Introduction
High mobility group box protein 1 (HMGB1) is the
non-chromosome-related group of proteins. It was first isolated in
1973 by Goodwin et al (1), and
it is named after its rapid rate of electrophoresis in a
polyacrylamide gel. HMGB1 is expressed in the nucleus of almost all
eukaryotic cells and is encoded by the human HMGB1 gene (13q12)
(2). HMGB1 is involved in stabilizing
chromosomal structure in the nucleus, and in regulating the
transcription of genes that are critical for maintaining basic life
processes. When released from the cell, HMGB1 binds to its specific
receptor under specific pathological or physiological conditions,
which can mediate multiple inflammatory and autoimmune diseases
(3).
In recent years, the high incidence of
cerebrovascular disease has markedly affected the lives of patients
(4). According to recently released
data, in hospitalized patients aged between 55 and 63 years in the
United States, the incidence of acute ischemic stroke is
202.5/10,000, the incidence of subarachnoid hemorrhage (SAH) is
11.9/10,000 and the incidence of intracerebral hemorrhage is
22.6/10,000 (4). Although treatment
methods have improved over time, treatment remains invasive
(5,6).
Therefore, it is important to investigate the pathogenesis of
cerebrovascular disease and to identify non-invasive treatment
methods.
An increasing number of studies have demonstrated
that the inflammatory response involving HMGB1 serves an important
role in the course of acute cerebrovascular disease. This review
summarized the structure, function, receptors and signaling
pathways of HMGB1, and retrospectively analyzed the role of HMGB1
in ischemic cerebrovascular disease, hemorrhagic cerebrovascular
disease and cerebral venous sinus thrombosis.
HMGB1
The structure of HMGB1
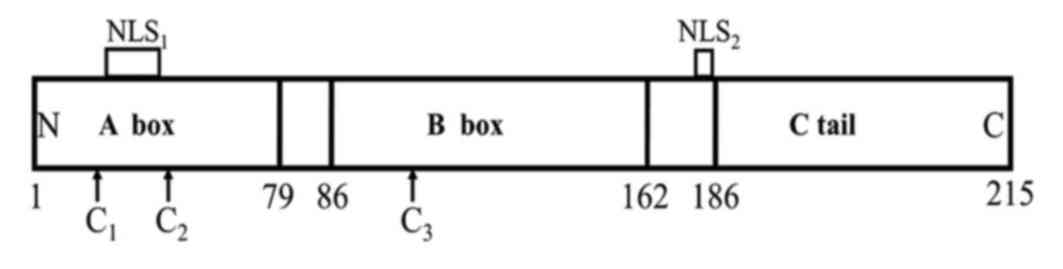
The sequence and structure of the HMGB1 protein are
highly evolutionarily conserved. HMGB1 is composed of 215 amino
acids, and has a molecular weight of ~25 kDa. HMGB1 includes three
structural domains: Two relatively rigid DNA binding domains (A and
B box) located at the N-terminal, which is termed the HMG box
field, and a negatively charged acidic tail comprising 30 glutamic
and aspartic acids (7,8). The A box is located at the 1–79 loci of
the HMGB1 molecular amino acid sequence and the B box is located at
the 86–162 loci, and the amino acid homology rate of the two is
>80%. The acidic tail between the B box and the C-terminal is
connected by a flexible connection containing 24 amino acids
(Fig. 1). Following HMGB1 being
released to the outside of the cell, the B box is the main
structural functional area that causes inflammation (7,9). The A box
has an antagonistic effect on the inflammatory response caused by
the B box, and this anti-inflammatory ability is enhanced following
the fusion of the acidic C-terminal.
The HMGB1 molecule contains two nuclear localization
sequences (NLS), respectively located in the A box (28–44) and
the junction area of box B and the C tail (179–185). It also
contains three cysteine residues, which are located separately at
the 23 and 45 sites of the A box and the 106 locus of box B
(Fig. 1) (8). Following stimulation, two cysteine
residues can form a disulfide bond, and thus HMGB1 exists as three
subtypes, the ‘disulfide HMGB1′, ‘thiol HMGB1′ and ‘oxidized HMGB1′
(10). Disulfide HMGB1 is the main
subtype involved in the acute and chronic inflammatory response in
the extracellular space and serum, which can further activate
macrophages/monocytes to amplify the inflammatory response. The
mechanism of HMGB1 is mainly involved in non-inflammatory responses
and its mechanism has yet to be elucidated. Thiol HMGB1 can be
released early and is able to repair cell damage by recruiting
inflammatory cells (11).
Secretion of HMGB1
HMGB1 is secreted by two modes: Passive release and
active secretion. The two secretory pathways differ in their
molecular mechanism, release kinetics and downstream signaling
pathway. Passive release occurs instantaneously upon the
destruction of cellular integrity, as HMGB1 is not associated with
nuclear DNA in living cells (8).
Under the stimulation of pathogen/microbe-associated
molecular patterns and endogenous inflammatory mediators, including
tumor necrosis factor (TNF), interleukin-1 (IL-1) and interferon-γ
(IFN-γ), macrophages, monocytes, dendritic cells, endothelial cells
and other immune cells can actively secrete HMGB1 (8). HMGB1 can also induce its own release
through pre-feedback regulation. Neurons, astrocytes, leukemia
cells and neuroblastoma cells can also promote the active secretion
of HMGB1 (8). Active secretion is
much slower than passive release, and can be divided into two
steps. To begin with, the HMGB1 in the nucleus is transferred to
the cytoplasm through the internuclear pore. This process relies on
the Janus kinase-signal transducer and activator of transcription
signaling pathway and the super-acetylation of two key lysine
residues in NLS (12), preventing
HMB1 from entering the cytoplasm and returning to the nucleus,
which may aid HMGB1 in accumulating in the cytoplasm. The second
stage gradually induces the programmed death of inflammatory cells;
alternatively, through secreted lysosomes, the intracellular HMGB1
is released from the cells (13).
