Introduction
Crouzon syndrome is a rare genetic disorder that is
characterized by a triad of premature craniosynostosis, orbital
proptosis and midfacial hypoplasia (1). Crouzon syndrome, which exhibits
autosomal dominant inheritance, was first described by Louis
Edouard Octave in 1912(2) and has
been identified as one of the most common craniosynostosis
syndromes (3). When compared with
Crouzon syndrome, Apert syndrome has a broad clinical spectrum,
including complex craniofacial involvement, and deformities of the
hands, feet and other joints that require multiple surgical
procedures to correct (4). In
addition to these severe dysmorphologic characteristics, patients
with Apert syndrome have been reported to exhibit variable degrees
of neurodevelopmental delay, cognitive deficit and mental
retardation, while most patients with Crouzon present with normal
mental development and intelligence (4). The severity of symptoms in Crouzon
syndrome may vary among affected people, even within a family. The
worldwide prevalence of Crouzon syndrome is from 1/60,000 to
1/1,000 live births based on several factors, including race,
region and ethnicity (5).
Fibroblast growth factors (FGFs) comprise a large
group of developmental and physiological signaling molecules. FGFs
serve critical roles in the proliferation, migration and
particularly in the differentiation of endothelial cells, and serve
to promote angiogenesis (6). The
specificity of FGFs-FGF receptor interactions and the factors that
affect the stability of this complex, have been the focus of many
studies (7,8). The human fibroblast growth factor
receptor (FGFRs) family is a subfamily of receptor tyrosine kinases
(RTKs), which comprise five members: FGFR1, FGFR2, FGFR3, FGFR4 and
FGFR5. The common structure of the FGFR comprises an intracellular
domain with tyrosine kinase activity, an extracellular ligand
domain with three immunoglobulin (Ig)-like domains (IgI, IgII and
IgIII) and a single transmembrane helix domain (6,9).
Crouzon syndrome formation is closely associated
with mutations of the FGFR2 gene (5,10). The
FGFR2 gene encodes the FGFR2 protein, which is located on
chromosome 10 and consists of >21 exons (11). FGFR2 is highly expressed in the
foetal brain and in important tissues, including the brain, retina,
spinal cord, salivary gland, skin, kidney and uterus of the adult
body (12). To date, ~60 different
mutations of FGFR2 have been linked to Crouzon syndrome formation
(11). Additionally, the majority of
patients (80%) with mutations in exon 8 and 10 are directly
associated with this syndrome (5).
However, few studies have assessed the influences of other distinct
exons that are linked to Crouzon syndrome.
The prenatal identification of the Crouzon
syndrome-causing FGFR2 mutation is crucial for the
subsequent pregnancies of affected patients. Prenatal real-time
ultrasonographic diagnosis of exophthalmus is possible at the 35th
week of gestation in a foetus of a patient affected with Crouzon
syndrome (13). Furthermore, the
successful diagnosis of Crouzon syndrome during pregnancy may occur
via chorionic villus biopsy using polymerase chain reaction (PCR)
by targeting FGFR2, a known mutation found within the
pregnant mother (14). However, the
diagnosis of Crouzon syndrome remains challenging due to the
relatively complicated phenotype and the difficulty and
availability of early diagnostic techniques.
The current study assessed the FGFR2 gene
associated with Crouzon syndrome in a Vietnamese family and
characterized the associated clinical features.
Materials and methods
Patient recruitment and clinical
evaluations
The current study was approved by the Institute of
Biotechnology and Hanoi Medical University Hospital (Ha Noi,
Vietnam), and informed consent was obtained from all family members
prior to blood sample collection. All family members from a
Vietnamese family of three generations and the 200 control subjects
were recruited at Hanoi Medical University Hospital (Ha Noi,
Vietnam) between June and July 2018. The samples were collected
from 100 males and 100 unpregnant females between 10-60 years of
age at the time of sample collection, and were tested within 1
month from the time of blood collection. A standard ophthalmic
test, which included the assessment of visual acuity, refractive
error and intraocular/fluid pressure was performed in patient
III-16, who was diagnosed with Crouzon syndrome. Visual acuity was
examined using the VISUSCREEN 100/500 acuity chart (Carl Zeiss AG,
Oberkochen, Germany). The degree of refractive error in the eye was
examined via VISUREF® 100 (Carl Zeiss AG) and the
intraocular and fluid pressure inside the eye was determined using
VISUPLAN 500 (Carl Zeiss AG). Computed tomography (CT) and physical
examinations including blood examination, urinalysis,
electrocardiogram, chest X-ray, blood biochemistry, blood lipid and
blood coagulation tests, were also performed to exclude systemic
diseases The biochemical tests were performed using the blood of
patient III-16 using a Biochemistry Analyzer Machine with
biochemistry test kit (cat. no. MSLBA28; MSL Electronic Technology
(Shenzhen) Co., Ltd., Shenzhen, China). This was utilized to
measure sodium, potassium, chloride, bicarbonate, blood urea
nitrogen, magnesium, creatinine, glucose and calcium. An ultrasound
examination was used to assess a 16-week-old fetus (III-18) of
mother (II-16).
