Introduction
Ovarian cancer is the seventh most common cancer
among women globally (1). Clinicians
are facing an increasing number of ovarian cancer cases diagnosed
each year. This disease is associated with a poor prognosis as the
majority of cases are diagnosed at an advanced stage (1). Clinical observations and molecular
analyses have divided ovarian cancer into two major subtypes: Type
1 cancer types, which are composed of low grade serous cancer,
endometrioid cancer, clear cell cancer and mucinous cancer; and
type 2 cancer types, which include high grade serous cancers
(HGSC), carcinosarcomas and undifferentiated carcinomas (2). Clear cell and endometrioid carcinomas
arise in endometriosis [endometriosis-associated ovarian cancer
(EAOC)] (2). HGSC was originally
thought to develop from the ovarian surface epithelium and/or
ovarian inclusion cysts (2). However,
recent research has contradicted this and suggests that the
majority of HGSC develops from fallopian tube fimbrial epithelium
(3).
Recent promising advances in the field of cancer
treatment have resulted in the development of successful strategies
for molecular-targeted medicine (including small-molecule
inhibitors and antibodies), clinical applications of immunotherapy
and identification of synthetic lethal partners (4). Substantial progress has been made in the
treatment of ovarian cancer through use of targeted therapies and
immunotherapy (5). Anti-angiogenic
therapy using bevacizumab, considered the most promising targeted
therapy for ovarian cancer, may improve the overall survival time
of patients with poor prognosis (6).
One previous study also demonstrated the safety and antitumor
activity of nivolumab, an anti-programmed cell death protein 1
(PD-1) antibody that blocks PD-1 signaling, in patients with
platinum-resistant ovarian cancer (7). Since observations have questioned the
prospect of using PD-ligand 1 (PD-L1) expression as a biomarker for
discriminating responders from non-responders, it must be
complemented by a superior predictive biomarker. Such a predictive
biomarker must be able to select sensitive patients in order to
reduce toxicity and costs and increase progression-free survival
and overall survival time. In addition, resistance to therapy is a
major problem due to molecular heterogeneity (4). The prioritized driver or passenger
genes, including p53, BRCA1/2, DNA repair associated (BRCA1/2),
AT-rich interaction domain 1A (ARID1A), phosphatase and tensin
homolog (PTEN), phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit α (PIK3CA) and MYC proto-oncogene, bHLH
transcription factor (c-MYC), exert loss- or gain-of-functions
(4). Functional losses in tumor
suppressor genes cannot be restored through small molecules or
specific antibodies (8). Furthermore,
the functions of intracellular oncogene products, including Ras and
c-MYC, may be difficult to modulate directly due to the increasing
cytotoxicity of normal cells (8).
The aim of the present report was to provide a
review of synthetic lethality-based therapies for patients with
ovarian cancer. Additionally, future perspectives on their
plausible applications for novel therapeutic methods of clear cell
carcinoma of the ovary (CCC) in the era of personalized medicine
are provided.
Literature search
A computerized literature search was conducted to
identify relevant studies reported in the English language. MEDLINE
submissions (https://www.ncbi.nlm.nih.gov/pubmed) were searched for
studies published between January 2000 and April 2018, combining
the keywords ‘ovarian cancer’, ‘ovarian clear cell carcinoma’,
‘carcinogenesis, ‘synthetic lethality’ and ‘DNA damage repair’. A
variety of combinations of these terms were used, depending on
which database was searched. Each gene was also linked to the
associated National Centre for Biotechnology Information Entrez
Gene pages (http://www.ncbi.nlm.nih.gov/sites/entrez).
Furthermore, the references of each article were searched to
identify potentially relevant studies. Publications of original
studies and review papers were included, while those documenting
opinions, points of view or anecdotes were discarded.
Mutations and molecular heterogeneity of
ovarian cancer
HGSC has been frequently associated with
loss-of-function genes [including tumor protein p53 (p53), BRCA1/2,
cyclin dependent kinase inhibitor 2A also known as
p16INK4a, RB transcriptional corepressor 1 and PTEN]
(3). Previous genomic study has
indicated that germline and somatic mutations in homologous
recombination genes including BRCA1/2 and p53 may increase the risk
of HGSC (9). Homologous recombination
is essential for DNA damage repair, prevention of various mutations
and maintenance of genomic integrity (10). HGSC tumor types also exhibit somatic
loss of function of BRCA and its associated genes (3). Certain sporadic cancer types that share
the molecular features of BRCA1/2-mutant tumor types may respond to
similar therapeutic strategies (11).
This trait is referred to as ‘BRCAness’. Furthermore, the key
oncogenic driver, c-MYC, and the G1/S phase regulatory gene, cyclin
E (CCNE1), are frequently amplified in HGSC and are major factors
in tumorigenesis (3). MYC-amplified
cancer cells have been associated with elevated levels of DNA
replication stress (12).
Amplification of CCNE1 and mutation of BRCA1/2 genes are reportedly
mutually exclusive in HGSC, suggesting that CCNE1 induces synthetic
lethality in BRCA1/2-mutated cells (3).