Biological functions of HMGB1
Under physiological conditions, HMGB1 is involved in
stabilizing chromosomal structure in the nucleus, maintaining gene
stability, and induces DNA bending. In this process, box A of the
two molecules of HMGB1 can form a special structure successively or
simultaneously with DNA, which causes the DNA to bend and
reconstruct (14). In addition, HMGB1
can directly participate in the repair process of DNA damage
following binding to DNA, namely in nucleotide, base excision,
mismatch and double chain fracture repairs (15). The absence of HMGB1 can lead to
increasing chromosomal instability (16).
Yanai et al (17), demonstrated that the absence of HMGB1
would weaken the response of the body to the extracellular signals
associated with viral invasion, Toll-like receptor (TLR) ligands
and PRRs. Therefore, HMGB1 may have a central role in early
immunization activities. HMGB1 was one of the first members of its
family to be identified. The downstream inflammatory ligands can
induce the oligomerization of NOD-like receptor (NLR) and the
assembly of inflammatory protein complexes (18). NLR molecules contain a leucine-rich
repeat (LRR) domain, which has the functions of a combination of
ligands. HMGB1 can also induce cell stress, mediating
double-stranded RNA-dependent protein kinase autophosphorylation
and promoting inflammatory cascade amplification in the process
(19).
The HMGB1 in the cytoplasm can initiate autophagy,
which is a self-protective process that removes damaged
mitochondria and microbial invasions in the cell by combining with
beclin-1 (20).
The HMGB1 signaling pathway
RAGE was the first identified receptor of HMGB1; it
is a multifunctional transmembrane receptor of the immunoglobulin
superfamily, which is involved in the maintenance of homeostasis
and the occurrence of inflammation, and is encoded by a gene on
chromosome 6p21.3 (21). When
combined with HMGB1, it can activate p38 mitogen-activated protein
kinase (MAPK), extracellular signal-regulated kinase 1 (ERK1) and
ERK2, which in turn causes phosphorylation and degradation of the
inhibitor of nuclear factor κB (IκB) to activate NF-κB (22). HMGB1/RAGE can also induce the
expression of MAPK, vascular cell adhesion molecule 1 and matrix
metalloproteinase (MMP) (23).
The TLR is a member of the type I transmembrane
superfamily, and consists of an extracellular LRR structural domain
and a Toll/interleukin-1 receptor (TIR) structural domain in the
cytoplasm (21). The TLR signaling
pathway is divided into MyD88-dependent and MyD88-independent
pathways. Following combining with a single ligand, TLR upregulates
MyD88 or other adaptive molecules to induce the activation of
downstream factors, including NF-κB, MAPK and IFN regulatory
factors (24). HMGB1 interacts with
TLR-2, TLR-4 and TLR-9, triggering the activation of NK-κB and IRF
pathways (25). Following the
stimulation of TLR2, the downstream signal can be mediated by Rac1
and PI3K, increasing the adhesion of CD11b/CD18 and intercellular
cell adhesion molecule-1 (ICAM-1), and Akt is activated directly by
the p65 transcription complex or by the IκB kinase pathway; both of
these pathways are able to activate NF-κB (22). Following the binding of TLR-4 to
HMGB1, the interleukin-1 receptor-related kinase 1 can be
phosphorylated, thereby activating the downstream signaling
molecule NF-κB, and promoting MAPK by phosphorylating JNK, ERK, p38
and IκB (26). TLR9, normally located
in the endoplasmic reticulum, can be transferred to the early
endosomes in an HMGB1-dependent manner (27). TLR9 in endosomes and/or lysosomes can
identify CpG DNA binding to HMGB1, and can then mediate NF-κB and
its downstream inflammatory response (28). In recent years, it has been
demonstrated that the combination of HMGB1 and TLR5 can also rely
on MyD88 to activate the downstream signal (29).
C-X-C chemokine receptor 4 (CXCR4) is a member of
the G protein-coupled receptor family, which has a low expression
in normal tissues, but a significantly higher expression in tumor
tissues (21). Recent studies have
demonstrated that CXCR4 is also involved in inflammatory responses,
and that thiol HMGB1 can form complexes with CXCL12 and activate
CXCR4 to promote the production of inflammatory cells and cytokines
(30).
HMGB1 and acute cerebrovascular disease
Acute ischemic cerebrovascular
disease
Worldwide, ischemic cerebrovascular disease is the
leading cause of disability and the third leading cause of
mortality, and therefore is a heavy social and economic burden
(31). Following cerebral ischemia,
neuroinflammation and stress serve important roles in the
pathogenesis of the disease (31).
The inflammatory response in ischemic stroke consists of two
stages: The early stage of destruction of the nerve tissue and the
late stage of organizational reconstruction.
Following acute ischemic stroke, damaged brain
tissue can release HMGB1, recruiting a variety of pro-inflammatory
cytokines and chemokines, and increasing adhesion molecule
expression, further activating brain cells and immune cells
(32). Following cerebral ischemia,
astrocytes and endothelial cells may be the early targets of HMGB1,
which can be activated in this process. The former can directly
transmit signals to neurons and blood vessels, and the latter
upregulates ICAM-1 expression, recruiting immune cells into the
ischemic area (33). The destruction
of the blood brain barrier (BBB) is an important stage of ischemic
brain injury, involving multiple cytokines. The activation of MMPs
and the expression of various proteases results in the
decomposition of the BBB, exacerbating leukocyte extravasation
(34). HMGB1 can increase vascular
permeability and promote BBB decomposition (35). Anti-HMGB1 antibody can inhibit the
morphological and functional changes in the BBB induced by HMGB1
(36). Tsukagawa et al
(32) demonstrated that quantitative
serum HMGB1 levels could be used to evaluate the prognosis of
ischemic stroke and may be more accurate than the existing
evaluation methods.