Sample collection
The peripheral blood leucocytes of all family
members (excluding the 16-week-old fetus) were separated from whole
blood using a Histopaque-1077 kit (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany; cat. no. 10771) according to manufacturer's
protocol. Genomic DNA samples were then extracted from the
peripheral blood leucocytes of all family members (excluding the
16-week-old foetus) using the Qiagen QIAamp DNA Mini kit (Qiagen
Inc., Valencia, CA, USA) according to manufacturer's protocol. A
foetal genomic DNA sample was carefully extracted from the amniotic
fluid of the mother using the QIAamp Circulating Nucleic Acid kit
(Qiagen Inc.) according to the manufacturer's protocol. In
addition, DNA samples collected from 200 patients in the same
population that did not present with diagnostic features of Crouzon
syndrome were used as controls. DNA concentration and purity was
measured using a NanoDrop™ ND-1000 spectrophotometer
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Genomic DNA
samples were preserved at -20˚C prior to use.
Mutation detection and analysis
Nearly all the FGRF2 gene coding sequences were
amplified in family members and controls using PCR with Taq DNA
polymerase (Thermo Fisher Scientific, Inc.) and specific primers
sequences previously designed by Kan et al (15) (listed in Table I). A different set of primers were
utilized for the amplification of exon 10 and part of the intron
between exon 10 and 11 for uncomplicated confirmation by
restriction enzyme. These primer sequences were as follows:
forward, 5'-CCTCCACAATCATTCCTGTGTC-3' and reverse,
5'-TATCGCAACATGCAGCAAGC-3' (product size 733 bp). DNA (100 ng) in a
50 µl reaction was amplified. All reagents used for PCR were
purchased from Thermo Fisher Scientific, Inc. The amplification
included a single 5 min step at 94˚C followed by 40 cycles of 94˚C
for 45 sec, annealing [at temperatures stated by Kan et al
(15), and listed in Table I] for 45 sec and 72˚C for 45 sec
followed by a final 10 min step at 72˚C. Products were separated
using 1% agarose gel electrophoresis and stained with ethidium
bromide. DNA products then were purified using a QIAquick PCR
Purification kit (Qiagen Inc.) and sequenced in each direction
using an ABI3100 Genetic Analyzer (Thermo Fisher Scientific, Inc.).
The sequencing results were analysed using SeqMan (version 2.3;
Technelysium Pty, Ltd., South Brisbane, QLD, Australia) and
compared against reference sequences obtained from the ENSEMBL
Human Genome Browser (http://asia.ensembl.org/Homo_sapiens/Info/Index) with
the code of ENST00000358487. The mutation in exon 10 of the FGFR2
gene was confirmed via BsoBI restriction-enzyme digestion
with a recognition site of C↓YCGR↑G. The location of exon 10 in the
FGFR2 gene where the heterozygous missense mutation occurred was
from 1010 to 1015 with the change of sequence from CTGGGG to
CTCGGG. The DNA from a patient containing this heterozygous
mutation was gain of the BsoBI site in exon 10 of the FGFR2
gene. Purified PCR products containing the FGFR2 exon 10 of patient
III-16, other family members and controls were digested with
BsoBI. Digestion products were separated via 2% agarose gel
electrophoresis and stained with ethidium bromide.
 | Table I.Primers for amplification of
FGFR2. |
Table I.
