Introduction
Left ventricular hypertrophy is characterized by
abnormal thickness of the ventricular wall in response to increased
heart workload, leading to heart failure and sudden death (1). The most common cause of ventricular
hypertrophy is high blood pressure (2). Angiotensin II (AngII) is a key signaling
molecule that induces ventricular myocyte hypertrophy through the
activation of angiotensin receptor type 1 (AT1R) (3). Unlike the AngII-dependent pathway,
stretch induces the translocation of β-arrestin2 in ventricular
myocytes. β-arrestin2 binds and activates AT1R, inducing
hypertrophic gene expression and the development of ventricular
hypertrophy (4).
Cysteine-rich transmembrane bone morphogenetic
protein regulator 1 (Crim1) is a type-I transmembrane protein, and
is expressed in multiple types of cell and tissue, including the
proepicardium, epicardium, central nervous system, vascular system
and the heart (5-10).
Crim1 serves an important role in vascular tube formation (6) and has cell-autonomous and paracrine
roles during heart development (10).
Deficiency of the CRIM1 gene causes perinatal mortality with
defects in multiple organs, including hemorrhagic necrosis,
enlargement of glomerular capillary lumens, congenital heart
defects, epicardial dysplasia and ventricular myocardium
densification dysplasia (11,12). However, whether Crim1 is involved in
left ventricular hypertrophy and whether the AT1R signaling pathway
affects the expression of Crim1 in ventricular myocytes remain
unclear. The present study investigated the suppressive function of
Crim1 on left ventricular hypertrophy and the effect of AT1R
signaling on the expression of Crim1 in ventricular myocytes.
Materials and methods
Isolation of primary ventricular
myocytes
Primary ventricular myocytes were isolated from the
left ventricles of neonatal Sprague-Dawley rats (4 male and 6
female, 1 day after birth) (purchased from Peking University
Medical Center Animal Center, Beijing, China; approval no.
SYXK2011-0039) as described previously (13). The rats were housed at room
temperature (20-25˚C) with 40-60% relative humidity and a day-night
cycle of 12 h. Food and water were provided ad libitum. The
rat ventricular muscle was digested with 0.01% trypsin and 0.03%
type II collagenase at 37˚C for 6 min (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), followed by differential adhesion and
5'-bromodeoxyuridine treatment. The attached cells were then
cultured at 37˚C for 48 h in DMEM (cat no. C11885500BT, Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal bovine serum (FBS; cat no. 10099141, Gibco; Thermo Fisher
Scientific, Inc.). Ventricular myocytes were identified by
immunohistochemical staining with rabbit anti-rat α-striated muscle
actin (α-SCA) antibodies (1:100; cat. no. bs-0189R, Shanghai Kemin
Bio-tech, Shanghai, China) for 1 h at room temperature, followed by
incubation with horseradish peroxidase-conjugated goat anti-rabbit
secondary antibodies (1:3,000; cat. no. SSA-004, Beijing Zhongyu
Jinqiao Company, Beijing, China) for 1 h at room temperature. For
immunofluorescent microscopy, the myocytes were fixed with 3.7%
paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at
room temperature, and treated with 0.5% Triton X-100
(Sigma-Aldrich; Merck KGaA) in PBS for 15 min at room temperature.
Following a wash with PBS, the cells were incubated with
FITC-conjugated α-SCA antibodies (Shanghai Kemin Bio-tech, China)
for 30 min at room temperature. Following further washing with PBS,
the cells were examined using an LSM 410 confocal microscope (Zeiss
AG, Oberkochen, Germany).