Oxidative stress induced by repeated hemorrhaging
increases the susceptibility of endometriotic cells to DNA damage
and subsequent malignant transformation, resulting in the
development of type 1 cancer, named EAOC (13). Type 1 cancer has a high mutational
burden: Somatic gene mutations identified include those in ARID1A
mutations (40-50%), PIK3CA (40%), PTEN (20%), catenin beta 1
(CTNNB1; 16-54%), protein phosphatase 2 scaffold subunit α (20%),
KRAS proto-oncogene, GTPase (KRAS; 4-5%), microsatellite
instability (13-50%) and overexpression of transcription factor
hepatocyte nuclear factor-1β (HNF; >90% in CCC) (14). ARID1A is the most frequent driver
mutation in EAOC (15).
PIK3CA/protein kinase B/mechanistic target of rapamycin (mTOR)
pathway is also commonly altered in CCC (16). Furthermore, the overexpression of HNF
has been identified in a large proportion of CCC (17). Checkpoint kinase 1 (Chk1) has been
revealed to be a downstream target of HNF and serve a role within
the cell cycle checkpoint pathways (18). DNA damage increases the persistent
phosphorylation of Chk1 and induction of G2/M phase cell cycle
arrest in cells overexpressing HNF (19,20). HNF
deletion, in turn, induces apoptosis, suggesting that enhanced
levels of HNF may be essential for survival in CCC (17,20).
Molecular changes, including those in HNF and Chk1, may be a
manifestation of essential alterations in cell cycle regulation,
detoxification and chemoresistance in CCC (13,18,21).
Targeted therapy with specific inhibitors of Chk1 or the HNF-Chk1
axis may be explored as a potential treatment modality for patients
with CCC (22). This knowledge may
result in novel potential combination therapeutic methods, for
instance chemotherapeutic agents and targeting of Chk1, which may
overcome drug resistance and produce more successful disease
treatment. In this regard, a number of important drivers have been
identified in ovarian cancer, including BRCA1/2, p53, CCNE1,
ARID1A, Chk1 and HNF. These alterations represent potential future
therapeutic opportunities for CCC.
Synthetic lethality
The concept of tumor-selective synthetic lethality
as a therapeutic strategy is as follows: Two genes are
‘synthetically lethal’ if concurrent loss of the two genes results
in cellular lethality or cell death, whereas the deficiency of
either alone is compatible with cellular viability (8). Specific inhibitors of the cancer driver
genes or oncogenes may contribute to development of idiosyncratic
toxicity (23). An example of
synthetic lethality is PARP inhibition in BRCA1/2-deficient ovarian
and/or breast cancer types (3,24,25). Since homologous
recombination-associated genes, including BRCA1, BRCA2, RAD51
recombinase (RAD51), Fanconi anemia complementation group and p53
genes, are essential for DNA damage repair and survival, cancer
cells may use an alternative approach (non-oncogenes) and become
dependent upon the parallel route (26). Therefore, targeting non-oncogenes may
result in the selective death of the cancer cells (27). If one gene, one paralog or one pathway
is inactive (e.g., germline BRCA mutations), the loss or inhibition
of a second functionally parallel gene or pathway (e.g., PARP
inhibitions) may result in cell death specifically (26). Pharmacological inhibition of PARP
alone may selectively kill homologous recombination deficient
cancer cells but leave normal cells intact (26).
Synthetic lethality screening
The identification of synthetic lethal interactions
is made possible by the SynLethDB database (http://histone.sce.ntu.edu.sg/SynLethDB/) (28). A comprehensive understanding of the
synthetic lethality-related genes and their corresponding
interactions is essential for developing novel therapeutics against
cancer using network-based research platforms (29). The high mutational burden of cancer
cells may contain genes that trigger susceptibility to synthetic
lethality and are synthetic lethal interactors that indicate a
therapeutic opportunity (30).
Despite the high number of mutated genes and the consequential
plethora of potential gene-gene interaction data in CCC,
identifying synthetically lethal genes and their partners is
challenging due to the difficulty of distinguishing true partners
from errors (31). Genomic
high-throughput experimental data may aid investigators to
understand the synthetic lethal screening, including from
high-throughput forward genetic screening approaches, genome-wide
small interfering RNA (siRNA) or clustered regularly interspaced
short palindromic repeat-based human cell line screening, and
comprehensive in silico pipeline computational approaches,
or a combination of these methods (32). Among these screenings, the dominant
approach is high-throughput screening using RNA interference or
compound libraries (32). At present,
a kinome siRNA library of >40,000 siRNAs targeting >10,000
genes (four siRNAs per gene) is commercially available. In a
previous study, a variety of human cancer cell lines with a
particular tumor suppressor gene or oncogene were plated in arrayed
siRNA library-coated wells using a library targeting protein
networks (8). The hit list from the
primary screen was then systematically narrowed. Candidate
synthetic lethality genes were validated to confirm whether
synthetic lethal pairs selectively kill cancer cells, but not
normal cells (8). However,
limitations for the screening-based approach are a high false
positive rate of computational predictions and high cost.
Synthetic lethal gene pairs
In the present review, the synthetic lethal pairs
were divided into four categories as described by Fang (8): i) Components of a backup or a parallel
pathway with overlapping functions; ii) components encoded by
paralogous pairs; iii) subunit components that form heteromeric
complexes and iv) components that are arranged in a single linear
pathway (Fig. 1).