Primers for amplification of
FGFR2.
| | Primer sequences | | |
|---|
| Target | Forward | Reverse | Fragment size
(bp) | Temperature (˚C)
annealing |
|---|
| Exon 2 |
TCCCTGACTCGCCAATCTCTTTC |
TGCCCCCAGACAAATCCCAAAAC | 341 | 55 |
| Exon 3 |
CACTGACCTTTGTTGGACGTTC |
GAGAAGAGAGAGCATAGTGCTGG | 380 | 64 |
| Exon 4 |
TGGAGAAGGTCTCAGTTGTAGAT |
AGACAGGTGACAGGCAGAACT | 232 | 55 |
| Exon 5 |
CAAAGCGAAATGATCTTACCTG |
AGAAATGTGATGTTCTGAAAGC | 291 | 62 |
| Exon 6 |
GCTAGGATTGTTAAATAACCGCC |
AAACGAGTCAAGCAAGAATGGG | 226 | 62 |
| Exon 7 (5') |
TGAGTTTGCCTCTCCTCGTGTG |
CCTTCTACAGTTGCCCTGTTGG | 390 | 62 |
| Exon 7(3') |
GATGTGCTGTAGCAGACCTTTGG |
ATCATCACAGGCAAAACCTGGG | 360 | 62 |
| Exon 8 |
GGTCTCTCATTCTCCCATCCC |
CCAACAGGAAATCAAAGAACC | 325 | 62 |
| Exon 9 |
AATGCTAAGACCTTCCTGGTTGG |
CAGTCTCCCAAAGCACCAAGTC | 284 | 55 |
| Exon 10 and part of
intron |
CCTCCACAATCATTCCTGTGTC |
TATCGCAACATGCAGCAAGC | 733 | 53 |
| Exon 11 |
TGCGTCAGTCTGGTGTGCTAAC |
AGGACAAGATCCACAAGCTGGC | 341 | 64 |
| Exon 12 |
TGACTTCCAGCCTTCTCAGATG |
AGTCTCCATCCTGGGACATGG | 252 | 64 |
| Exon 13 |
CCCCATCACCAGATGCTATGTG |
TTGATAAGACTCTCCACCCAGCC | 221 | 55 |
| Exon 14 |
TAGCTGCCCATGAGTTAGAGG |
ATCTGGAAGCCCAGCCATTTC | 250 | 62 |
| Exon 15 |
TGTTTTGCTGAATTGCCCAAG |
TCCACCCAGCCAAGTAGAATG | 294 | 55 |
| Exon 16 |
CTGGCGGTGTTTTGAAATTAG |
CCTTTCTTCCTGGAACATTCTG | 242 | 60 |
| Exon 17 |
AGCCCTATTGAGCCTGCTAAG |
CCAGGAAAAAGCCAGAGAAAAG | 177 | 62 |
| Exon 18 |
GGTTTTGGCAACGTGGATGGG |
GGTATTACTGGTGTGGCAAGTCC | 250 | 60 |
| Exon 19 |
ACACCACGTCCCCATATTGCC |
CTCACAAGACAACCAAGGACAAG | 243 | 60 |
| Exon 20 |
TCTGCCAAAATTGTTGTTTCTAGT |
GGTCTGGAACTCCTGACCTCA | 208 | 60 |
| Exon 21 |
TCCCACGTCCAATACCCACATC |
TACTGTTCGAGAGGTTGGCTGAG | 196 | 62 |
| Exon 22 |
CGTCCAATACCCACATCTCAAG |
TTCCCAGTGCTGTCCTGTTTGG | 363 | 60 |
The possible impact of the G338R exon 10 mutation on
the structure and function of FGFR2 was predicted using PolyPhen-2
(Polymorphism Phenotyping v2; http://genetics.bwh.harvard.edu/pph2/dbsearch.shtml).
The effect of the G338R exon 10 mutation on FGFR2 function, based
on sequence homology and the physical properties of amino acids,
was predicted by sorting intolerant from tolerant (SIFT, http://sift.bii.a-star.edu.sg/). Substitutions
with a score <0.05 are predicted to affect protein function.
