Introduction
Ridaifens (RIDs) are novel tamoxifen derivatives
(1,2).
First generation RIDs possess common triphenylethylene structure,
which is similar to tamoxifen, and various amine side chains
connected to para-positions of the aromatic rings. Although
tamoxifen reportedly induces anti-tumor effects by competitive
inhibition of estrogen receptors (ERs) expressed in tumor cells,
RIDs exhibit a growth-inhibitory effect on numerous tumor cell
types regardless of the expression of ERs, suggesting that the
mechanism underlying the anti-tumor effect of RIDs differs from
that of tamoxifen (3). In previous
study, among 48 RIDs, 40 exhibited greater growth inhibitory effect
than tamoxifen, which was evaluated by a JFCR39 panel assay of 39
tumor cell lines, including breast cancer, glioma, colorectal
cancer, lung cancer, melanoma, ovarian cancer, renal cancer,
gastric cancer and prostate cancer (4). Furthermore, the mechanism of
RID-mediated cancer cell growth inhibition may differ from that of
currently used anti-cancer drugs, indicated by COMPARE analysis
(4). One of the RIDs, RID-G, could
induce caspase-independent atypical cell death involving
mitochondrial dysfunction in human neoplastic hematopoietic cell
lines (5), and has been indicated to
interact with calmodulin, heterogeneous nuclear ribonucleoproteins
A2/B1 and zinc finger protein 638 during its cancer cell growth
inhibition (6). RID-F may serve as a
proteasome inhibitor, and inhibit chymotrypsin-like, trypsin-like
and peptidylglutamyl peptide hydrolase activities (7,8). These
findings suggest that the various mechanisms of RID-mediated cancer
cell growth inhibition should be considered in future study.
Anti-cancer drugs and their metabolites work through
various mechanisms to induce damage to cancer cells. Certain
metabolites of 5-fluorouracil disrupt RNA function by
misincorporation into RNA and/or cause DNA damage by binding
thymidylate synthase (9), while
cisplatin crosslinks DNA by binding to guanines bases (10). These findings suggest the possibility
of binding of RID-B and double-stranded DNA in cancer cells.
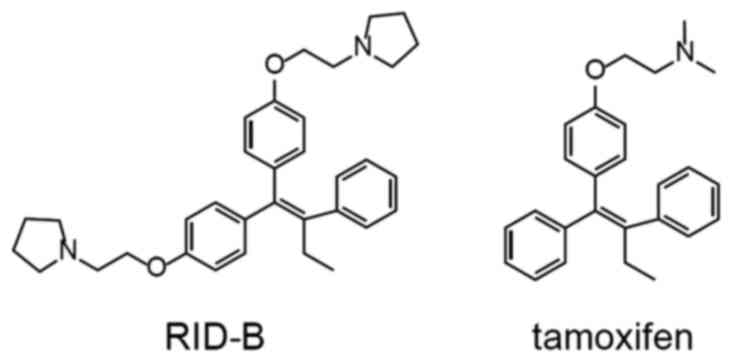
RID-B
(1,1-bis[4-[2-(pyrrolidin-1-yl)ethoxy]phenyl]-2-phenyl-1-butene),
one of the first generation RIDs, contains pyrrolidine rings at the
end of its alkyl side chains (Fig.
1), and has been observed to elicit marked cellular damage
against both ER-positive and -negative tumor cells (11). It has also been reported that RID-B
induces autophagy in the ER-negative human leukemia Jurkat cell
line (12). RID-B may bind to Grb10
interacting GYF protein 2 (GIGYF2) and inhibits GIGYF2-mediated Akt
phosphorylation (13). By previous
JFCR39 panel assay, it was determined that the mean value of the
concentration at which cell growth was inhibited by 50%
(GI50; designated as MG-MID) of RID-B was 1.17 µM, which
was 6.3 times lower than the MG-MID of tamoxifen (4). However, to the best of our knowledge,
the anti-proliferative effect of RID-B on hepatoma cells has not
yet been investigated. Therefore, the aim of the current study was
to evaluate the anti-proliferative effect of RID-B on hepatoma
cells. The mechanism underlying the anti-proliferative effect of
RID-B was also examined.
Materials and methods
Materials
RID-B was synthesized as described previously
(1,2).
Z-VAD-FMK, the caspase-1 and −3 inhibitor, was purchased from
Promega Corporation (Madison, WI, USA), and all other general
reagents not specified in the following text were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany), Kanto Chemical Co.,
Inc. (Tokyo, Japan), Nacalai Tesque, Inc. (Kyoto, Japan) and Wako
Pure Chemical Industries, Ltd. (Osaka, Japan).