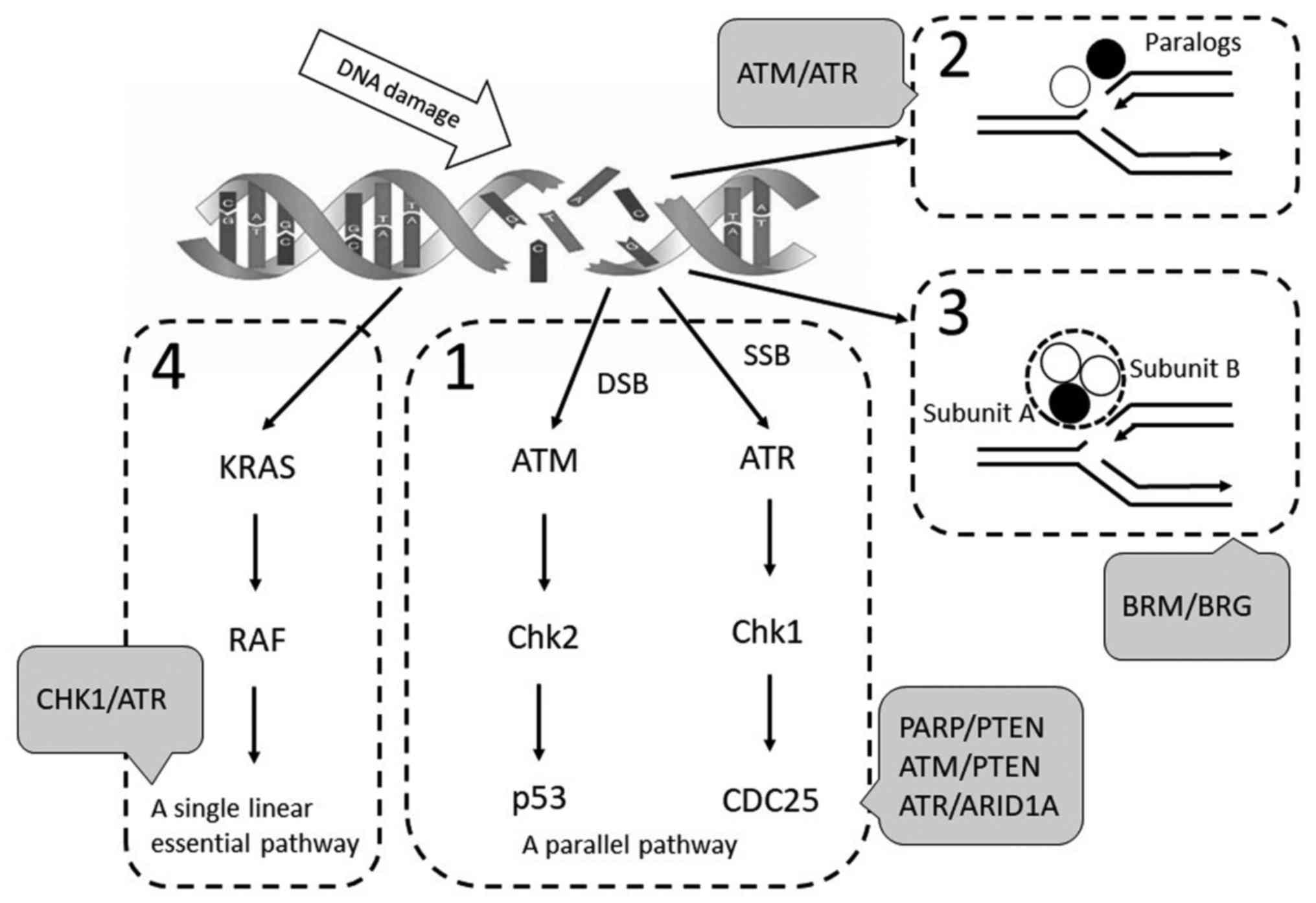 | Figure 1.Landscape of synthetic lethal pairs.
Synthetic lethal pairs were divided into four categories: (1) Components of a backup or a parallel
pathway with overlapping functions; (2) components encoded by paralogous pairs;
(3) Subunit components that form
heteromeric complexes; and (4)
components that are arranged in a single linear pathway. KRAS, KRAS
proto-oncogene, GTPase; Chk, checkpoint kinase; ATR, ATR
serine/threonine kinase; ATM, ATM serine/threonine kinase; p53,
tumor protein p53; PARP, poly(ADP-ribose) polymerases; PTEN,
phosphatase and tensin homolog; ARID1A, AT-rich interaction domain
1A; BRM, SWItch/Sucrose non-fermentable-related, matrix associated,
actin dependent regulator of chromatin, subfamily a, member 2; BRG,
SWItch/Sucrose non-fermentable-related, matrix associated, actin
dependent regulator of chromatin, subfamily a, member 4. |
Category 1
Components of a backup or a parallel pathway with
overlapping functions. Mutational screening studies of synthetic
lethality have been reported (8,33–36). The majority of the category 1 genes
are associated with DNA damage repair and cell survival. A high
number of DNA double-strand breaks are repaired by BRCA-dependent
homologous recombination (37). PARP1
may serve as an alternative or a backup pair (38). Preclinical studies revealed that
proteins involved in DNA repair and response to damage, including
BRCA1/2, X-ray repair cross complementing (XRCC)1, XRCC2, XRCC3,
ATM serine/threonine kinase (ATM), ATR serine/threonine kinase
(ATR), Chk1, Chk2, proliferating cell nuclear antigen, RAD51, MRE11
homolog, double strand break repair nuclease, ERCC excision repair
1, endonuclease non-catalytic subunit, p53, cyclin dependent kinase
12 (CDK12) and PTEN, in addition to the Fanconi anemia pathway,
induce synthetic lethality with PARP inhibitors and are implicated
as potential predictive markers for tumor cell response to PARP
inhibitors (8,33). As presented in Table I, PARP inhibitors (Table I; A synthetic lethal pair) are
currently under investigations as therapeutic agents for the
treatment of cancer types with deficiencies in BRCA, CDK12, PTEN,
ATM, ATR or Cohesin genes (Table I;
Germline and/or somatic mutations) (34,35).