Results
Clinical presentation
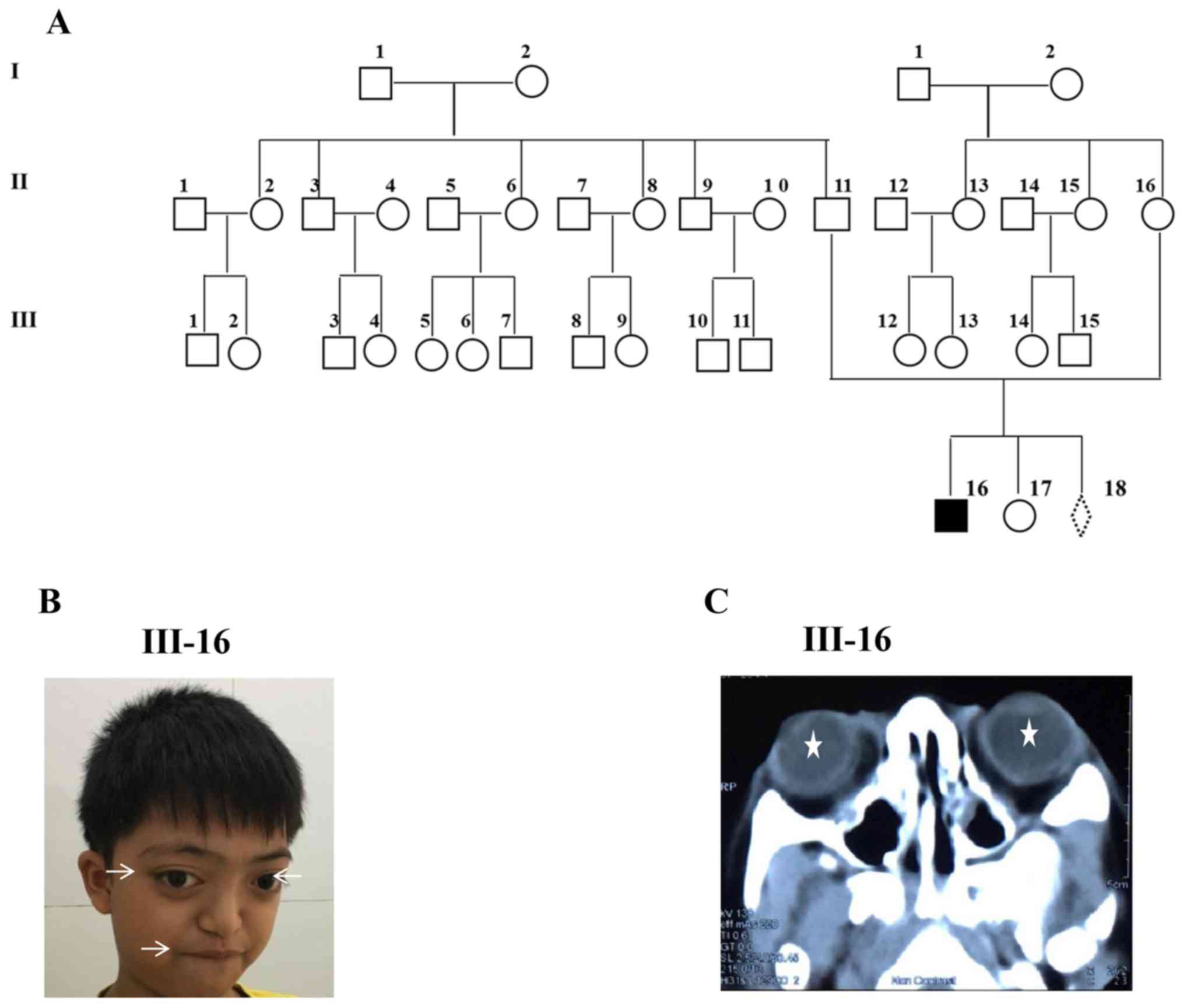
Personal and family histories were reviewed for each
member of the three-generation Vietnamese family (Fig. 1A). Systemic diseases were excluded
upon examination. Based on the results of all blood biochemical
tests, blood lipid, blood coagulation, urinalysis,
electrocardiogram and X-rays all appeared normal/within the normal
range. All results of these tests were within the normal range
(data not shown). Patient III-16 was a 10-year-old boy of two
healthy parents. He was born following a normal pregnancy and was
diagnosed with Crouzon syndrome at 6 years of age due to his
prominent eyes. Clinical examination of this patient indicated that
he had shallow orbits and ocular proptosis, mid-face hypoplasia and
craniosynostosis (Fig. 1B). He also
exhibited bulging eyes, leading to a reduction in his vision, a
prominent nasal bridge, an underdeveloped upper jaw and a
protruding lower jaw. In addition, his teeth were overcrowded and
he underwent surgery to resolve this. Furthermore, his skull
appeared ‘too tall’ and overly flat from the middle part of the
face upward. No abnormalities were detected in his hands and feet.