Preparation of normal primary rat
hepatocytes
Female Sprague-Dawley rats aged 8 weeks (n=3; weight
range, 150–190 g) were obtained from Sankyo Labo Service
Corporation, Inc. (Tokyo, Japan). The animals were housed in
individual cages supplied with HEPA-filtered air. The ambient
temperature in the animal room was maintained at 23±1°C, and the
relative humidity at 50–60%. A 12-h light/dark cycle was maintained
in the animal room, and food and water were provided ad
libitum.
Rat hepatocytes were isolated as described
previously (14–16) with some modifications. The liver of
the anesthetized rats was perfused in situ via a cannulation of the
portal vein. The liver was perfused for calcium removal with 50 ml
of 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer
containing 3.6 mM NaHCO3, 5.6 mM glucose and 6.0 mM
EGTA, following collagenase perfusion with 50 ml phosphate-buffered
saline (PBS) containing 0.5 mg/ml collagenase, 4 mM
NaHCO3, 0.9 mM MgSO4 and 4.5 mM
CaCl2. The hepatocytes were dispersed and washed three
times with Dulbecco's modified Eagle's medium (DMEM) supplemented
with antibiotic-antimycotic (100 U/ml penicillin, 100 µg/ml
streptomycin, 0.25 µg/ml amphotericin B; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and gentamicin (10 µg/ml, Thermo Fisher
Scientific, Inc.). Cells were then resuspended with DMEM
supplemented with antibiotic-antimycotic (100 U/ml penicillin, 100
µg/ml streptomycin, 0.25 µg/ml amphotericin B), gentamicin (10
µg/ml) and 10% heat-inactivated fetal bovine serum (FBS), and
seeded into 96-well plates (1,000 cells/well). After 24 h of
incubation at 37°C with 5% CO2, the culture medium was
replenished, and cells were used for a proliferation assay. All
animal procedures were conducted according to a protocol approved
by the Tokyo University of Science Institutional Animal Care and
Use Committee (Tokyo, Japan).
Cell proliferation assay
Human hepatoma Huh-7 cells (Japanese Collection of
Research Bioresources Cell Bank, Osaka, Japan) were maintained at
37°C with 5% CO2 in DMEM supplemented with
antibiotic-antimycotic (100 U/ml penicillin, 100 µg/ml
streptomycin, 0.25 µg/ml amphotericin B), gentamicin (10 µg/ml) and
10% heat-inactivated FBS. Cells of passage number 70–80 were used
throughout the study. Cells were grew to confluence for 2 days
following passage.
The Huh-7 cells and normal primary rat hepatocytes
were seeded in 96-well plates (1,000 cells/well) and incubated for
24 h at 37°C. Cells were then incubated with RID-B (0.5, 1 and 2
µM) for 48 h, and cell number was estimated by using WST-1 cell
proliferation reagent (Cell Counting Kit; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) according to the
manufacturer's protocol. The GI50 value of RID-B was
calculated using JMP 9 software (SAS Institute, Inc., Cary, NC,
USA) using the 4-parameter logistic model (17).
Plasmid DNA and salmon sperm DNA were used
subsequently to competitively inhibit the effect of RID-B on Huh-7
cells. Huh-7 cells were seeded in 24-well plates (4×104
cells/well) and incubated for 24 h. Cells were then incubated with
RID-B (1 µM) in the presence of pSecTag2B plasmid DNA (0.125, 0.25
and 0.5 mg/ml, respectively; Thermo Fisher Scientific, Inc.) or
salmon sperm DNA (0.125, 0.25 and 0.5 mg/ml, respectively; Wako
Pure Chemical Industries, Ltd.) overnight at 37°C. Following
incubation, cells were stained with trypan blue and live cells were
counted immediately under a Nikon TMS Inverted Microscope (Nikon
Corporation, Tokyo, Japan) using a Neubauer hemocytometer. The
number of cells was determined as the mean of all live cells
counted in eight fields (0.1 mm3/field) of view.
Determination of caspase-3
activity
Huh-7 cells were seeded in 24-well plates
(8×104 cells/well) and incubated for 24 h at 37°C. Cells
were then incubated with RID-B (0.5 and 5 µM) for 24 h at 37°C.
Following incubation, cells were washed with PBS and lysed with 10
mM phosphate containing 0.15 M NaCl, 5 mM EDTA, 0.1% Nonidet P-40,
0.1% SDS and 0.1% sodium deoxycholate. Cell lysate was incubated
with 0.1 mM caspase-3 substrate
(acetyl-Asp-Glu-Val-Asp-4-methyl-coumaryl-7-amide; Peptide
Institute, Inc., Osaka, Japan) for 3 min at room temperature, and
the increase of fluorescence intensity was monitored by using an
FP-6200 spectrofluorometer (λexcitation =380 nm,
λemission =460 nm; JASCO Corporation, Tokyo, Japan).