Furthermore, comprehensive experiments have identified novel
synthetic lethal gene pairs, including p53-ATM, p53-ATR, TP53-mTOR,
p53-vascular endothelial growth factor receptor 2, p53-epidermal
growth factor receptor, p53-transforming acidic coiled-coil
containing protein 3, p53-MYC, p53-CTNNB1, p53-casein kinase 1
epsilon (CSNK1E), ATR-ARID1A, ATR-XRCC1, ATR-DNA topoisomerase I,
ATR-DNA polymerase δ1, catalytic subunit, ATM-protein kinase C α,
KRAS-CDK1, KRAS-polo like kinase 1 (PLK1), CDC28 protein kinase
regulatory subunit 1-PLK1, APC-CTNNB1, APC-casein kinase 1 α1,
APC-tryptophan 2,3-dioxygenase, CSNK1E-MYC, CTNNB1-hepatocyte
growth factor-regulated tyrosine kinase substrate, PTEN-nemo like
kinase and mutS homolog 2-DNA polymerase beta (32,34,36).
 | Table I.List of candidate synthetic lethality
gene pairs those with germline and/or somatic mutation in four
categories. |
Table I.
List of candidate synthetic lethality
gene pairs those with germline and/or somatic mutation in four
categories.
| Components of a
backup or a parallel pathway with overlapping functions. |
|---|
| PARP | BRCA |
| PARP | CDK12 |
| PARP | PTEN |
| PARP | ATM |
| PARP | Cohesin |
| mTOR | TP53 |
| VEGFR2 | TP53 |
| EGFR | TP53 |
| CSNK1E | TP53 |
| CTNNB1 | TP53 |
| CSNK1A1 | TP53 |
| CDK6 | TP53 |
| CSNK1A1 | APC |
| TDO2 | APC |
| CTNNB1 | APC |
| BRAF | APC |
| IGF1R | APC |
| WNK1 | APC |
| ATM | ATR |
| ATM | TP53 |
| ATM | PTEN |
| ATM | PRKCA |
| ATR | XRCC1 |
| ATR | TOP1 |
| ATR | POLD1 |
| ATR | ARID1A |
| CDK1 | KRAS |
| MAPK14 | KRAS |
| PRKDC | MYC |
| CSNK1E | MYC |
| CKS1B | PLK1 |
| KRAS | PLK1 |
| TRRAP | PIK3CA |
| HH | PIK3CA |
| HGS | CTNNB1 |
| NLK | PTEN |
| POLB | MSH2 |
| POLD1 | ATR |
| Components encoded
by paralogous pairs. |
| ATM | ATR |
| TRRAP | PIK3CA |
| CBP | EP300 |
| BMR | BRG |
| Subunit components
that form heteromeric complexes. |
| RAD52 | BRCA |
| PARP1 | RAD54B |
| PARP1 | RAD51 |
| BMR | BRG |
| Components that are
arrangedin a single linear pathway. |
| Chk1 | ATR |
| RAF1 | KRAS |
Category 2
Components encoded by paralogous pairs (paralogs).
DNA damage response pathway factors, including ATM and ATR, result
in a parallel signaling cascade that is activated by replication
stress and uses a complex and coordinated set of proteins to
maintain genomic stability (39). ATR
is considered among the top candidates of the category 2 genes, as
ATR inhibitors are entering clinical trials and have therapeutic
utility (34). cAMP-response element
binding protein (CREBBP) serves a role in growth by coupling
chromatin remodeling to transcription factor recognition (40). The CREBBP paralog, p300/EP300, is a
synthetic-lethal gene in CREBBP-deficient cancer types (41). SWItch/Sucrose non-fermentable
(SWI/SNF)-related, matrix associated, actin dependent regulator of
chromatin, subfamily a, member 2 (BRM) and SWI/SNF-related, matrix
associated, actin dependent regulator of chromatin, subfamily a,
member 4 (BRG) are DNA-dependent ATPases of the SWI/SNF chromatin
remodeling complex (42). Synthetic
lethality may be explained by BRM or BRG paralog insufficiency
(42). Two pair genes are organized
in the networks of paralogs (42).
Category 3
Subunit components that form heteromeric complexes.
The SWI/SNF complex facilitates the homologous
recombination-dependent processes, including the recruitment of
BRM, BRG, ARID and RAD molecules to DNA double-strand breaks
(43). In a similar manner to PARP1
inhibitors, the suppression of RAD52 reduces homologous
recombination and results in synthetic lethality in cells deficient
in BRCA1/2 or associated proteins (including partner and localizer
of BRCA2, RAD51B/C/D and XRCC2/3), whilst sparing normal cells
(44). RAD51/RAD54B and PARP1 have
been reported to function as synthetic lethal interactors, where it
was thus revealed that PARP1 may be a novel candidate drug target
in RAD51/RAD54B-deficient cells (45). Chromatin remodeling proteins are often
contained within multiprotein complexes (43).