Ischemic reperfusion injury can further aggravate
functional metabolic disorders and structural damage in ischemic
tissues. Apoptosis is strictly regulated by the MAPK family, and
the c-Jun N-terminal kinase (JNK), ERK1/2 and p38 protein family in
ischemia reperfusion injury is activated (37). The continuous activation of MAPK is
associated with the death or apoptosis of neurons in the
post-stroke stage of ischemic stroke (37). A study by Gong et al (38), demonstrated that glycyrrhizin can be
used as a HMGB1 inhibitor to inhibit the JNK and p38 pathways in
rats. Umahara et al (39),
revealed that in the chronic stages of cerebral infarction, certain
macrophages that were located in the ischemic region were positive
for HMGB1. They hypothesized that there may be two reasons for
this. One is that the HMGB1-associated inflammatory response in
chronic cerebral infarction develops from acute cerebral
infarction. Alternatively, HMGB1-positive macrophages may induce
autophagy in the area of chronic ischemic injury, possibly due to
the fact that HMGB1 can maintain autophagy (20).
In the recovery phase of ischemic stroke, HMGB1 may
promote brain remodeling. Chen et al (40) and Wu et al (41), demonstrated that IL-6 and vascular
endothelial growth factor (VEGF) mediate the reactive astrocyte
release of HMGB1, and participate in the angiogenesis and
neurogenesis in the late phase of stroke, promoting brain
remodeling and neurological function recovery. Brain remodeling was
inhibited following the administration of HMGB1 inhibitors. These
studies have aided in improving the prognosis of ischemic stroke at
different stages.
Acute hemorrhagic cerebrovascular
disease
Intracranial aneurysm and SAH
Intracranial aneurysms are pathological local
dilations caused by changes in local intracranial vessels. Among
the various causes of SAH, spontaneous aneurysm rupture is the most
common and requires attention. Zhang et al (42), demonstrated that HMGB1 was highly
expressed on ruptured and unruptured aneurysm walls and, compared
with the latter, the former had a higher level of expression.
However, there is no significant association between the size of
the aneurysm and the expression level of HMGB1. Through double
immunofluorescence staining, Chalouhi et al (43) demonstrated that HMGB1 was expressed in
the nucleus of smooth muscle cells, macrophages, lymphocytes and
endothelial cells; these cells were involved in the remodeling of
the aneurysm wall. NF-κB is highly expressed in the aneurysm wall,
and the formation of the aneurysm is hindered by the application of
NF-κB inhibitors (44). The incidence
of intracranial aneurysms is associated with atherosclerosis and
HMGB1 is involved in the formation of atheromatous plaques;
following endothelial cell injury, activated NF-κB induces the
activation of leukocyte adhesion molecules and a variety of
cytokines, including HMGB1, and these signals can collect
macrophages in the wall of blood vessels. Macrophages immersed in
vessel walls can transform into foam cells and release HMGB1 again,
forming a positive feedback inflammatory pathway (45). It has been demonstrated that in the
formation and development of intracranial aneurysms, HMGB1 mediates
the inflammatory response and participates in the formation of
atherosclerotic plaques; as a result, the vascular wall is
thickened or narrowed, and vascular remodeling increases the risk
of rupture. Therefore, inhibition of HMGB1 may alleviate
atheromatous plaque formation and reduce the risk of aneurysm
rupture.
SAH is a life-threatening central nervous system
disease. Cerebral vasospasm is one of the most important causes of
the high morbidity and mortality of SAH. Previous study have
demonstrated that 30–70% of patients with aneurysms and SAH will
have vasospasm (46). It has been
observed that this pathophysiological process is associated with
inflammatory reactions, including leukocyte recruitment,
infiltration and activation. Following SAH, HMGB1 participates in
the inflammatory response and serves an important role in apoptosis
and vascular spasm (47). Zhao et
al (48), revealed that the
artery endothelial cells and smooth muscle cells in the damaged
brain region following SAH were activated to stimulate the
secretion of HMGB1, which may promote intracranial arterial spasms.
Umahara et al (39), conducted
autopsies of patients with SAH and revealed that an HMGB1-like
immunoreaction was observed in the cytoplasm of vascular smooth
muscle cells in the hematoma. HMGB1 could not only promote the
occurrence of cerebral vasospasm, but also increase the gene and
protein expression levels of RAGE in neurons and microglia around
the hematoma, and the number of microglia was also significantly
increased. As a main downstream factor of RAGE, the main p65
subunit of NF-κB was significantly increased, indicating that RAGE
promoted the activation of NF-κB at the early stage of SAH
(49). Resveratrol, as an inhibitor
of HMGB1, can relieve this pathophysiological change; the reason
for this is that resveratrol may be involved in SAH-induced
neuronal apoptosis, brain edema and nerve injury via inhibition of
the HMGB1-mediated TLR4/MyD88/NF-κB pathway in the early stage of
SAH (50). Clinically, patients with
aneurysmal SAH may develop cerebral vasospasm and delayed cerebral
ischemia, increased expression of HMGB1 during the course of
disease, increased cerebral vasospasms and eventually an increased
risk of cerebral infarction (51).
Therefore, HMGB1 rapidly interacts with nerve cells and glial cells
following SAH, and participates in cerebral vasospasm and
apoptosis.
In the recovery phase of SAH, Tian et al
(52) suggested that HMGB1 can
promote neurological recovery and blood vessel regeneration via
RAGE mediation, which further demonstrated that HMGB1 serves
different roles at different disease stages.
Intracranial hemorrhage (ICH)
ICH accounts for ~15% of all stroke cases and has a
mortality rate close to 50% (53).
Even patients who survive ICH often experience severe disability
(53). The reaction that follows ICH
is a complex process and involves a series of pathophysiological
reactions, including excitotoxicity, free radical injury and
inflammatory reactions (54).
Hematoma can cause inflammatory reactions in the surrounding
tissue, and can activate glial cells and neurons to aggravate
cerebral injury (55). Following
acute ICH, HMGB1 is released into the extracellular space by the
damaged cell and, as a proinflammatory cytokine, it immediately
causes a downstream inflammatory reaction (56), leading to brain damage, affecting
neurobehavioral functioning, increasing the permeability of the BBB
and aggravating brain edema (57). It
has been reported that an anti-HMGB1 antibody can improve brain
injury and nerve function defects following ICH in rats (58). In this process, HMGB1 triggers three
specific downstream receptors, RAGE, TLR-2 and TLR-4, of which RAGE
may be the most important. The use of RAGE antagonists alone
hinders ICH-induced inflammatory cell infiltration, and decreases
its downstream factors IL-1β and MMP-9 in the brain tissue around
the site of the ICH (57). As a
downstream factor of HMGB1, NF-κB is highly sensitive to oxidative
stress in the surrounding area following ICH, and mediates
downstream IL-1β and ICAM-1, which serve a key role in cell death
following ICH, particularly in apoptosis (55).
