abnormally in
microglia (3,5). Although little is known about the role
of iron in microglial function, a previous study by our group
demonstrated that the addition of iron to cultures of LPS-activated
microglia lead to intracellular iron accumulation and alteration of
gene expression and function (6). In
the present study, an in vitro model of iron-loaded
activated microglia was established using the mouse microglial cell
line BV2. The concentration of iron used in this study was
approximately the concentration of iron observed in amyloid plaques
in AD (27). This concentration was
not toxic to the cells, as determined from the cell viability
assay. Furthermore, the addition of iron to the medium, in the
presence of LPS, was sufficient to generate iron-rich activated
microglia resembling the iron-laden phenotype of activated
microglia in many neurodegenerative diseases (5,6). With this
model, it was demonstrated that NO and IL-1β production, as well as
the transcript levels of iNOS and IL-1β, were significantly
increased compared with untreated control levels. Notably, with
this model it was also demonstrated that VPA treatment decreased NO
and IL-1β production by decreasing the mRNA expression of iNOS and
IL-1β in LPS-activated microglial cells under iron-rich conditions.
NF-κB is among the transcription factors that serve a critical role
in regulating inflammatory mediators and inflammatory cytokines
(20). Under normal physiological
conditions, NF-κB is localized in the cytosol, bound by members of
the IκB family of inhibitory proteins (28). On stimulation with specific inducers,
including LPS or TNF-α, the IκB kinase complex is activated, which
in turn phosphorylates IκB, triggering its degradation by the
proteasome and allowing free NF-κB to translocate to the nucleus
and activate gene expression of proinflammatory genes including
iNOS and IL-1β (20,29–31). In
the present study, it was demonstrated that microglial activation
under iron-rich conditions increased the level of NF-κB nuclear
translocation, which was suppressed following VPA treatment.
However, the mechanisms by which VPA reduced the level of NF-κB
nuclear translocation under iron-rich conditions were not
determined, and require investigation in future studies.
Furthermore, the lack of an LPS group was a limitation, as there
was no comparison with activated microglia alone as untreated
controls.
In conclusion, the present results indicate that VPA
treatment decreases NO and IL-1β production and iNOS and IL-1β mRNA
expression by suppressing NF-κB nuclear translocation in
LPS-activated microglia under iron-rich conditions. These findings
suggest that VPA may be used as a therapy for inhibiting the
inflammation that occurs under the iron-rich conditions observed in
neurodegenerative diseases such as PD and AD.
Acknowledgements
The authors are thankful to Dr Tim Cushnie at the
Faculty of Medicine at Mahasarakham University (Maha Sarakham,
Thailand) for the language editing assistance.
Funding
The current study was supported by grants from the
Chulalongkorn University Faculty of Medicine (Bangkok, Thailand)
and Mahasarakham University Faculty of Medicine.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
NM and PC were responsible for conception and design
of the study, data analysis and interpretation, and revision of the
manuscript. NM was responsible for acquisition of data and drafting
of the manuscript. The final version of the manuscript has been
read and approved by both authors.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wallis LI, Paley MN, Graham JM, Grünewald
RA, Wignall EL, Joy HM and Griffiths PD: MRI assessment of basal
ganglia iron deposition in Parkinson's disease. J Magn Reson
Imaging. 28:1061–1067. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
House MJ, St Pierre TG, Foster JK, Martins
RN and Clarnette R: Quantitative MR imaging R2 relaxometry in
elderly participants reporting memory loss. AJNR Am J Neuroradiol.
27:430–439. 2006.PubMed/NCBI
|
|
3
|
Connor JR, Menzies SL, St Martin SM and
Mufson EJ: A histochemical study of iron, transferrin, and ferritin
in Alzheimer's diseased brains. J Neurosci Res. 31:75–83. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zecca L, Youdim MB, Riederer P, Connor JR
and Crichton RR: Iron, brain ageing and neurodegenerative
disorders. Nat Rev Neurosci. 5:863–873. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Duijn S, Bulk M, van Duinen SG,
Nabuurs RJA, van Buchem MA, van der Weerd L and Natté R: Cortical
Iron Reflects Severity of Alzheimer's Disease. J Alzheimers Dis.
60:1533–1545. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mairuae N, Connor JR and Cheepsunthorn P:
Increased cellular iron levels affect matrix metalloproteinase
expression and phagocytosis in activated microglia. Neurosci Lett.
500:36–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kanai H, Sawa A, Chen RW, Leeds P and
Chuang DM: Valproic acid inhibits histone deacetylase activity and
suppresses excitotoxicity-induced GAPDH nuclear accumulation and
apoptotic death in neurons. Pharmacogenomics J. 4:336–344. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li R and El-Mallahk RS: A novel evidence
of different mechanisms of lithium and valproate neuroprotective
action on human SY5Y neuroblastoma cells: Caspase-3 dependency.
Neurosci Lett. 294:147–150. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mora A, Gonzalez Polo RA, Fuentes JM,
Soler G and Centeno F: Different mechanisms of protection against
apoptosis by valproate and Li+. Eur J Biochem. 266:886–891. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ximenes JC, Neves KR, Leal LK, do Carmo
MR, Brito GA, Naffah-Mazzacoratti MG, Cavalheiro ÉA and Viana GS:
Valproic Acid Neuroprotection in the 6-OHDA Model of Parkinson's
Disease Is Possibly Related to Its Anti-Inflammatory and HDAC
Inhibitory Properties. J Neurodegener Dis.