Category 4
Components that are arranged in a single linear
pathway. The ATR and Chk1 kinases, critical mediators of the DNA
damage response pathway, maintain cell survival and protect cells
from replication stress (46). These
two kinases are currently the focus of ongoing clinical trials
(47). Inhibition of Raf-1
proto-oncogene serine threonine kinase (RAF1) is a synthetic lethal
target in KRAS mutant tumor types (48). The ATR-Chk1 and KRAS-RAF1 pathways
function as a cancer-specific synthetic lethality between two
proteins in the same pathway (47,48).
The present review discussed recent developments in
the synthetic lethality gene pairs in four categories, which
includes licensed inhibitors in preclinical and clinical trials: i)
PARP inhibitors for homologous recombination deficient cancer
types; ii) disruption of the ATM paralog ATR; iii) deficiency of
BRM or BRG in an essential multiprotein complex and iv) two
proteins in the same pathway, ATR and Chk1. Recent clinical studies
reported the evidence for the use of PARP inhibitors in high-grade
serous ovarian cancer with germline or somatic BRCA1/2 mutations
(49).
Synthetic lethal partners in CCC
Preclinical and clinical data of targeting synthetic
lethal partners for the treatment of CCC cell lines and patients
with CCC, respectively, are discussed as follows. Firstly, previous
literature suggests a lack of prognostic and early detection
markers available for CCC (21).
However, Arakawa et al (50)
reported that tissue factor pathway inhibitor 2 may be a useful
serum marker for the detection of CCC. In addition, the
overexpression of transcription factor HNF (>90%) (17) and somatic mutations of ARID1A
(15) and PIK3CA (~50%) (51) are common genetic changes reported in
CCC (52). In previous study,
suppression of HNF expression resulted in apoptotic cell death in
CCC cell lines, indicating that HNF may be associated with tumor
survival (18). Thus, HNF may be
suggested as a desirable candidate for targeted cancer therapy.
However, the pharmacological inhibition of HNF may be particularly
toxic for normal cells as this transcription factor is expressed in
the liver, digestive tract, pancreas and kidneys (18). Chk1 has been identified as a
downstream target of HNF, demonstrating that the overexpression or
persistent phosphorylation of Chk1 is a characteristic feature of
the HNF-overexpressing CCC (18). The
HNF-Chk1 axis serves a critical role in the cell cycle arrest at
G2/M phase and DNA repair (20). CCC
is sensitive to Chk1 inhibitors as they induce a high degree of
HNF-induced replication stress (22).
The HNF-Chk1 axis is a main pathway for cell survival in CCC
(20). These data suggest that the
inhibition of Chk1 may be lethal in HNF-overexpressing CCC
cells.
Secondly, ATR and Chk1 are a synthetic lethal pair
of two proteins in the same pathway and maintain cancer cell
survival under replication stress (47). Inhibitors of ATR and Chk1 are
currently undergoing clinical trials and may represent a promising
strategy for cancer therapy (47).
Furthermore, a randomized phase II trial study has been conducted
to assess whether adding ATR kinase inhibitor M6620 to standard
treatment with gemcitabine hydrochloride is more effective than
gemcitabine hydrochloride alone in treating patients with ovarian,
primary peritoneal or fallopian tube cancer (trial number,
NCT02595892) (53). A phase II single
arm pilot study has been conducted to determine the objective
response rate of single agent Chk1/2 inhibitor (LY2606368) in
patients with germline BRCA mutation-associated breast or ovarian
cancer, high-grade serous cancer and triple-negative breast cancer
(trial number, NCT02203513) (54).
The latter study provided positive proof of concept of the efficacy
and tolerability of the Chk1/2 inhibitor in patients with ovarian
cancer (54). From such studies it
may be assumed that ovarian cancer cells, including CCC cells,
exhibit an increased reliance on the ATR-Chk1 axis for cell
survival.
Thirdly, CCC cells commonly lack compensatory DNA
damage response machinery, as mutations in ARID1A represent one of
the most common molecular alterations in CCC (15). ARID1A deficiency reportedly triggers
genomic instability and apoptosis in ATR-deficient cells (32). These data suggest that ARID1A may be a
synthetic lethal partner of ATR.
Finally, the PTEN mutations are also commonly
observed in CCC (31). siRNA
screening strategies identified PTEN and ATM as synthetically
lethal partners (55). One
preclinical study demonstrated that ATM inhibition resulted in DNA
damage, cell cycle arrest and apoptosis specifically in
PTEN-deficient cells (55). PARP
inhibition also sensitized cancer cells to genotoxic stress in
PTEN-deficient cells (56). Although
a variety of loss of function genes, including in HNF, ARID1A,
PIK3CA and PTEN, in CCC are driver or passenger events, these genes
may function as synthetic lethal pairs under replication stress
conditions (16).
Conclusions
The present article summarized recent advances in
molecular understanding of synthetic lethal partners. CCC, the most
common type 1 ovarian cancer in Japan, demonstrated chemoresistance
resulting in poor prognosis (57).
Novel potential targets in CCC therapy were additionally discussed.
The development of novel cancer drugs is a time-consuming and
expensive process with a high rate of failure in late-stage
clinical trials (58). The synthetic
lethality strategy to identify novel partners for potential genetic
mutations has emerged to overcome this challenge (5,8).