At the end of the course of the disease, the brain
begins to undergo a remodeling process, involving synaptic and
vascular regeneration, which may aid in the recovery of nerve
function following ICH. In this process, the progenitor cells of
the subventricular region of the hippocampus migrate into the
damaged brain region and differentiate into mature neurons and
glial cells (59). HMGB1 serves an
important role in promoting tissue recovery and remodeling at the
late stage of disease. Following ICH, the expression levels of
HMGB1 and VEGF are increased in surrounding brain tissue, and the
inhibition of HMGB1 activity could significantly reduce the
upregulation of VEGF (56). The ICH
rat model induced by collagenase indicates that HMGB1 promotes
angiogenesis mainly by mediating RAGE (56). In addition, HMGB1 can promote the
expression of MMP-9, improve brain damage and restore nerve
function (54). Therefore, when
inhibiting the HMGB1-RAGE signaling pathway for ICH treatment,
attention should be paid to the fact that early inhibition of this
pathway can improve the treatment of ICH, while later inhibition of
this pathway could hinder the recovery of neurological function
(57). These studies indicated that
HMGB1 exerted different effects at different phases of intracranial
hemorrhage; therefore, future studies need to focus on determining
time frames.
Cerebral venous thrombosis
Cerebral venous sinus thrombosis is a relatively
rare cerebrovascular disease, which accounts for 0.5–1% of the
causes of stroke. A total of 50% of patients have venous cerebral
infarction with a series of clinical manifestations, including
headache, hemiplegia, epileptic seizures and intracranial
hypertension. Its pathophysiological mechanism can be explained by
the fact that, following thrombosis, the pressure in the veins and
capillaries is higher and blood return is limited, leading to
tissue hypoxia, which contributes toward brain edema and ICH. The
increase in venous pressure also hinders the reabsorption of the
cerebrospinal fluid and further aggravates the intracranial
hypertension (60). Although an
increasing amount of attention has been paid to CVST in recent
years, little is known regarding the exact pathogenesis and
progression of the disease.
In recent years, a number of studies have
demonstrated that HMGB1 may be involved in the pathogenesis of CVST
(Fig. 2): i) In a study undertaken by
Gu et al (61), the protein
and mRNA expression levels of HMGB1-RAGE were upregulated in the
cerebral infarction area in rats following CVST. Recombinant human
soluble (rhs)-TM was added to the model, which reduced the nerve
injury and infarct volume, and reduced the expression level of
HMGB1-RAGE and the proinflammatory cytokines TNF-α, IL-1β and IL-6
in the ischemic penumbra; and ii) in the pathogenesis of CVST,
venous return is blocked, but in view of the abundant collateral
circulation of the cerebral venous system, there may be other
mechanisms involved in the occurrence of cerebral edema following
CVST. Nagai et al (62),
demonstrated that following the occurrence of CVST, the
concentration of monocyte chemoattractant protein-1 (MCP-1)
increased. Meanwhile, HMGB1 could activate the expression of MCP-1
(23); therefore, HMGB1 may also be
involved in the inflammatory response process of CVST in this way.
A study undertaken by Yang et al (63), also demonstrated that apoptosis
participated in the pathogenesis of CVST: caspase-3 served an
important role in cellular apoptosis, and caspase-3 participated in
the pathogenesis of CVST. Bax is a type of pro-apoptotic protein