Cell stretch and transfection in
vitro
Following culture in DMEM containing 10% FBS at 37˚C
for 48 h, the primary left ventricular myocytes were cultured in
serum-free DMEM and treated with 10 µM telmisartan (Medicinal
product name: Micardis; AT1R inhibitor; Sigma-Aldrich; Merck KGaA)
at 37˚C for 2 h, followed by 24 h of cell stretch. Cell stretch was
performed in silicone dishes (Strex Cell; 105
cells/dish) coated with collagen (150 µg/ml). The cells were
exposed to 20% cyclic stretch in uniaxial strain at 30 cycles/min
using a computer-controlled stepping motor (Strex Cell). Cells
without exposure to stretch or telmisartan treatment served as a
control. Alternatively, primary ventricular myocytes
(104 cells/well) were seeded in wells of 12-well tissue
culture plates and transfected with recombinant adenovirus
expressing Crim1 (Ad-Crim1; Thermo Fisher Scientific, Inc.) in DMEM
with 10% FBS at 37˚C for 36 h using Lipofectamine® 2000 (cat. no.
18324-111, Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocols. The multiplicity of infection (MOI)
of Ad-Crim1 was 25, 100 or 200 (active viral particles per
myocyte). Empty virus (Ad-null) at MOI=100 served as a control. The
transfected cells were then harvested, and washed with
centrifugation at 178 x g for 5 min at room temperature. The cell
pellet was suspended in PBS with 0.25% SDS, and aliquoted for
protein or DNA measurement. Protein was quantified using the
bicinchoninic acid method (Sigma-Aldrich; Merck KGaA). DNA was
measured using a Quant-iTTM dsDNA assay kit (Invitrogen; Thermo
Fisher Scientific, Inc.). The protein/DNA ratio was then calculated
as an indicator of myocyte hypertrophy.
Histochemistry
The left ventricular myocytes were stained with
crystal violet, as described previously (14). Changes in cell size were analyzed with
ImageJ software (Version 1.4, National Institutes of Health) using
a Leica DM300 binocular microscope (Leica Microsystems GmbH,
Wetzlar, Germany). Alternatively, paraffin-embedded myocardial
sections (5 µm) were subjected to hematoxylin and eosin (H&E)
staining, and images were captured under a microscope. The
cross-sectional area of the ventricular myocytes was analyzed using
ImageJ software. A total of 50 ventricular myocytes per group were
analyzed and the mean value was calculated.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RNA was isolated from the rat left ventricle or
ventricular myocytes using TRIzol reagent, as described previously
(15). The mRNA expression of Crim1
was measured by RT-qPCR analysis (15). Briefly, total RNA (1.5 µg) was
subjected to reverse transcription with TransScript® First-Strand
cDNA Synthesis SuperMix. The reverse transcribed products (2 µg)
were used for qPCR with TransStart® PT Green qPCR
SuperMix and primers (Beijing Golden Biotechnology Co., Ltd.,
Beijing, China; Table I). The PCR
program was as follows: 95˚C pre-denaturation for 10 min, followed
by 40 cycles of 95˚C denaturation for 30 sec, 60˚C annealing for 30
sec, and 72˚C extension for 30 sec. The glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) gene was used as an internal control.
Relative gene expression was calculated using the 2-ΔΔCq
method (16).
 | Table IReverse transcription-quantitative
polymerase chain reaction primers. |
Table I
Reverse transcription-quantitative
polymerase chain reaction primers.
| Gene | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| Crim1 |
GTCTTTCCCCGGCGATCA |
TTGTTGCAGGTTCGGATGGT |
| GAPDH |
GTCAGTGGTGGACCTGGACCT |
AGGGGAGATTCAGTGTGGTG |
Western blot analysis
Total protein was extracted from the left ventricle
or ventricular myocytes with RIPA lysis buffer containing protease
inhibitors, as described previously (17). Protein concentration was quantified
using the bicinchoninic acid method. The proteins isolated from
myocytes (40 µg) or the left ventricle (100 µg) were separated on
6% SDS-PAGE and transferred onto PVDF membranes (Bio-Rad
Laboratories, Inc. Hercules, CA, USA). The membranes were incubated
with rabbit anti-rat Crim1 (1:100; cat. no. bs-2034R, Beijing Boao
Sen Company, Beijing, China) or anti-GAPDH (1:200; cat. no.
sc-365062, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) primary
antibodies for 1 h at room temperature, followed by incubation with
horseradish peroxidase-#173;conjugated goat anti-rabbit secondary
antibodies (1:3,000; cat. no. SSA-004, Beijing Zhongyu Jinqiao
Company) for 1 h at room temperature. The protein bands were
detected with a Bio-Rad chemiluminescence detector and analyzed
with ImageJ software.