Terminal deoxynucleotidyl transferase
dUTP nick-end labelling (TUNEL) assay
Huh-7 cells were seeded on 8-well culture slides
(4×104 cells/well, BD Biosciences, San Jose, CA, USA)
and cultured for 24 h at 37°C. Following cultivation, cells were
pre-incubated with or without 40 µM Z-VAD-FMK for 2 h at 37°C, and
were then incubated with RID-B (1 µM) in the presence or absence of
0.5 mg/ml pSecTag2B for 24 h at 37°C. Following a thorough wash
with PBS, cells were fixed with 3.7% formaldehyde/PBS for 30 min at
room temperature and permeabilized with 0.1% sodium citrate
containing 0.1% Triton X-100 for 2 min on ice. Cells were stained
using a MEBSTAIN Apoptosis TUNEL Kit Direct (Medical and Biological
Laboratories Co., Ltd., Nagoya, Japan), and TUNEL-positive cells
were visualized by confocal laser scanning microscopy (LSM710; Carl
Zeiss AG, Oberkochen, Germany).
Detection of RID-B and double-stranded
DNA binding
For the preparation of biotinylated double-stranded
DNA, complementary oligonucleotides (biotin-TTTTTTATATAT and
ATATATAAAAAA; 1 µg/ml each) in distilled water were annealed for 30
min at 25°C. The double-stranded DNA was incubated with RID-B (10
µM) in distilled water for 1.5 h at room temperature, and the
reaction product was then incubated with streptoavidin-conjugated
magnetic beads (APRO Science, Tokushima, Japan) for 1 h at room
temperature. Magnetic beads were washed three times with distilled
water to remove RID-B unbound to DNA, and the beads were then
boiled for 5 min to separate RID-B from DNA. The supernatant
containing RID-B was collected in a 4-tube magnetic rack (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Betaine (5 µg/ml) was added
to the supernatant for internal standard, and RID-B, which had
bound to DNA, was detected by electrospray ionization
time-of-flight mass spectrometry (ESI-TOF-MS; micrOTOF-NR-focus;
Bruker Daltonik GmbH, Bremen, Germany). The mass spectrometer
settings were as follows: Ionization mode, positive; nitrogen gas
temperature, 180°C; nebulizer pressure, 5.8 psi; flow rate: 4.0