and Bcl-2 is one of the most active inhibitors of apoptosis.
Bcl-2/Bax has been demonstrated to be essential for determining
whether apoptosis has occurred, and the ratio of Bcl-2/Bax
decreases significantly following the occurrence of CVST. HMGB1 can
induce apoptosis via NF-κB by activating TLR-4 (47). The results of these studies suggested
that HMGB1 serves an important role in the pathogenesis of
CVST.
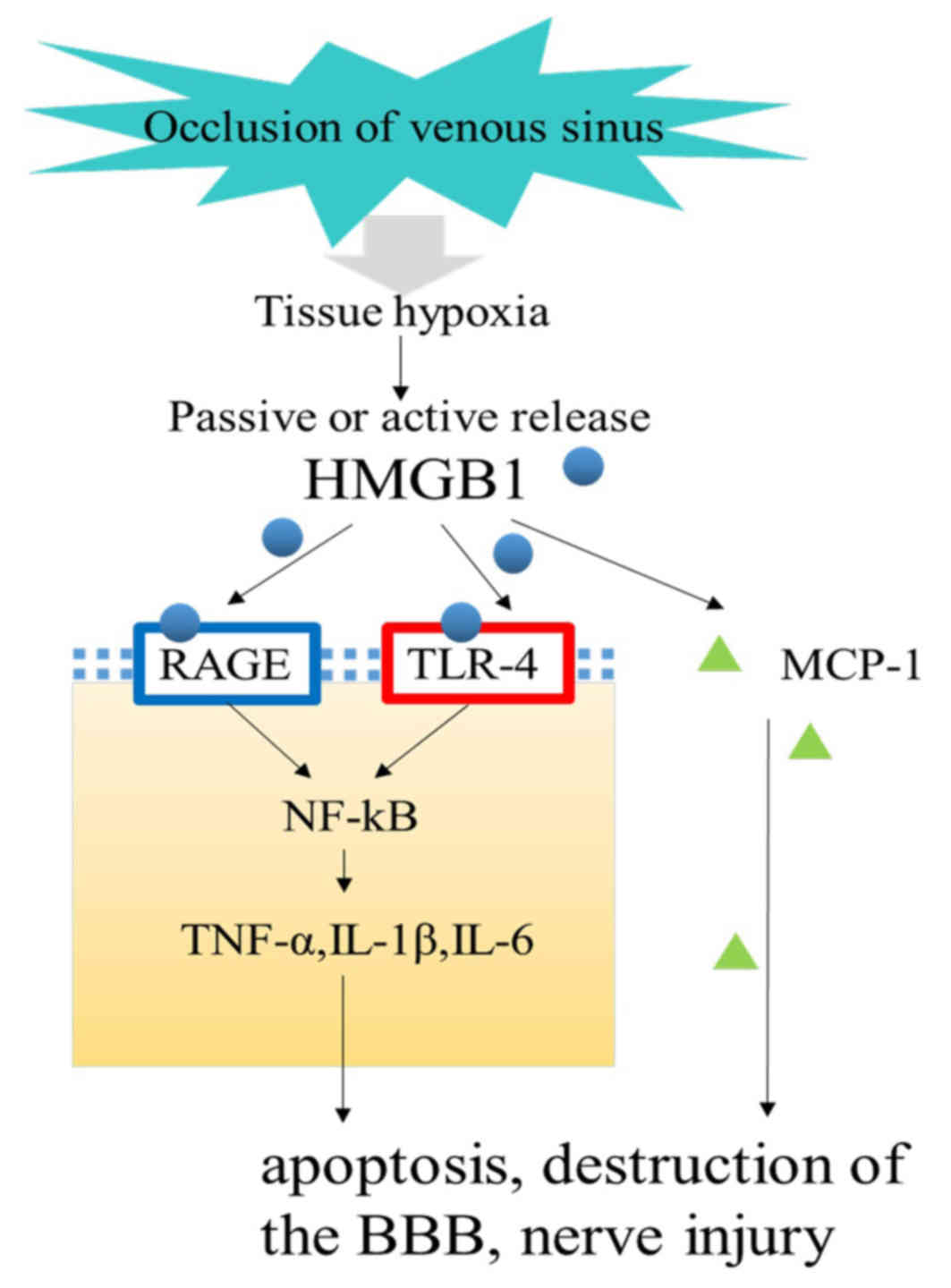 | Figure 2.Signaling pathway of HMGB1 in
cerebral venous sinus thrombosis. Following thrombosis, the
pressure in the veins and capillaries is higher and blood return is
limited, leading to tissue hypoxia. Subsequently, damaged tissue
can actively or passively release HMGB1, triggering downstream
inflammatory responses, participating in BBB disruption, apoptosis
and nerve damage, and further aggravating brain damage. HMGB1, high
mobility group box B1; BBB, blood brain barrier; TNF-α, tumor
necrosis factor α; IL, interleukin; MCP-1, monocyte chemoattractant
protein-1; TLR-4, Toll-like receptor 4. |
In general, following acute cerebrovascular
incident, HMGB1 interacts with neurons, endothelial cells and glial
cells to participate in BBB disruption, vasospasm and apoptosis
through mediating downstream inflammatory factors, which contribute
toward cerebral edema and nerve injury. On the other hand, during
the recovery phase, HMGB1 can promote brain repair and remodeling,
contributing toward the recovery of neurological function.
Therefore, targeted therapy for HMGB1 may have a positive effect on
acute cerebrovascular disease (Fig.
3).
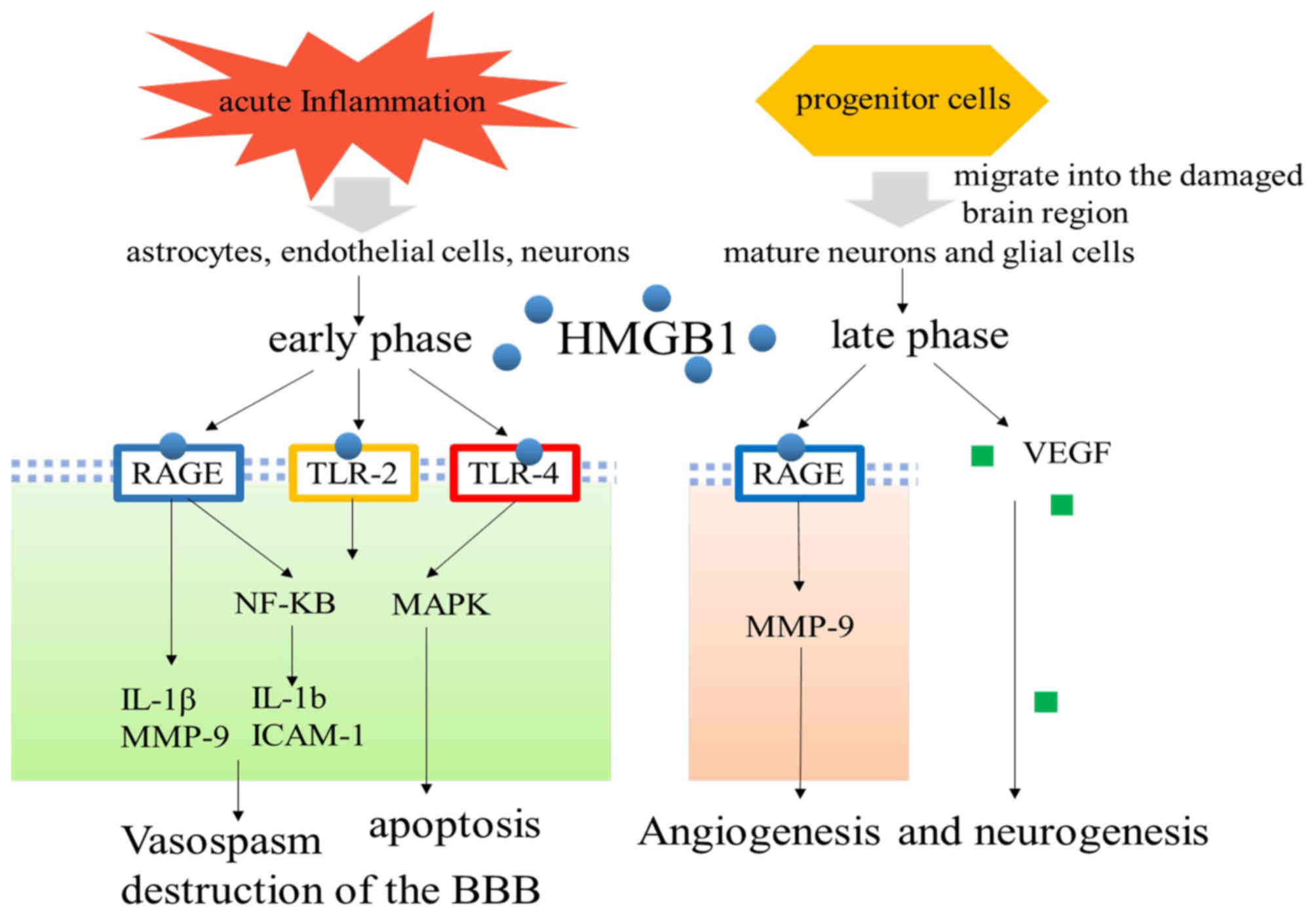 | Figure 3.Signaling pathway of HMGB1 in the
early and late phases of cerebrovascular disease. Following acute
cerebrovascular disease, under the stimulation of inflammation,
astrocytes, endothelial cells and neurons can actively secrete
HMGB1, while passive release occurs instantaneously upon the
destruction of cellular integrity. Extracellular HMGB1 interacts
with neurons, endothelial cells and glial cells to participate in
BBB disruption, vasospasm and apoptosis through mediating
downstream inflammatory factors, which contribute toward cerebral
edema and nerve injury. On the other hand, during the late phase,
HMGB1 can promote brain repair and remodeling, participating in
angiogenesis and neurogenesis, contributing toward the recovery of
neurological function. HMGB1, high mobility group box B1; BBB,
blood brain barrier; TLR, Toll-like receptor; VEGF, vascular
endothelial growth factor; NF-κB, nuclear factor-κB; MAPK,
mitogen-activated protein kinase; IL, interleukin; MMP, matrix
metalloproteinase; ICAM-1, intercellular cell adhesion
molecule-1. |
Conclusion
Due to the diversity of acute cerebrovascular
diseases and complex pathophysiological mechanisms, it is very
difficult to fundamentally cure acute cerebrovascular diseases by
relying on the existing treatment methods. Gene therapy has the
characteristics of high specificity, high biological activity and
low toxicity, and can be targeted to improve or inhibit the
expression of the target gene in vivo, so as to achieve the
purpose of treating a disease. HMGB1 serves an important role in
promoting the inflammatory response in acute cerebrovascular
diseases. Gene therapy targeting HMGB1 may achieve satisfactory
results in patients with acute cerebrovascular diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Fujian Province (grant no. 2017J01323).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SWM researched literatures, and was a major
contributor in writing the manuscript. YD researched literatures