His hearing and his physical examination results were normal. He
presented with no mental retardation and had been in primary school
at the appropriate age. The CT scan confirmed shallow orbits and
exotropia in each eye of the patient (Fig. 1C). Ultrasound examination of a
16-week-old fetus revealed a normal appearance (data not shown).
Both parents (II-11 and II-16) and daughter (III-17) exhibited
normal visual acuity, with unremarkable eye examinations and all
family members had no known history of learning difficulties or
genetic problems.
Mutation screening
All coding sequences of the FGRF2 gene were
amplified using PCR and directly sequenced to identify the
mutations in all family members and controls. Sequencing results
indicated that there were no mutations identified in other exons of
the FGFR2 gene, excluding exon 10 and 11 (data not
shown).
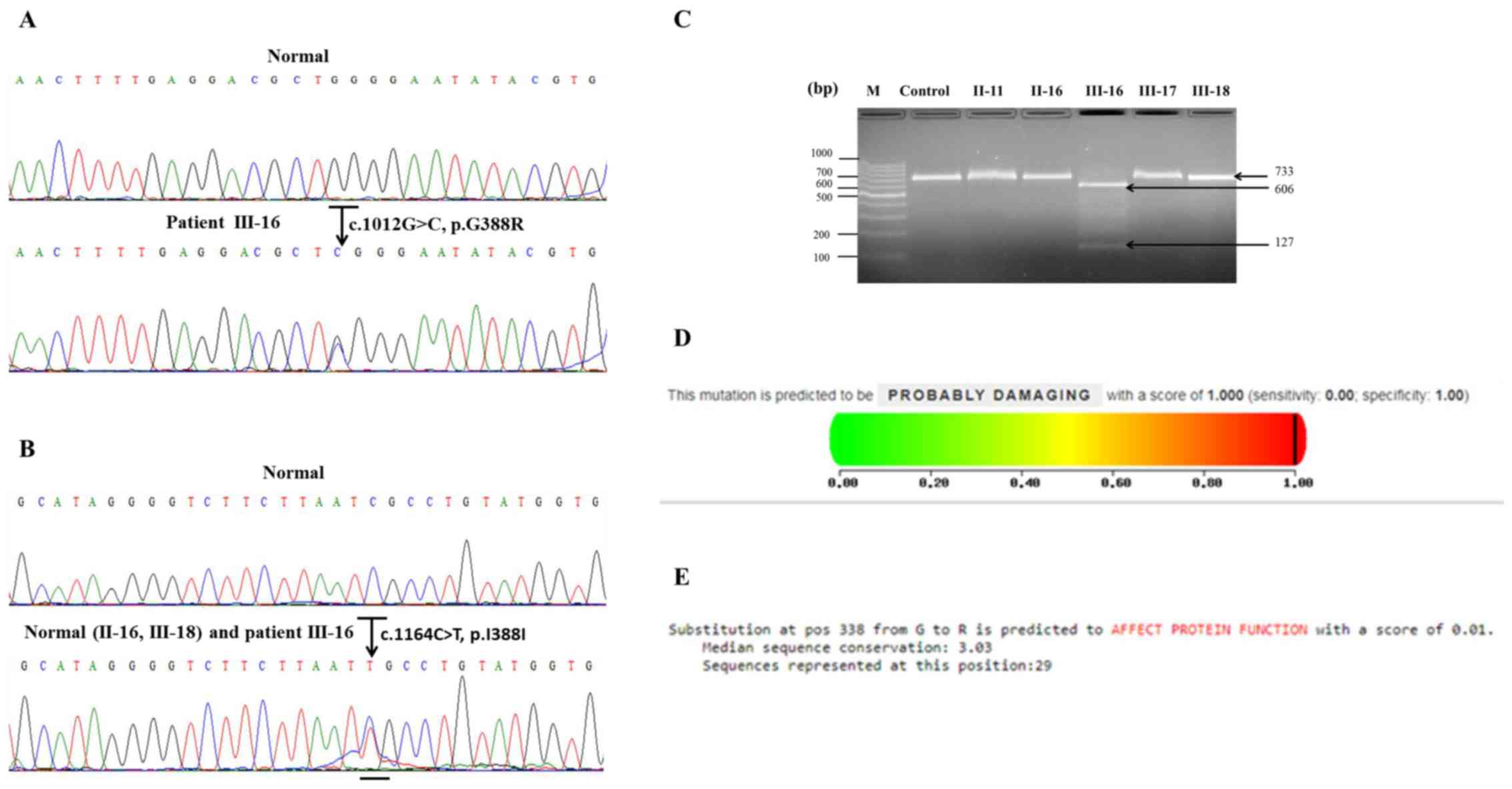
Patient III-16 carried a heterozygous missense
mutation (c.1012G>C; p.G338R) in exon 10 of the FGFR2
gene (Fig. 2A). This mutation was
observed in patient III-16 only and was not present in the
unaffected family members, the 16-week-old foetus or the unrelated
controls. The mutation was located at the coding region of Ig-like
domain 3 and caused the replacement of glycine by arginine at amino
acid position 338. This region is a very highly conserved segment
of the FGFR2 gene.