l/min. The calculated mass (M+H+) for
C34H43N2O2 (RID-B) is
511.3319; the mass for RID-B identified in the experiments was
511.3628 (positive control) and 511.3584 (DNA (+)), respectively.
The calculated mass (M+H+) for
C5H11NO2 (betaine) is 118.0868;
the mass for betaine identified in the experiments was 118.0991
(positive control), 118.0988 (DNA (+)) and 118.0976 (DNA (−)),
respectively.
Statistical analysis
Data are expressed as the mean ± standard deviation.
All experiments were performed at least three times. Two-way
analysis of variance with Fisher's protected least significant
difference was employed to compare mean values between groups.
P<0.05 was considered to indicate a statistically significant
difference. Data analysis was performed using JMP 9 software.
Results
RID-B inhibits the growth of human
hepatoma Huh-7 cells but not normal primary rat hepatocytes
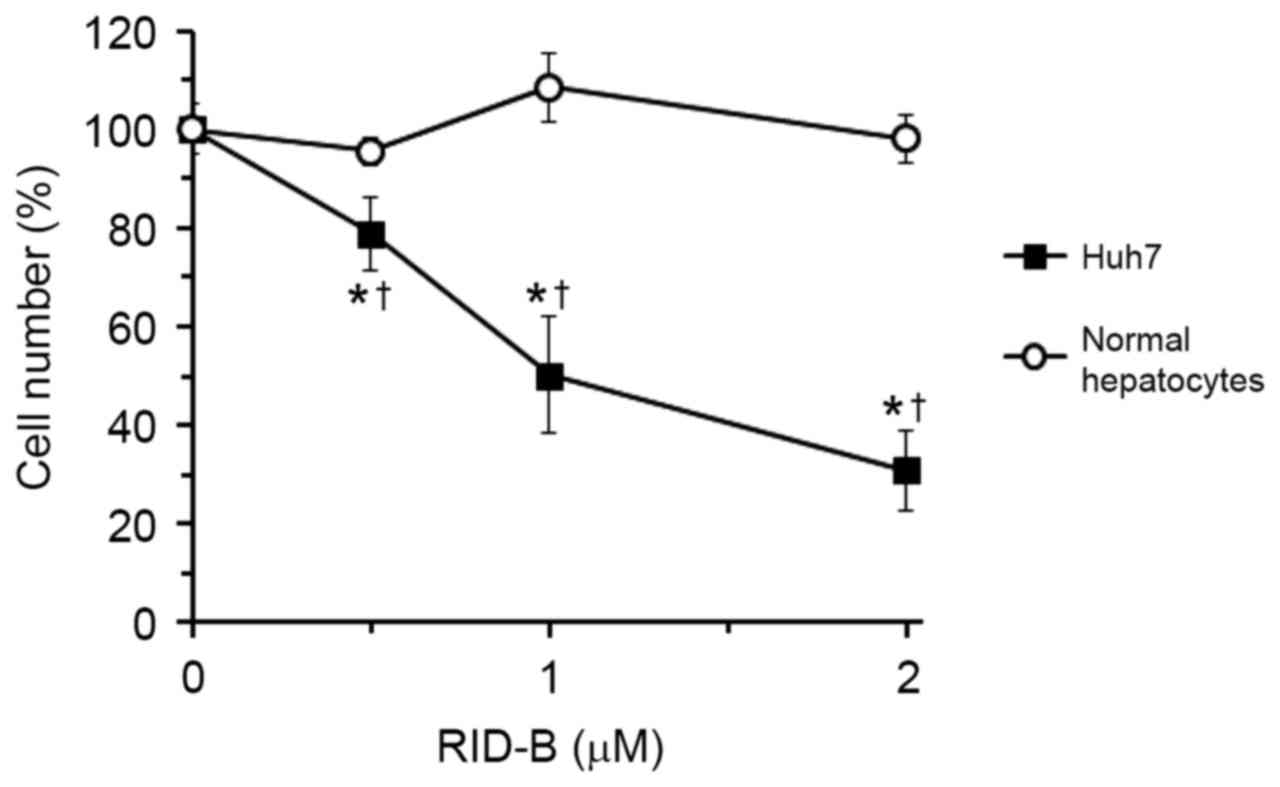
To evaluate the anti-proliferative effect of RID-B
in hepatoma cells, human hepatoma Huh-7 cells and normal primary
rat hepatocytes were incubated with RID-B, and cell numbers were
estimated using WST-1 reagent (Fig.
2). Previous study indicated that the mean GI50, for
RID-B in 39 human cancer cell lines was 1.17 µM (4). Therefore, the RID-B concentration range
of 0.5–2 µM was selected in the current study. RID-B inhibited the
growth of Huh-7 cells between the concentrations of 0.5–2 µM
(P<0.05 vs. 0 µM RID-B), in an apparent dose-dependent manner;
however, RID-B did not inhibit the growth of normal hepatocytes
within the same range. The GI50 of RID-B in Huh-7 cells
was 1.00±0.16 µM. The GI50 of RID-B in rat hepatocytes
could not be calculated as cell number did not fall below 90%
(Fig. 2) and was therefore estimated
as >2 µM. These results suggested that RID-B was more effective
at inhibiting the growth of hepatoma cells than normal
hepatocytes.
RID-B induces apoptosis in Huh-7
cells
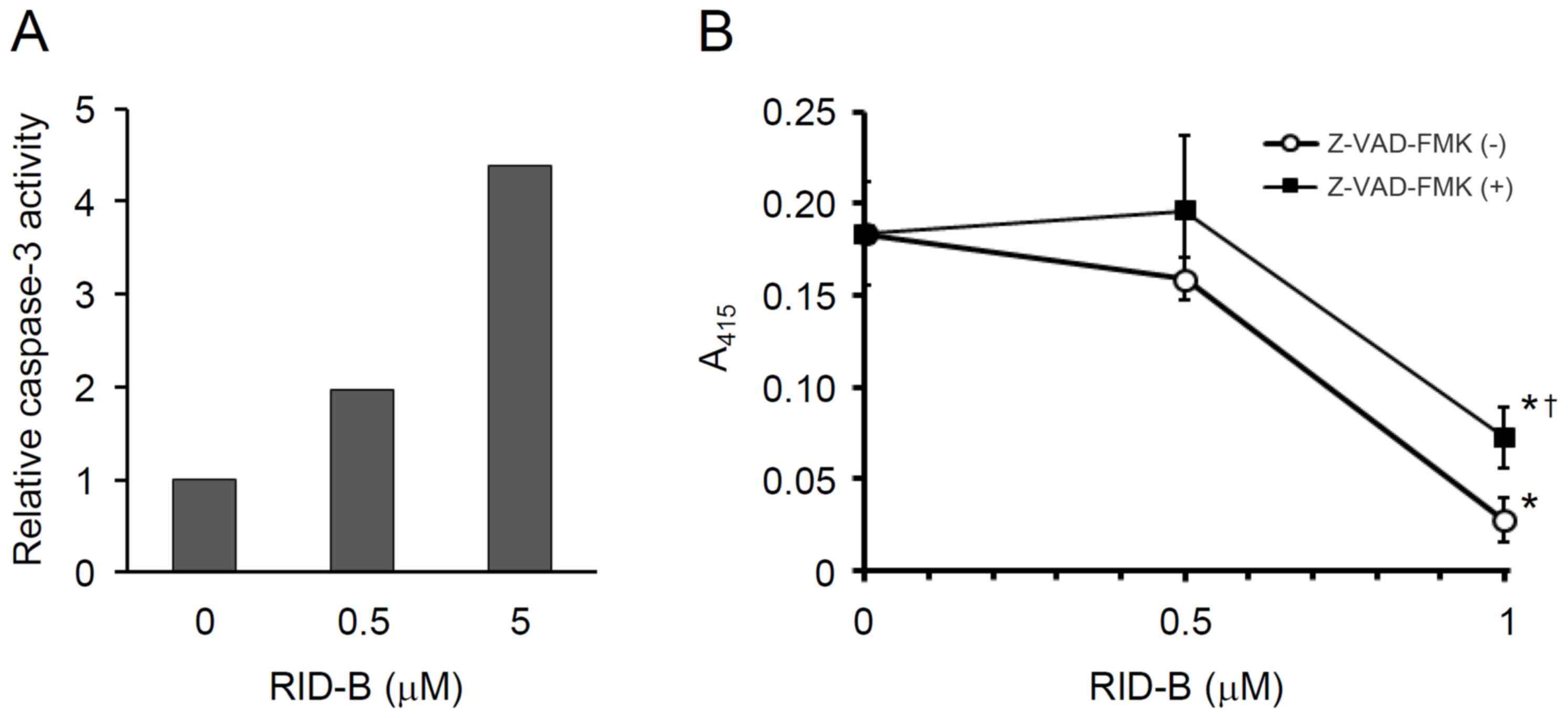
To clarify the mechanism of cancer cell growth
inhibition by RID-B, the effect of caspase inhibitor, Z-VAD-FMK, on