and edited the manuscript. SSW reviewed the manuscript. JJG
reviewed the manuscript and approved the final version. All authors
read and approved the final manuscript.
Ethics statement and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Goodwin GH, Sanders C and Johns EW: A new
group of chromatin-associated proteins with a high content of
acidic and basic amino acids. Eur J Biochem. 38:14–19. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sohun M and Shen H: The implication and
potential applications of high-mobility group box 1 protein in
breast cancer. Ann Transl Med. 4:2172016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bertheloot D and Latz E: HMGB1, IL-1α,
IL-33 and S100 proteins: Dual-function alarmins. Cell Mol Immunol.
14:43–64. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
George MG, Tong X and Bowman BA:
Prevalence of cardiovascular risk factors and strokes in younger
adults. JAMA Neurol. 74:695–703. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fotakopoulos G, Tsianaka E, Fountas K,
Makris D, Spyrou M and Hernesniemi J: Clipping versus coiling in
anterior circulation ruptured intracranial aneurysms: A
meta-analysis. World Neurosurg. 104:482–488. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ilyas A, Chen CJ, Raper DM, Ding D, Buell
T, Mastorakos P and Liu KC: Endovascular mechanical thrombectomy
for cerebral venous sinus thrombosis: A systematic review. J
Neurointerv Surg. 9:1086–1092. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Musumeci D, Roviello GN and Montesarchio
D: An overview on HMGB1 inhibitors as potential therapeutic agents
in HMGB1-related pathologies. Pharmacol Ther. 141:347–357. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andersson U and Tracey KJ: HMGB1 is a
therapeutic target for sterile inflammation and infection. Annu Rev
Immunol. 29:139–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang H, Wang H, Chavan SS and Andersson U:
High mobility group box protein 1 (HMGB1): The prototypical
endogenous danger molecule. Mol Med. 21 Suppl 1:S6–S12. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Antoine DJ, Harris HE, Andersson U, Tracey
KJ and Bianchi ME: A systematic nomenclature for the redox states
of high mobility group box (HMGB) proteins. Mol Med. 20:135–137.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andersson U, Antoine DJ and Tracey KJ: The
functions of HMGB1 depend on molecular localization and
post-translational modifications. J Intern Med. 276:420–424. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu B, Antoine DJ, Kwan K, Lundbäck P,
Wähämaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J,
et al: JAK/STAT1 signaling promotes HMGB1 hyperacetylation and
nuclear translocation. Proc Natl Acad Sci USA. 111:3068–3073. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu B, Nakamura T, Inouye K, Li J, Tang Y,
Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, et al:
Novel role of PKR in inflammasome activation and HMGB1 release.
Nature. 488:670–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sánchez-Giraldo R, Acosta-Reyes FJ,
Malarkey CS, Saperas N, Churchill ME and Campos JL: Two
high-mobility group box domains act together to underwind and kink
DNA. Acta Crystallogr D Biol Crystallogr. 71:1423–1432. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martinotti S, Patrone M and Ranzato E:
Emerging roles for HMGB1 protein in immunity, inflammation, and
cancer. Immunotargets Ther. 4:101–109. 2015.PubMed/NCBI
|
|
16
|
Kang R, Zhang Q, Zeh HJ III, Lotze MT and
Tang D: HMGB1 in cancer: Good, bad, or both? Clin Cancer Res.