Homologous recombination deficiency including BRCA1/2 or p53
mutations combined with an increase in replication stress may
result in a tumor being more susceptible to PARP inhibition
(38,45,49,56). Since
these particular genes are rarely observed to be mutated in CCC,
there is considerable interest in identifying alternative
therapeutic options of PARP inhibitor sensitivity (2). Synthetic lethal interactions have a
variety of types, involving genes that function as a critical
backup, paralog genes derived from the same ancestral gene, subunit
genes of a multiprotein complex or genes that belong to a single
linear signaling pathway (8). The
present review highlighted promising candidate genes, including p53
and BRCA1-regulated stress-inducible genes and genes involved in
the regulation of replication initiation, relevant DNA repair
factors and cell cycle distribution, which may be associated with
the observed deficiency in homologous recombination capacity. Among
these synthetic lethal pairs, the genetic ablation of the DNA
repair response genes located in parallel signaling pathways may be
one of the important patterns resulting in synthetic lethality.
Investigators have unraveled a set of candidate gene targets
hypothetically resulting in synthetic lethality. HNF, ARID1A, ATR,
ATM, Chk1 and PTEN may be the key genes in CCC (18,32,47,18).
Notably, the HNF-Chk1 axis was specifically upregulated in CCC
(18). ARID1A mutation-driven cancer
cells may rely on HNF overexpression for survival, predicting the
potential association between ARID1A and HNF determining synthetic
lethality. The pharmacological inhibition of Chk1 is also likely to
be synthetically lethal to ARID1A. These predicted genes are
promising candidates for in-depth investigation and experimental
validation to accumulate significant data for the application of
clinical trials. Further clinical studies are required to
investigate the safety and therapeutic effects of synthetic
lethality pairs in CCC tumor types with replication stress.
In conclusion, the development of synthetic lethal
approaches may expedite the formation of novel concepts regarding
targeted cancer therapeutics in the future, and this strategy
represents a potentially paradigm-shifting initiative. An
understanding of high-confidence synthetic lethal interaction pairs
may aid in the development of cancer therapies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the grant-in-aid
for scientific research from the Ministry of Education, Science and
Culture of Japan to the Department of Obstetrics and Gynecology,
Nara Medical University, Nara, Japan (awarded to Dr Hiroshi
Kobayashi; grant no. 26293361).
Availability of data and materials
Not applicable.
Authors' contributions
SM collected data regarding the signaling pathways
and underlying mechanism of synthetic lethality using the SynLethDB
database. KI, EN and SM performed the literature search and
supervised the study. YY, KO and NK made substantial contribution
to conception of the study. HK contributed to the study design and
interpretation of included research studies. The final version of
the manuscript has been read and approved by all authors.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted
in the absence of any commercial or financial relationships that
could be construed as a potential conflict of interest.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurman RJ and IeM Shih: The origin and
pathogenesis of epithelial ovarian cancer: A proposed unifying
theory. Am J Surg Pathol. 34:433–443. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kroeger PT Jr and Drapkin R: Pathogenesis
and heterogeneity of ovarian cancer. Curr Opin Obstet Gynecol.
29:26–34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saijo N: Present status and problems on
molecular targeted therapy of cancer. Cancer Res Treat. 44:1–10.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thompson N, Adams DJ and Ranzani M:
Synthetic lethality: Emerging targets and opportunities in
melanoma. Pigment Cell Melanoma Res. 30:183–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Duan P, Fan L, Gao Q, Silwal BM, Ren M,
Shen Y and Qu W: Targeted Therapy of Ovarian Cancer with
Angiogenesis Inhibitors. Curr Drug Targets. 18:1171–1178. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hamanishi J, Mandai M, Ikeda T, Minami M,
Kawaguchi A, Murayama T, Kanai M, Mori Y, Matsumoto S, Chikuma S,
et al: Safety and Antitumor Activity of Anti-PD-1 Antibody,
Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J
Clin Oncol. 33:4015–4022. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fang B: Development of synthetic lethality
anticancer therapeutics. J Med Chem. 57:7859–7873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Perets R, Wyant GA, Muto KW, Bijron JG,
Poole BB, Chin KT, Chen JY, Ohman AW, Stepule CD, Kwak S, et al:
Transformation of the fallopian tube secretory epithelium leads to
high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer
Cell. 24:751–765. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scully R: Role of BRCA gene dysfunction in
breast and ovarian cancer predisposition. Breast Cancer Res.
2:324–330. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Turner N, Tutt A and Ashworth A: Hallmarks
of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 4:814–819. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dominguez-Sola D and Gautier J: MYC and
the control of DNA replication. Cold Spring Harb Perspect Med.
4:pii: a014423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kobayashi H, Shigetomi H and Yoshimoto C:
Checkpoint kinase 1 inhibitors as targeted molecular agents for
clear cell carcinoma of the ovary. Oncol Lett. 10:571–576. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lyttle B, Bernardi L and Pavone ME:
Ovarian cancer in endometriosis: Clinical and molecular aspects.
Minerva Ginecol. 66:155–164. 2014.PubMed/NCBI
|
|
15
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y,
Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et
al: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin Y, Li Y and Pan L: The target therapy
of ovarian clear cell carcinoma. OncoTargets Ther. 7:1647–1652.