RID-B-mediated cell growth inhibition was examined. As depicted in
Fig. 3A, the caspase-3 activity of
Huh-7 cells was apparently increased dose-dependently between the
RID-B concentrations 0.5 and 5 µM. Furthermore, by the pretreatment
with Z-VAD-FMK, RID-B-mediated Huh-7 cell growth was significantly
attenuated at the RID-B concentration of 1 µM (P<0.05; Fig. 3B). It appears evident from these data
that RID-B induced the apoptosis of Huh-7 cells via the activation
of caspase-3.
Involvement of RID-B and DNA binding
in Huh-7 cell apoptosis
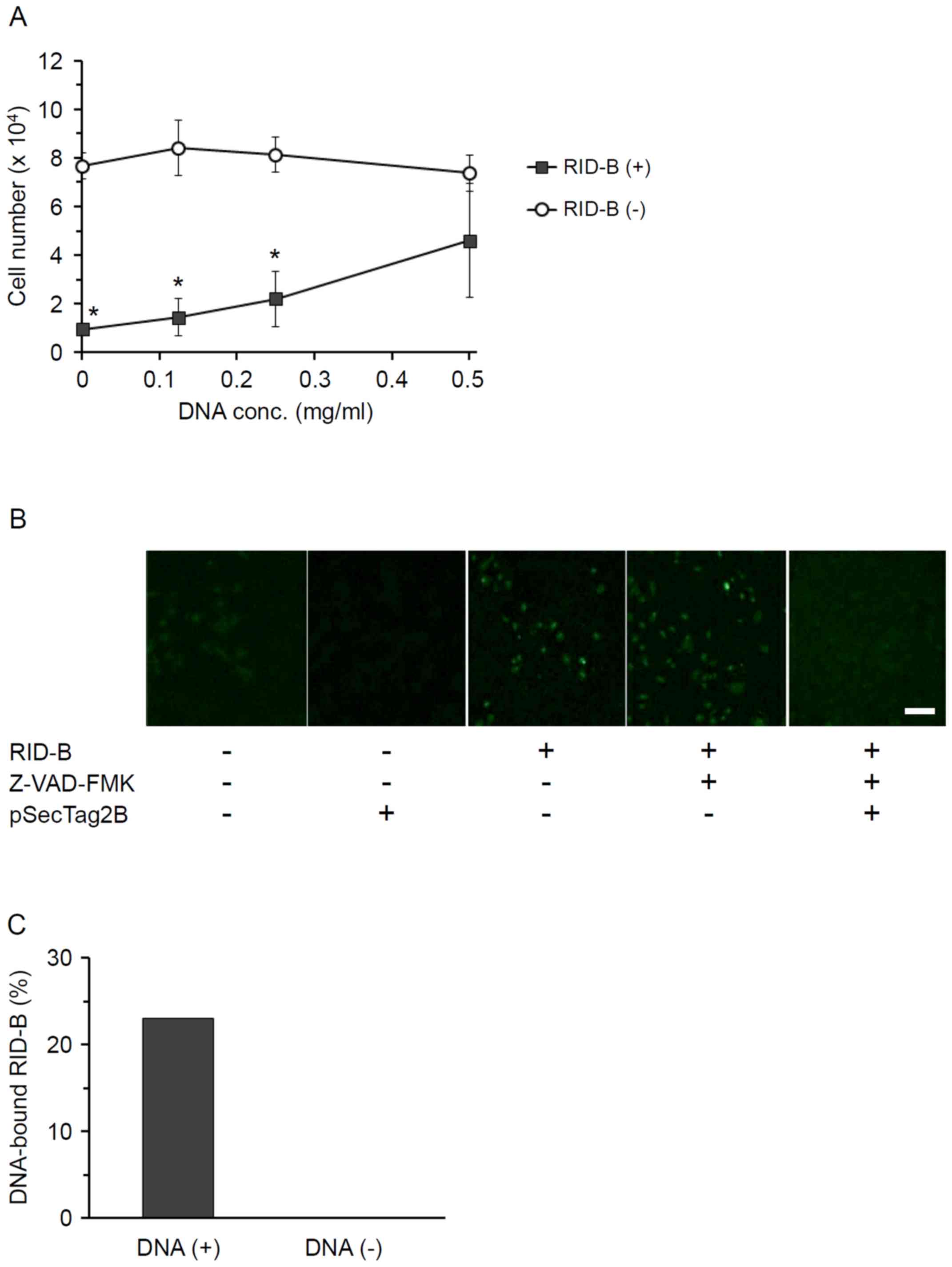
To clarify the mechanism of RID-B-induced apoptosis
in cancer cells, the effect of DNA on RID-B-mediated Huh-7 cell
growth inhibition was examined. Huh-7 cells were incubated with
RID-B in the presence of plasmid DNA (pSecTag2B), and live cells
were then counted (Fig. 4A). The
addition of plasmid DNA suppressed RID-B-mediated Huh-7 cell growth
inhibition; notably, this competitive inhibition by plasmid DNA
appeared dose-dependent, as no significant difference was
identified between the number of Huh-7 cells incubated with and
without RID-B at the DNA concentration of 0.5 mg/ml. Similar
results were observed when using salmon sperm DNA instead of
plasmid DNA in the same DNA concentration range (data not shown).
From these data, it may be suggested that RID-B binds to DNA to
induce cancer cell apoptosis.
As in Fig. 3, RID-B
induced apoptosis in Huh-7 cells seemingly via the activation of
caspase-3. To confirm whether DNA inhibited RID-B-induced apoptosis
in cancer cells, Huh-7 cells were pre-incubated with Z-VAD-FMK, and
the cells were then incubated with RID-B in the presence of plasmid
DNA. By TUNEL assay, it was observed that RID-B caused DNA
fragmentation of Huh-7 cells, suggesting that RID-B induced cancer
cell apoptosis by causing DNA fragmentation. Z-VAD-FMK had no
obvious effect on DNA fragmentation in RID-B-treated Huh-7 cells;
whereas the addition of plasmid DNA appeared to completely suppress
DNA fragmentation (Fig. 4B).
From the data in Fig. 4A