Patient III-16, his mother II-16 and the 16-week-old
foetus III-18 carried a heterozygous silent mutation (c.1164C>T;
p.I388I) in exon 11 of the FGFR2 gene (Fig. 2B). However, other unaffected family
members and unrelated controls did not carry any heterozygous
silent mutations in this exon.
The mutation in exon 10 of the FGFR2 gene was
confirmed by restriction enzyme digestion. The results revealed
that a mutation was present in exon 10 of the FGFR2 gene of
patient III-16 due to the gain of the BsoBI site, which
resulted in the appearance of two bands in the agarose gel (606 bp
and 127 bp; Fig 2C). In contrast, no
BsoBI site was exhibited in the other family members and
controls (data not shown), producing only one band in the agarose
gel (733 bp; Fig. 2C).
A possible impact of the G388R exon 10 mutation on
the structure and function of FGFR2 by PolyPhen-2 was predicted.
The G338 mutation was predicted to be ‘probably damaging’ to the
structure and function of FGFR2, with a score of 1.000 (Fig. 2D). Furthermore, the effect of the
G338R exon 10 mutation on FGFR2 function was predicted by SIFT
based on sequence homology and the physical properties of amino
acids. The G338R mutation was predicted to affect FGFR2 function
with score of 0.01 (Fig. 2E).
Discussion
Crouzon syndrome is an autosomal dominant disorder
with variable expressivity that is characterized by
craniosynostosis, shallow orbits, ocular proptosis and midface
hypoplasia (16). In the current
study, the patient III-16 was clinically examined and presented
with typical features of Crouzon syndrome, but not Apert syndrome.
The current study excluded the diagnosis of Apert syndrome, as the
patient exhibited no abnormalities in the hands and feet, normal
mental development and normal intelligence. FGFR2 mutations are a
well-known cause of Crouzon and Apert syndrome, but each are caused
by a different mutation in FGFR2. Two heterozygous gain-of-function
substitutions (Ser252Trp and Pro253Arg) in exon IIIa of FGFR2 are
responsible for >98% of Apert syndrome cases (17). To date, the G338R mutation in FGFR2
has not been detected in patients with Apert syndrome.
The current study identified a heterozygous missense
mutation (c.1012G>C; p.G338R) in a Vietnamese family with
autosomal dominant Crouzon syndrome. This mutation had been
previously detected in British and Chinese cases (15,18) but
not in the Vietnamese population. In comparison to the clinical
manifestations of patients in a previous study (18), patient III-16 exhibited similar
symptoms, including shallow orbits and ocular proptosis,
accompanied with craniosynostosis and mid-face hypoplasia. However,
some clinical manifestations of patient III-16 were different,
including normal hands, normal feet and a normal hearing ability.
Furthermore, his teeth were overcrowded, which required surgery to
resolve. Patient III-16, also did not present with mental
retardation, had a normal intelligence and did not exhibit any
other abnormality following physical examination. Therefore, the
severity of Crouzon syndrome signs and symptoms can vary among
affected patients, even with the same FGFR2 mutation.
Previous studies have indicated that the most common
genetic mutation of FGFR2 is localized to the third Ig-like
domain coded by exons IIIa (exon 8) and IIIc (exon 10) (15,19,20). The
G338R mutation identified in the family included the present study
was localized to exon IIIc. Most of the mutations in exon 10 of
FGFR2 results in a gain or loss of cysteine residues associated
with Crouzon syndrome (5,11,15).