2014. View Article : Google Scholar
|
|
17
|
Tsuchiya A, Sakamoto M, Yasuda J, Chuma M,
Ohta T, Ohki M, Yasugi T, Taketani Y and Hirohashi S: Expression
profiling in ovarian clear cell carcinoma: Identification of
hepatocyte nuclear factor-1 beta as a molecular marker and a
possible molecular target for therapy of ovarian clear cell
carcinoma. Am J Pathol. 163:2503–2512. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shigetomi H, Sudo T, Shimada K, Uekuri C,
Tsuji Y, Kanayama S, Naruse K, Yamada Y, Konishi N and Kobayashi H:
Inhibition of cell death and induction of G2 arrest accumulation in
human ovarian clear cells by HNF-1β transcription factor:
Chemosensitivity is regulated by checkpoint kinase CHK1. Int J
Gynecol Cancer. 24:838–843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng ZG, Yao YB, Yang J, Tang YL and Huang
X: Mangiferin induces cell cycle arrest at G2/M phase through
ATR-Chk1 pathway in HL-60 leukemia cells. Genet Mol Res.
14:4989–5002. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ito F, Yoshimoto C, Yamada Y, Sudo T and
Kobayashi H: The HNF-1β-USP28-Claspin pathway upregulates DNA
damage-induced Chk1 activation in ovarian clear cell carcinoma.
Oncotarget. 9:17512–17522. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kobayashi H, Sugimoto H, Onishi S and
Nakano K: Novel biomarker candidates for the diagnosis of ovarian
clear cell carcinoma. Oncol Lett. 10:612–618. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Itamochi H, Nishimura M, Oumi N, Kato M,
Oishi T, Shimada M, Sato S, Naniwa J, Sato S, Kudoh A, et al:
Checkpoint kinase inhibitor AZD7762 overcomes cisplatin resistance
in clear cell carcinoma of the ovary. Int J Gynecol Cancer.
24:61–69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang S, Wu L, Li X, Huang J, Zhong J and
Chen X: Crizotinib-associated toxic epidermal necrolysis in an
ALK-positive advanced NSCLC patient. Mol Clin Oncol. 8:457–459.
2018.PubMed/NCBI
|
|
24
|
Mirza MR, Monk BJ, Herrstedt J, Oza AM,
Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I,
et al ENGOT-OV16/NOVA Investigators: Niraparib Maintenance Therapy
in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med.
375:2154–2164. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Papa A, Caruso D, Strudel M, Tomao S and
Tomao F: Update on Poly-ADP-ribose polymerase inhibition for
ovarian cancer treatment. J Transl Med. 14:2672016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Raj L, Ide T, Gurkar AU, Foley M, Schenone
M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al:
Selective killing of cancer cells by a small molecule targeting the
stress response to ROS. Nature. 475:231–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo J, Liu H and Zheng J: SynLethDB:
Synthetic lethality database toward discovery of selective and
sensitive anticancer drug targets. Nucleic Acids Res. 44(D1):
D1011–D1017. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pathak HB, Zhou Y, Sethi G, Hirst J,
Schilder RJ, Golemis EA and Godwin AK: A Synthetic Lethality Screen
Using a Focused siRNA Library to Identify Sensitizers to Dasatinib
Therapy for the Treatment of Epithelial Ovarian Cancer. PLoS One.
10:e01441262015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Crespan E, Garbelli A, Amoroso A and Maga
G: Exploiting the nucleotide substrate specificity of repair DNA
polymerases to develop novel anticancer agents. Molecules.
16:7994–8019. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bitler BG, Fatkhutdinov N and Zhang R:
Potential therapeutic targets in ARID1A-mutated cancers. Expert
Opin Ther Targets. 19:1419–1422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Williamson CT, Miller R, Pemberton HN,
Jones SE, Campbell J, Konde A, Badham N, Rafiq R, Brough R, Gulati
A, et al: ATR inhibitors as a synthetic lethal therapy for tumours
deficient in ARID1A. Nat Commun. 7:138372016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jdey W, Thierry S, Russo C, Devun F, Al
Abo M, Noguiez-Hellin P, Sun JS, Barillot E, Zinovyev A, Kuperstein
I, et al: Drug-Driven Synthetic Lethality: Bypassing Tumor Cell
Genetics with a Combination of AsiDNA and PARP Inhibitors. Clin
Cancer Res. 23:1001–1011. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Toledo LI, Murga M, Zur R, Soria R,
Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR and
Fernandez-Capetillo O: A cell-based screen identifies ATR
inhibitors with synthetic lethal properties for cancer-associated
mutations. Nat Struct Mol Biol. 18:721–727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Subhash VV, Tan SH, Yeo MS, Yan FL,
Peethala PC, Liem N, Krishnan V and Yong WP: ATM Expression
Predicts Veliparib and Irinotecan Sensitivity in Gastric Cancer by
Mediating P53-Independent Regulation of Cell Cycle and Apoptosis.
Mol Cancer Ther. 15:3087–3096. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang X, Zhang Y, Han ZG and He KY:
Malignancy of Cancers and Synthetic Lethal Interactions Associated
With Mutations of Cancer Driver Genes. Medicine (Baltimore).