and B, it was apparent that RID-B directly bound to DNA. To
verify this hypothesis, RID-B was incubated with biotinylated
double-stranded DNA, and DNA-bound RID-B was then collected by
streptoavidin-conjugated magnetic beads. DNA-bound RID-B was
detected by ESI-TOF-MS (Fig. 4C). The
peak derived from RID-B was detected only from the sample incubated
with double-stranded DNA, and not from that incubated without DNA,
indicating that RID-B directly bound to double-stranded DNA. The
ratio of RID-B bound to DNA was 23.0%.
Discussion
In the present study, the anti-proliferative effect
of RID-B on human hepatoma Huh-7 cells was evaluated. Although
RID-B did not inhibit the proliferation of rat primary normal
hepatocytes, RID-B inhibited cell growth and induced apoptosis
seemingly by activating caspase-3 and inducing DNA fragmentation in
Huh-7 cells. The binding of RID-B to double-stranded DNA indicated
that RID-B may directly interact with genomic DNA. To the best of
our knowledge, this is the first paper to report that RID-B could
directly bind to DNA. Although the detailed mechanism for
RID-B-mediated cancer cell growth inhibition and apoptosis is
unknown, the following mechanism was indicated from the present
findings: RID-B may activate caspase-3 to induce DNA fragmentation,
and RID-B may directly bind to DNA to inhibit DNA synthesis. It has
been reported that RID-B reduced mitochondrial membrane potential
during the induction of apoptosis in the human lymphoid helper
T-cell line Jurkat (11), and that
RID-B induced microtubule-associated protein 1A/1B-light chain 3
and lysosome colocalization resulting in autophagy in Jurkat cells
(12). Additionally, the current data
revealed that caspase inhibitor, Z-VAD-FMK, partially suppressed
RID-B-mediated growth inhibition and DNA fragmentation in Huh-7
cells. Taken together, the findings indicate the presence of
diverse pathways involved in RID-B-mediated cancer cell growth
inhibition and apoptosis. Since RID-B also influenced the
proliferation of Huh-7 cells, our group is now investigating the
effects of RID-B on the cell cycle.