19:4046–4057. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yanai H, Ban T, Wang Z, Choi MK, Kawamura
T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, et al: HMGB
proteins function as universal sentinels for nucleic-acid-mediated
innate immune responses. Nature. 462:99–103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franchi L, Muñoz-Planillo R and Núñez G:
Sensing and reacting to microbes through the inflammasomes. Nat
Immunol. 13:325–332. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu B, Wang H, Andersson U and Tracey KJ:
Regulation of HMGB1 release by inflammasomes. Protein Cell.
4:163–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yanai H, Matsuda A, An J, Koshiba R,
Nishio J, Negishi H, Ikushima H, Onoe T, Ohdan H, Yoshida N, et al:
Conditional ablation of HMGB1 in mice reveals its protective
function against endotoxemia and bacterial infection. Proc Natl
Acad Sci USA. 110:20699–20704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He SJ, Cheng J, Feng X, Yu Y, Tian L and
Huang Q: The dual role and therapeutic potential of high-mobility
group box 1 in cancer. Oncotarget. 8:64534–64550. 2017.PubMed/NCBI
|
|
22
|
van Beijnum JR, Buurman WA and Griffioen
AW: Convergence and amplification of toll-like receptor (TLR) and
receptor for advanced glycation end products (RAGE) signaling
pathways via high mobility group B1 (HMGB1). Angiogenesis.
11:91–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fiuza C, Bustin M, Talwar S, Tropea M,
Gerstenberger E, Shelhamer JH and Suffredini AF:
Inflammation-promoting activity of HMGB1 on human microvascular
endothelial cells. Blood. 101:2652–2660. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pradere JP, Dapito DH and Schwabe RF: The
Yin and Yang of Toll-like receptors in cancer. Oncogene.
33:3485–3495. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xi Y, Shao F, Bai XY, Cai G, Lv Y and Chen
X: Changes in the expression of the Toll-like receptor system in
the aging rat kidneys. PLoS One. 9:e963512014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang G, Zhang L, Ma L, Jiang R, Kuang G,
Li K, Tie H, Wang B, Chen X, Xie T, et al: Glycyrrhetinic acid
prevents acetaminophen-induced acute liver injury via the
inhibition of CYP2E1 expression and HMGB1-TLR4 signal activation in
mice. Int Immunopharmacol. 50:186–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian J, Avalos AM, Mao SY, Chen B, Senthil
K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al:
Toll-like receptor 9-dependent activation by DNA-containing immune
complexes is mediated by HMGB1 and RAGE. Nat Immunol. 8:487–496.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hiraku Y, Guo F, Ma N, Yamada T, Wang S,
Kawanishi S and Murata M: Multi-walled carbon nanotube induces
nitrative DNA damage in human lung epithelial cells via HMGB1-RAGE
interaction and Toll-like receptor 9 activation. Part Fibre
Toxicol. 13:162016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Das N, Dewan V, Grace PM, Gunn RJ, Tamura
R, Tzarum N, Watkins LR, Wilson IA and Yin H: HMGB1 activates
proinflammatory signaling via TLR5 leading to allodynia. Cell
Reports. 17:1128–1140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Magna M and Pisetsky DS: The role of HMGB1
in the pathogenesis of inflammatory and autoimmune diseases. Mol
Med. 20:138–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Godinho J, de Oliveira RMW, de
Sa-Nakanishi AB, Bacarin CC, Huzita CH, Longhini R, Mello JCP,
Nakamura CV, Previdelli IS, Dal Molin Ribeiro MH, et al:
Ethyl-acetate fraction of Trichilia catigua restores long-term
retrograde memory and reduces oxidative stress and inflammation
after global cerebral ischemia in rats. Behav Brain Res.
337:173–182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tsukagawa T, Katsumata R, Fujita M, Yasui
K, Akhoon C, Ono K, Dohi K and Aruga T: Elevated serum
high-mobility group box-1 protein level Is associated with poor
functional outcome in ischemic stroke. J Stroke Cerebrovasc Dis.
26:2404–2411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiu J, Nishimura M, Wang Y, Sims JR, Qiu
S, Savitz SI, Salomone S and Moskowitz MA: Early release of HMGB-1
from neurons after the onset of brain ischemia. J Cereb Blood Flow
Metab. 28:927–938. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shichita T, Sakaguchi R, Suzuki M and
Yoshimura A: Post-ischemic inflammation in the brain. Front
Immunol. 3:1322012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Takahashi HK, Liu K, Wake H, Liu
R, Maruo T, Date I, Yoshino T, Ohtsuka A, Mori S, et al: Anti-high
mobility group box-1 monoclonal antibody protects the blood-brain
barrier from ischemia-induced disruption in rats. Stroke.
42:1420–1428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li WA, Moore-Langston S, Chakraborty T,
Rafols JA, Conti AC and Ding Y: Hyperglycemia in stroke and
possible treatments. Neurol Res. 35:479–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang J, Wu Y, Weng Z, Zhou T, Feng T and
Lin Y: Glycyrrhizin protects brain against ischemia-reperfusion
injury in mice through HMGB1-TLR4-IL-17A signaling pathway. Brain
Res. 1582:176–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gong G, Xiang L, Yuan L, Hu L, Wu W, Cai
L, Yin L and Dong H: Protective effect of glycyrrhizin, a direct
HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced
inflammation, oxidative stress, and apoptosis in rats. PLoS One.
9:e894502014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Umahara T, Uchihara T, Hirokawa K, Hirao
K, Shimizu S, Hashimoto T, Terasi H and Hanyu H: Time-dependent and
lesion-dependent HMGB1-selective localization in brains of patients
with cerebrovascular diseases. Histol Histopathol. 33:215–222.