95:e26972016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Carvalho JF and Kanaar R: Targeting
homologous recombination-mediated DNA repair in cancer. Expert Opin
Ther Targets. 18:427–458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu FW and Tewari KS: New Targeted Agents
in Gynecologic Cancers: Synthetic Lethality, Homologous
Recombination Deficiency, and PARP Inhibitors. Curr Treat Options
Oncol. 17:122016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH,
Lee JE, Wang C, Brindle PK, Dent SY and Ge K: Distinct roles of
GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in
nuclear receptor transactivation. EMBO J. 30:249–262. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ogiwara H, Sasaki M, Mitachi T, Oike T,
Higuchi S, Tominaga Y and Kohno T: Targeting p300 Addiction in
CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell
Death due to Abrogation of MYC Expression. Cancer Discov.
6:430–445. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hoffman GR, Rahal R, Buxton F, Xiang K,
McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D,
et al: Functional epigenetics approach identifies BRM/SMARCA2 as a
critical synthetic lethal target in BRG1-deficient cancers. Proc
Natl Acad Sci USA. 111:3128–3133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Raab JR, Runge JS, Spear CC and Magnuson
T: Co-regulation of transcription by BRG1 and BRM, two mutually
exclusive SWI/SNF ATPase subunits. Epigenetics Chromatin.
10:622017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chandramouly G, McDevitt S, Sullivan K,
Kent T, Luz A, Glickman JF, Andrake M, Skorski T and Pomerantz RT:
Small-Molecule Disruption of RAD52 Rings as a Mechanism for
Precision Medicine in BRCA-Deficient Cancers. Chem Biol.
22:1491–1504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wiegmans AP, Yap PY, Ward A, Lim YC and
Khanna KK: Differences in Expression of Key DNA Damage Repair Genes
after Epigenetic-Induced BRCAness Dictate Synthetic Lethality with
PARP1 Inhibition. Mol Cancer Ther. 14:2321–2331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gatei M, Kijas AW, Biard D, Dörk T and
Lavin MF: RAD50 phosphorylation promotes ATR downstream signaling
and DNA restart following replication stress. Hum Mol Genet.
23:4232–4248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sanjiv K, Hagenkort A, Calderón-Montaño
JM, Koolmeister T, Reaper PM, Mortusewicz O, Jacques SA, Kuiper RV,
Schultz N, Scobie M, et al: Cancer-Specific Synthetic Lethality
between ATR and CHK1 Kinase Activities. Cell Reports. 14:298–309.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lamba S, Russo M, Sun C, Lazzari L,
Cancelliere C, Grernrum W, Lieftink C, Bernards R, Di Nicolantonio
F and Bardelli A: RAF suppression synergizes with MEK inhibition in
KRAS mutant cancer cells. Cell Reports. 8:1475–1483. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Morgan RD, Clamp AR, Evans DGR, Edmondson
RJ and Jayson GC: PARP inhibitors in platinum-sensitive high-grade
serous ovarian cancer. Cancer Chemother Pharmacol. 81:647–658.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Arakawa N, Kobayashi H, Yonemoto N,
Masuishi Y, Ino Y, Shigetomi H, Furukawa N, Ohtake N, Miyagi Y,
Hirahara F, et al: Clinical Significance of Tissue Factor Pathway
Inhibitor 2, a Serum Biomarker Candidate for Ovarian Clear Cell
Carcinoma. PLoS One. 11:e01656092016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang Y, Helland A, Holm R, Kristensen GB
and Børresen-Dale AL: PIK3CA mutations in advanced ovarian
carcinomas. Hum Mutat. 25:3222005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matsuzaki S, Yoshino K, Ueda Y, Matsuzaki
S, Kakuda M, Okazawa A, Egawa-Takata T, Kobayashi E and Kimura T:
Potential targets for ovarian clear cell carcinoma: A review of
updates and future perspectives. Cancer Cell Int. 15:1172015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fokas E, Prevo R, Pollard JR, Reaper PM,
Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM,
Gillies McKenna W, et al: Targeting ATR in vivo using the novel
inhibitor VE-822 results in selective sensitization of pancreatic
tumors to radiation. Cell Death Dis. 3:e4412012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee JM, Nair J, Zimmer A, Lipkowitz S,
Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee
MJ, et al: Prexasertib, a cell cycle checkpoint kinase 1 and 2
inhibitor, in BRCA wild-type recurrent high-grade serous ovarian
cancer: A first-in-class proof-of-concept phase 2 study. Lancet
Oncol. 19:207–215. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
McCabe N, Hanna C, Walker SM, Gonda D, Li
J, Wikstrom K, Savage KI, Butterworth KT, Chen C, Harkin DP, et al:
Mechanistic Rationale to Target PTEN-Deficient Tumor Cells with
Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res.
75:2159–2165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dedes KJ, Wetterskog D, Mendes-Pereira AM,
Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A,
Lord CJ, et al: PTEN deficiency in endometrioid endometrial
adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl
Med. 2:53ra75. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sugiyama T, Kamura T, Kigawa J, Terakawa
N, Kikuchi Y, Kita T, Suzuki M, Sato I and Taguchi K: Clinical
characteristics of clear cell carcinoma of the ovary: A distinct
histologic type with poor prognosis and resistance to
platinum-based chemotherapy. Cancer. 88:2584–2589. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Basith S, Cui M, Macalino SJY and Choi S:
Expediting the Design, Discovery and Development of Anticancer
Drugs using Computational Approaches. Curr Med Chem. 24:4753–4778.
2017.PubMed/NCBI
|