Additionally, the results indicated that hepatoma
cells were more sensitive to RID-B compared with normal rat
hepatocytes. It has been reported that RID-SB8 preferentially
induced cell death in human breast cancer cell lines, MCF-7 and
MDA-MB-231, over normal human mammary epithelial cells (18). Although the detailed mechanism for the
anti-proliferative effect of RID-B on cancer cells is as yet
unclear, the efficacy of RID-B may be dependent on cell growth
rate. Cancer cells generally grow more rapidly than normal cells,
and the rate of DNA replication is particularly elevated in these
cells (19). DNA is unstable in its
replication process (20), and thus
RID-B may more easily bind to DNA in cancer cells than in normal
cells, causing cancer cell death. Taken together, it may be
proposed that cancer cells are more sensitive to RIDs compared with
normal cells.
The current study used primary rat hepatocytes as a
normal counterpart for comparison of the anti-proliferative effect
of RID-B on the human hepatoma Huh-7 line. Although human cell
lines of normal hepatocytes, THLE-2 and THLE-3, express some
phenotypic characteristics of normal adult liver epithelial cells,
these cells were established as immortal lines by infection with
simian virus 40 (SV40) large T antigen (21). SV40 large T antigen-transfected cell
lines usually exhibit enhanced proliferative rate compared with
primary normal cells (22,23), suggesting that the proliferative
characteristics of SV40 large T antigen-transfected cells may
differ markedly from that of primary normal cells. For these
reasons, primary rat hepatocytes appeared more appropriate than
immortalized human hepatocyte cell lines as a normal counterpart in
the current study. The detailed mechanism of cancer cell death
induced by RID-B will be investigated further in our upcoming
studies.
In conclusion, the results of the current study
indicate that RID-B may be useful as a liver cancer therapeutic, by
inducing apoptosis via activation of caspase-3 and directly binding
to DNA to lead to DNA fragmentation. The current data further
suggest that RID-B affects hepatoma cells without or with little
side-effect against normal cells. Further studies are required to
validate its effectiveness and safety in prospective clinical
application as a liver cancer therapy.
Acknowledgements
The authors thank Professor Yukitoshi Nagahara
(Division of Life Science and Engineering, School of Science and
Engineering, Tokyo Denki University, Tokyo, Japan) for their advice
and sharing of materials throughout this study.
Funding
The current study was supported by a Health and
Labour Sciences Research Grant from the Ministry of Health, Labour
and Welfare, Japan (grant no. 11103425 to IS).
Availability of data and materials
The datasets used and/or analyzed during this study
are available from the corresponding author on reasonable
request.
Authors' contributions
GH and MS designed the study. AM, NO, NS, KT and IS
synthesized RID-B. GH, KA, KH, KS, YY and MH performed the cell
proliferation assay. KH measured caspase-3 activity. KS conducted
the TUNEL assay. GH, KA, YY, AM, NO and IS performed the RID-B-DNA
binding assay. GH analyzed the data. GH, IS and MS wrote the
manuscript. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All animal procedures were conducted by a protocol
approved by the Tokyo University of Science Institutional Animal
Care and Use Committee (Tokyo, Japan).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shiina I, Sano Y, Nakata K, Kikuchi T,
Sasaki A, Ikekita M and Hasome Y: Synthesis of the new
pseudo-symmetrical tamoxifen derivatives and their anti-tumor
activity. Bioorg Med Chem Lett. 17:2421–2424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shiina I, Sano Y, Nakata K, Kikuchi T,
Sasaki A, Ikekita M, Nagahara Y, Hasome Y, Yamori T and Yamazaki K:
Synthesis and pharmacological evaluation of the novel
pseudo-symmetrical tamoxifen derivatives as anti-tumor agents.
Biochem Pharmacol. 75:1014–1026. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shagufta and Ahmad I: Tamoxifen a
pioneering drug: An update on the therapeutic potential of
tamoxifen derivatives. Eur J Med Chem. 143:515–531. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo WZ, Wang Y, Umeda E, Shiina I, Dan S
and Yamori T: Search for novel anti-tumor agents from ridaifens
using JFCR39, a panel of human cancer cell lines. Biol Pharm Bull.