Furthermore, the cysteine residues of each site in exon 10 of the
FGFR2 gene form the disulfide bond in the third Ig-like domain,
which controls FGFR2 receptor activity to the FGFR2 protein
(11,21,22).
Therefore, mutations may lead to a conformational change in the
extracellular Ig-III loop or may disrupt the intra-Ig domain
disulfide bond, resulting in changes to FGF signaling and the
activation or down regulation of FGFR2 (11,21,22). Fan
et al (18) utilized a
three-dimensional structural model to assess the position of the
missense mutation, G338R. The results of the aforementioned study
revealed the presence of a hydrogen bond between Tyr328 and Arg330
in mutant-type FGFR2, but not in wild-type FGFR2. This bond is
situated near the Gly338 amino acid residue, which may reduce the
stability of the FGFR2 protein (18).
Furthermore, the expression of two osteoblast specific genes,
osteocalcin and alkaline phosphatase, were significantly increased
in the orbital bone of patients compared to normal individual
(18).
The current study identified a heterozygous silent
mutation (c.1164C>T) in exon 11 of the FGFR2 gene in
patient III-16, II-16 and III-18 but not in II-11, III-17 and other
family members. However, this mutation did not lead to changes in
amino acids and may therefore be a single nucleotide polymorphism.
Furthermore, different silent mutations in exon 11 and others of
the FGFR2 gene were identified in previous studies (15,23).
In summary, a heterozygous missense mutation in exon
10 of the FGFR2 gene associated with Crouzon syndrome and a
novel heterozygous silent mutation in exon 11 of the FGFR2
gene were identified in a Vietnamese family of three generations.
These results may not only expand the reported mutation spectrum of
FGFR2, but may also provide useful information to aid
clinicians to confirm the diagnosis of Crouzon syndrome.
Furthermore, this molecular analysis may also have a considerable
impact on prenatal diagnoses.
Acknowledgements
The authors would like to thank the patient and
family members for their participation.
Funding
The present study was supported by the National Key
Laboratory of Gene Technology, Institute of Biotechnology (Vietnam
Academy of Science and Technology; grant no. NV03-PTNTD2017) and
Hanoi Medical University Hospital (Vietnam) grant no.
KHCN-33.05.01/11-15.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TTH, NTN and Ha HC designed the current study. AL,
TQN, TCH and PDC performed clinical examinations. HH extracted DNA.
TTH and HH amplified all exons of FGFR2 using PCR and purified the
PCR products. Ha H performed sequencing. TTH analysed the
sequencing results, and Thuy TH performed restriction enzyme
digestion. TTH and AL wrote the manuscript. NTN and HHC revised the
manuscript. HHC is the corresponding author and holds all the
responsibilities associated with this manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures were performed in a bioassay
laboratory and approved by the local ethics committee of Hanoi
Medical University Hospital and Institute of biotechnology, Vietnam
Academy of Science and Technology (Ha Noi, Vietnam).
Patient consent for publication
The patient and parents provided written informed
consent for the publication of any associated data and accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Giordano BP, Tuli SS, Ryan SF, Stern M and
Tuli SY: Crouzon Syndrome: Visual Diagnosis. J Pediatr Health Care.
30:270–273. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mitulla B, Hinkel GK and Lorenz P: Crouzon
syndrome (Mc K 12350). Kinderarztl Prax. 59:278–280. 1991.(In
German). PubMed/NCBI
|
|
3
|
Liu J, Kwon TG, Nam HK and Hatch NE:
Craniosynostosis- associated Fgfr2(C342Y) mutant bone marrow
stromal cells exhibit cell autonomous abnormalities in osteoblast
differentiation and bone formation. Biomed Res Int.
2013(292506)2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Raposo-Amaral CE, Neto JG, Denadai R,
Raposo-Amaral CM and Raposo-Amaral CA: Patient-reported quality of
life in highest-functioning Apert and Crouzon syndromes: A
comparative study. Plast Reconstr Surg. 133:182e–191e.
2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lin Y, Gao H, Ai S, Eswarakumar JVP, Zhu
Y, Chen C, Li T, Liu B, Jiang H, Liu Y, et al: FGFR2 mutations and
associated clinical observations in two Chinese patients with
Crouzon syndrome. Mol Med Rep. 16:5841–5846. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Korc M and Friesel RE: The role of
fibroblast growth factors in tumor growth. Curr Cancer Drug
Targets. 9:639–651. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chae YK, Ranganath K, Hammerman PS,
Vaklavas C, Mohindra N, Kalyan A, Matsangou M, Costa R, Carneiro B,
Villaflor VM, et al: Inhibition of the fibroblast growth factor
receptor (FGFR) pathway: The current landscape and barriers to
clinical application. Oncotarget. 8:16052–16074. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Touat M, Ileana E, Postel-Vinay S, André F
and Soria JC: Targeting FGFR signaling in cancer. Clin Cancer Res.