2018.PubMed/NCBI
|
|
40
|
Chen JY, Yu Y, Yuan Y, Zhang YJ, Fan XP,
Yuan SY, Zhang JC and Yao SL: Enriched housing promotes post-stroke
functional recovery through astrocytic HMGB1-IL-6-mediated
angiogenesis. Cell Death Discov. 3:170542017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu X, Liu S, Hu Z, Zhu G, Zheng G and Wang
G: Enriched housing promotes post-stroke neurogenesis through
calpain 1-STAT3/HIF-1α/VEGF signaling. Brain Res Bull. 139133–143.
(201841)2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang D, Wu W, Yan H, Jiang T, Liu M, Yu
Z, Li H and Hang C: Upregulation of HMGB1 in wall of ruptured and
unruptured human cerebral aneurysms: Preliminary results. Neurol
Sci. 37:219–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chalouhi N, Ali MS, Jabbour PM,
Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ and Dumont AS:
Biology of intracranial aneurysms: Role of inflammation. J Cereb
Blood Flow Metab. 32:1659–1676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Aoki T, Kataoka H, Nishimura M, Ishibashi
R, Morishita R and Miyamoto S: Regression of intracranial aneurysms
by simultaneous inhibition of nuclear factor-κB and Ets with
chimeric decoy oligodeoxynucleotide treatment. Neurosurgery.
70:1534–1543; discussion 1543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bianchi ME and Manfredi AA: High-mobility
group box 1 (HMGB1) protein at the crossroads between innate and
adaptive immunity. Immunol Rev. 220:35–46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Przybycien-Szymanska MM and Ashley WW Jr:
Biomarker discovery in cerebral vasospasm after aneurysmal
subarachnoid hemorrhage. J Stroke Cerebrovasc Dis. 24:1453–1464.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chang CZ, Wu SC, Kwan AL and Lin CL:
Rhinacanthin-C, A fat-soluble extract from Rhinacanthus
nasutus, modulates high-mobility group box 1-related
neuro-inflammation and subarachnoid hemorrhage-induced brain
apoptosis in a rat model. World Neurosurg. 86:349–360. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao XD, Mao HY, Lv J and Lu XJ:
Expression of high-mobility group box-1 (HMGB1) in the basilar
artery after experimental subarachnoid hemorrhage. J Clin Neurosci.
27:161–165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li H, Wu W, Sun Q, Liu M, Li W, Zhang XS,
Zhou ML and Hang CH: Expression and cell distribution of receptor
for advanced glycation end-products in the rat cortex following
experimental subarachnoid hemorrhage. Brain Res. 1543:315–323.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang XS, Li W, Wu Q, Wu LY, Ye ZN, Liu
JP, Zhuang Z, Zhou ML, Zhang X and Hang CH: Resveratrol attenuates
acute inflammatory injury in experimental subarachnoid hemorrhage
in rats via inhibition of TLR4 pathway. Int J Mol Sci.
17:172016.
|
|
51
|
Hendrix P, Foreman PM, Harrigan MR, Fisher
WSR III, Vyas NA, Lipsky RH, Lin M, Walters BC, Tubbs RS, Shoja MM,
et al: Impact of high-mobility group box 1 polymorphism on delayed
cerebral ischemia after aneurysmal subarachnoid hemorrhage. World
Neurosurg. 101:325–330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tian X, Sun L, Feng D, Sun Q, Dou Y, Liu
C, Zhou F, Li H, Shen H, Wang Z, et al: HMGB1 promotes
neurovascular remodeling via Rage in the late phase of subarachnoid
hemorrhage. Brain Res. 1670:135–145. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Qureshi AI, Mendelow AD and Hanley DF:
Intracerebral haemorrhage. Lancet. 373:1632–1644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lei C, Wu B, Cao T, Zhang S and Liu M:
Activation of the high-mobility group box 1 protein-receptor for
advanced glycation end-products signaling pathway in rats during
neurogenesis after intracerebral hemorrhage. Stroke. 46:500–506.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang YX, Yan A, Ma ZH, Wang Z, Zhang B,
Ping JL, Zhu JS, Zhou Y and Dai L: Nuclear factor-κB and apoptosis
in patients with intracerebral hemorrhage. J Clin Neurosci.
18:1392–1395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lei C, Zhang S, Cao T, Tao W, Liu M and Wu
B: HMGB1 may act via RAGE to promote angiogenesis in the later
phase after intracerebral hemorrhage. Neuroscience. 295:39–47.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li D, Lei C, Zhang S, Zhang S, Liu M and
Wu B: Blockade of high mobility group box-1 signaling via the
receptor for advanced glycation end-products ameliorates
inflammatory damage after acute intracerebral hemorrhage. Neurosci
Lett. 609:109–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang D, Liu K, Wake H, Teshigawara K, Mori
S and Nishibori M: Anti-high mobility group box-1 (HMGB1) antibody
inhibits hemorrhage-induced brain injury and improved neurological
deficits in rats. Sci Rep. 7:462432017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shang J, Deguchi K, Ohta Y, Liu N, Zhang
X, Tian F, Yamashita T, Ikeda Y, Matsuura T, Funakoshi H, et al:
Strong neurogenesis, angiogenesis, synaptogenesis, and antifibrosis
of hepatocyte growth factor in rats brain after transient middle
cerebral artery occlusion. J Neurosci Res. 89:86–95. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sharma KM and Ahn J: Cerebral venous sinus
thrombophlebitis as a complication of acute otitis media. J Emerg
Med. 48:e9–e13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gu JJ, Chen JB, Zhang JH, Zhang H and Wang
SS: Recombinant human soluble thrombomodulin protects against brain
injury in a CVST rat model, via downregulation of the HMGB1-RAGE
axis. Mol Med Rep. 14:5217–5222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nagai M, Terao S, Yilmaz G, Yilmaz CE,
Esmon CT, Watanabe E and Granger DN: Roles of inflammation and the
activated protein C pathway in the brain edema associated with
cerebral venous sinus thrombosis. Stroke. 41:147–152. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yang H, Meng Z, Zhang C, Zhang P and Wang
Q: Establishing a new rat model of central venous sinus thrombosis
and analyzing its pathophysiological and apoptotic changes. J
Neurosci Methods. 203:130–135. 2012. View Article : Google Scholar : PubMed/NCBI
|