36:1008–1016. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anlifeire A, Hatori M, Morita A, Shiina I,
Nakata K, Tosaki Y, Wang Y-W, Ikekita M and Li G: Ridaifen G
induces caspase independent atypical cell death. Chin J Cell Biol.
33:635–644. 2011.
|
|
6
|
Ikeda K, Kamisuki S, Uetake S, Mizusawa A,
Ota N, Sasaki T, Tsukuda S, Kusayanagi T, Takakusagi Y, Morohashi
K, et al: Ridaifen G, tamoxifen analog, is a potent anticancer drug
working through a combinatorial association with multiple cellular
factors. Bioorg Med Chem. 23:6118–6124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hasegawa M, Yasuda Y, Tanaka M, Nakata K,
Umeda E, Wang Y, Watanabe C, Uetake S, Kunoh T, Shionyu M, et al: A
novel tamoxifen derivative, ridaifen-F, is a nonpeptidic
small-molecule proteasome inhibitor. Eur J Med Chem. 71:290–305.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tanaka M, Zhu Y, Shionyu M, Ota N, Shibata
N, Watanabe C, Mizusawa A, Sasaki R, Mizukami T, Shiina I, et al:
Ridaifen-F conjugated with cell-penetrating peptides inhibits
intracellular proteasome activities and induces drug-resistant cell
death. Eur J Med Chem. 146:636–650. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Todd RC and Lippard SJ: Inhibition of
transcription by platinum antitumor compounds. Metallomics.
1:280–291. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nagahara Y, Shiina I, Nakata K, Sasaki A,
Miyamoto T and Ikekita M: Induction of mitochondria-involved
apoptosis in estrogen receptor-negative cells by a novel tamoxifen
derivative, ridaifen-B. Cancer Sci. 99:608–614. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagahara Y, Takeyoshi M, Sakemoto S,
Shiina I, Nakata K, Fujimori K, Wang Y, Umeda E, Watanabe C, Uetake
S, et al: Novel tamoxifen derivative Ridaifen-B induces Bcl-2
independent autophagy without estrogen receptor involvement.
Biochem Biophys Res Commun. 435:657–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsukuda S, Kusayanagi T, Umeda E, Watanabe
C, Tosaki YT, Kamisuki S, Takeuchi T, Takakusagi Y, Shiina I and
Sugawara F: Ridaifen B, a tamoxifen derivative, directly binds to
Grb10 interacting GYF protein 2. Bioorg Med Chem. 21:311–320. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Seglen PO: Preparation of isolated rat
liver cells. Methods Cell Biol. 13:29–83. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quistorff B, Dich J and Grunnet N:
Preparation of Isolated Rat Liver HepatocytesAnimal Cell Culture.
Walker JM and Pollard JW: Humana Press; Totowa, NJ: pp. 151–160.
1990, View Article : Google Scholar
|
|
16
|
Francavilla A, Ove P, Polimeno L, Sciascia
C, Coetzee ML and Starzl TE: Epidermal growth factor and
proliferation in rat hepatocytes in primary culture isolated at
different times after partial hepatectomy. Cancer Res.
46:1318–1323. 1986.PubMed/NCBI
|
|
17
|
Sebaugh JL: Guidelines for accurate
EC50/IC50 estimation. Pharm Stat. 10:128–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo WZ, Shiina I, Wang Y, Umeda E,
Watanabe C, Uetake S, Ohashi Y, Yamori T and Dan S: Ridaifen-SB8, a
novel tamoxifen derivative, induces apoptosis via reactive oxygen
species-dependent signaling pathway. Biochem Pharmacol.
86:1272–1284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fadaka A, Ajiboye B, Ojo O, Adewale O,
Olayide I and Emuowhochere R: Biology of glucose metabolization in
cancer cells. J Oncol Sci. 3:45–51. 2017.
|
|
20
|
Jeggo PA, Pearl LH and Carr AM: DNA
repair, genome stability and cancer: A historical perspective. Nat
Rev Cancer. 16:35–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pfeifer AM, Cole KE, Smoot DT, Weston A,
Groopman JD, Shields PG, Vignaud JM, Juillerat M, Lipsky MM and
Trump BF: Simian virus 40 large tumor antigen-immortalized normal
human liver epithelial cells express hepatocyte characteristics and
metabolize chemical carcinogens. Proc Natl Acad Sci USA.
90:5123–5127. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reddel RR, De Silva R, Duncan EL, Rogan
EM, Whitaker NJ, Zahra DG, Ke Y, McMenamin MG, Gerwin BI and Harris
CC: SV40-induced immortalization and ras-transformation of human
bronchial epithelial cells. Int J Cancer. 61:199–205. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martin RG and Oppenheim A: Initiation
points for DNA replication in nontransformed and simian virus
40-transformed Chinese hamster lung cells. Cell. 11:859–869. 1977.
View Article : Google Scholar : PubMed/NCBI
|