21:2684–2694. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Belov AA and Mohammadi M: Molecular
mechanisms of fibroblast growth factor signaling in physiology and
pathology. Cold Spring Harb Perspect Biol. 5(pii): a015958.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jabs EW, Li X, Scott AF, Meyers G, Chen W,
Eccles M, Mao JI, Charnas LR, Jackson CE and Jaye M: Jackson-Weiss
and Crouzon syndromes are allelic with mutations in fibroblast
growth factor receptor 2. Nat Genet. 8:275–279. 1994.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li ZL, Chen X, Zhuang WJ, Zhao W, Liu YN,
Zhang FX, Ha RS, Wu JH, Zhao C and Sheng XL: FGFR2 mutation in a
Chinese family with unusual Crouzon syndrome. Int J Ophthalmol.
9:1403–1408. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Katoh M: (2008). FGFR 2 (fibroblast growth
factor receptor 2). Atlas of Genetics and Cytogenetics in Oncology
and Haematology. http://atlasgeneticsoncology.org/Genes/GC_FGFR2.html#LINKS.
Accessed 18 December 2018.
|
|
13
|
Menashe Y, Ben Baruch G, Rabinovitch O,
Shalev Y, Katzenlson MB and Shalev E: Exophthalmus - prenatal
ultrasonic features for diagnosis of Crouzon syndrome. Prenat
Diagn. 9:805–808. 1989.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Schwartz M, Kreiborg S and Skovby F:
First-trimester prenatal diagnosis of Crouzon syndrome. Prenat
Diagn. 16:155–158. 1996.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kan SH, Elanko N, Johnson D,
Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR,
Rannan-Eliya S, et al: Genomic screening of fibroblast
growth-factor receptor 2 reveals a wide spectrum of mutations in
patients with syndromic craniosynostosis. Am J Hum Genet.
70:472–486. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Reardon W, Winter RM, Rutland P, Pulleyn
LJ, Jones BM and Malcolm S: Mutations in the fibroblast growth
factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 8:98–103.
1994.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bochukova EG, Roscioli T, Hedges DJ,
Taylor IB, Johnson D, David DJ, Deininger PL and Wilkie AO: Rare
mutations of FGFR2 causing apert syndrome: Identification of the
first partial gene deletion, and an Alu element insertion from a
new subfamily. Hum Mutat. 30:204–211. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fan J, Li Y, Jia R and Fan X: An inherited
FGFR2 mutation increased osteogenesis gene expression and result in
Crouzon syndrome. BMC Med Genet. 19(91)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tartaglia M, Valeri S, Velardi F, Di Rocco
C and Battaglia PA: Trp290Cys mutation in exon IIIa of the
fibroblast growth factor receptor 2 (FGFR2) gene is associated with
Pfeiffer syndrome. Hum Genet. 99:602–606. 1997.PubMed/NCBI
|
|
20
|
Ke R, Yang X, Tianyi C, Ge M, Lei J and Mu
X: The C342R mutation in FGFR2 causes Crouzon syndrome with elbow
deformity. J Craniofac Surg. 26:584–586. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lapunzina P, Fernández A, Sánchez Romero
JM, Delicado A, Sáenz de Pipaon M, López Pajares I and Molano J: A
novel insertion in the FGFR2 gene in a patient with Crouzon
phenotype and sacrococcygeal tail. Birth Defects Res A Clin Mol
Teratol. 73:61–64. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Piccione M, Antona V, Niceta M, Fabiano C,
Martines M, Bianchi A and Corsello G: Q289P mutation in the FGFR2
gene: First report in a patient with type 1 Pfeiffer syndrome. Eur
J Pediatr. 168:1135–1139. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li X, Park WJ, Pyeritz RE and Jabs EW:
Effect on splicing of a silent FGFR2 mutation in Crouzon syndrome.
Nat Genet. 9:232–233. 1995.PubMed/NCBI View Article : Google Scholar
|