2015:3137022015.PubMed/NCBI
|
|
11
|
Xuan AG, Pan XB, Wei P, Ji WD, Zhang WJ,
Liu JH, Hong LP, Chen WL and Long DH: Valproic acid alleviates
memory deficits and attenuates amyloid-β deposition in transgenic
mouse model of Alzheimer's disease. Mol Neurobiol. 51:300–312.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suh HS, Choi S, Khattar P, Choi N and Lee
SC: Histone deacetylase inhibitors suppress the expression of
inflammatory and innate immune response genes in human microglia
and astrocytes. Journal of neuroimmune pharmacology: the official
journal of the Society on NeuroImmune Pharmacology. 5:521–532.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lauterbach EC: Repurposing psychiatric
medicines to target activated microglia in anxious mild cognitive
impairment and early Parkinson's disease. Am J Neurodegener Dis.
5:29–51. 2016.PubMed/NCBI
|
|
14
|
Smith AM, Gibbons HM and Dragunow M:
Valproic acid enhances microglial phagocytosis of
amyloid-beta(1–42). Neuroscience. 169:505–515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brown GC and Vilalta A: How microglia kill
neurons. Brain Res. 1628(Pt B): 1–297. 2015.PubMed/NCBI
|
|
16
|
Brown GC: Nitric oxide and neuronal death.
Nitric Oxide. 23:153–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brown GC and Bal-Price A: Inflammatory
neurodegeneration mediated by nitric oxide, glutamate, and
mitochondria. Mol Neurobiol. 27:325–355. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thornton P, Pinteaux E, Gibson RM, Allan
SM and Rothwell NJ: Interleukin-1-induced neurotoxicity is mediated
by glia and requires caspase activation and free radical release. J
Neurochem. 98:258–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou
Y, Qian G and Zheng JC: IL-1β and TNF-α induce neurotoxicity
through glutamate production: A potential role for neuronal
glutaminase. J Neurochem. 125:897–908. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tak PP and Firestein GS: NF-kappaB: A key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Breuer W, Epsztejn S and Cabantchik ZI:
Iron acquired from transferrin by K562 cells is delivered into a
cytoplasmic pool of chelatable iron(II). J Biol Chem.
270:24209–24215. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mairuae N, Connor JR, Lee SY,
Cheepsunthorn P and Tongjaroenbuangam W: The effects of okra
(Abelmoschus esculentus Linn.) on the cellular events associated
with Alzheimer's disease in a stably expressed HFE neuroblastoma
SH-SY5Y cell line. Neurosci Lett. 603:6–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tenopoulou M, Kurz T, Doulias PT, Galaris
D and Brunk UT: Does the calcein-AM method assay the total cellular
‘labile iron pool’ or only a fraction of it? Biochem J.
403:261–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Epsztejn S, Kakhlon O, Glickstein H,
Breuer W and Cabantchik I: Fluorescence analysis of the labile iron
pool of mammalian cells. Anal Biochem. 248:31–40. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stäubli A and Boelsterli UA: The labile
iron pool in hepatocytes: Prooxidant-induced increase in free iron
precedes oxidative cell injury. Am J Physiol. 274:G1031–G1037.
1998.PubMed/NCBI
|
|
26
|
Lipiński P, Drapier JC, Oliveira L,
Retmańska H, Sochanowicz B and Kruszewski M: Intracellular iron
status as a hallmark of mammalian cell susceptibility to oxidative
stress: A study of L5178Y mouse lymphoma cell lines differentially
sensitive to H(2)O(2). Blood. 95:2960–2966. 2000.PubMed/NCBI
|
|
27
|
Lovell MA, Robertson JD, Teesdale WJ,
Campbell JL and Markesbery WR: Copper, iron and zinc in Alzheimer's
disease senile plaques. J Neurol Sci. 158:47–52. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bharti AC, Donato N, Singh S and Aggarwal
BB: Curcumin (diferuloylmethane) down-regulates the constitutive
activation of nuclear factor-kappa B and IkappaBalpha kinase in
human multiple myeloma cells, leading to suppression of
proliferation and induction of apoptosis. Blood. 101:1053–1062.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park MH and Hong JT: Roles of NF-κB in
Cancer and Inflammatory Diseases and Their Therapeutic Approaches.
Cells. 5:52016. View Article : Google Scholar
|
|
30
|
Beg AA, Finco TS, Nantermet PV and Baldwin
AS Jr: Tumor necrosis factor and interleukin-1 lead to
phosphorylation and loss of I kappa B alpha: A mechanism for
NF-kappa B activation. Mol Cell Biol. 13:3301–3310. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Palombella VJ, Rando OJ, Goldberg AL and
Maniatis T: The ubiquitin-proteasome pathway is required for
processing the NF-kappa B1 precursor protein and the activation of
NF-kappa B. Cell. 78:773–785. 1994. View Article : Google Scholar : PubMed/NCBI
|