Animal experiments
Male Sprague-Dawley rats (8 weeks, weight 220-250 g)
were purchased from the Peking University Medical Center Animal
Center (Beijing, China), and housed at room temperature (20-25˚C)
with 40-60% relative humidity under a 12 hlight/dark cycle. Food
and water were provided ad libitum. Rats were weighed and
subjected to sham operation with saline or telmisartan (3.57
mg/kg/d; Sigma-Aldrich; Merck KGaA) gavage, abdominal aortic
coarctation (AAC) (18) with saline
gavage, or AAC with telmisartan gavage (3.57 mg/kg/d). The duration
of abdominal aortic coarctation was 1 week, and treatment with
telmisartan lasted for 8 weeks. Alternatively, the rats received a
sham operation with saline intramuscular injection, AAC + saline
intramuscular injection, AAC + Ad-null or Ad-Crim1 intramuscular
injection. Each group included 10 rats. All rats were provided by
the Peking University Medical Center Animal Center (Beijing,
China). The use of rats for experiments was approved by the Ethics
Review Committee of Guizhou Provincial People's Hospital (Guizhou,
China).
AAC
The rats were fasted for 8 h and subjected to
abdominal anesthesia with 10% chloral hydrate (300 mg/kg of body
weight) and laparotomy. Ligation was performed on the abdominal
aorta above the renal artery bifurcation (~1 cm long) by placing a
blunt needle (22-G). The duration of AAC was 7 days, and the
abdominal aorta was partially narrowed to ~0.7 mm diameter. Animals
in the sham operation group underwent laparotomy only, without
abdominal aorta ligation.
Intramuscular injection of
adenovirus
The rats were anesthetized 1 week following
abdominal surgery, and were subjected to intramuscular injection of
Ad-null or Ad-Crim1 (3x109 PFU in 200 µl saline) into
the left ventricle. The rats were subjected to echocardiography 8
weeks following viral injection.
Echocardiography
The rats were anesthetized with 1.0-1.5% isoflurane
9 weeks following abdominal surgery, and subjected to M-mode
echocardiography using a high-resolution echocardiographic system
(Sequoia 512; Acuson, Siemens, Munich, Germany). The thickness of
the left ventricular wall and the diameter of the left ventricle
were measured using the Vevo2100 ultrasound system. The weight of
the left ventricle was calculated using the following formula:
(IVSd + LVDd + PWDd)3-LVDd3, in which IVSd is the ventricular
diastolic thickness, LVDd is the left ventricular diastolic
diameter, and PWDd is the left ventricular posterior wall diastolic
thickness.
Left ventricle harvesting
The heart was immediately harvested following
cardiac ultrasound. The left ventricle was weighed following
removal of the atrium and the right ventricle (remaining room
interval). The left ventricle was fixed with 4% paraformaldehyde
for paraffin embedding, or frozen for the extraction of proteins or
RNA.
Statistical analysis
All data are presented as the mean ±standard
deviation, and were analyzed by Student's t-test or Tukey's
honestly significant difference post hoc test following one-way
ANOVA with GraphPad Prism 6 software (GraphPad Software, Inc.).
Each experiment was performed with three repetitions. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of Crim1 is decreased in
hypertrophic ventricular myocytes
Immunohistochemical staining and confocal microscopy
demonstrated that primary ventricular myocytes were α-SCA-positive
(Fig. 1A). Quantification of protein
and DNA in ventricular myocytes treated with or without stretch
(19) revealed that hypertrophic
myocytes induced by stretch had a significantly higher protein/DNA
ratio than those without stretch (Fig.
1B). Analysis of the mRNA expression of Crim1 by RT-qPCR
analysis (Fig. 1C) and protein
expression by western blot analysis (Fig.
1D) demonstrated that the mRNA and protein expression levels of
Crim1 were significantly lower in the hypertrophic ventricular
myocytes compared with those in the unstretched cells (Fig. 1C and D).
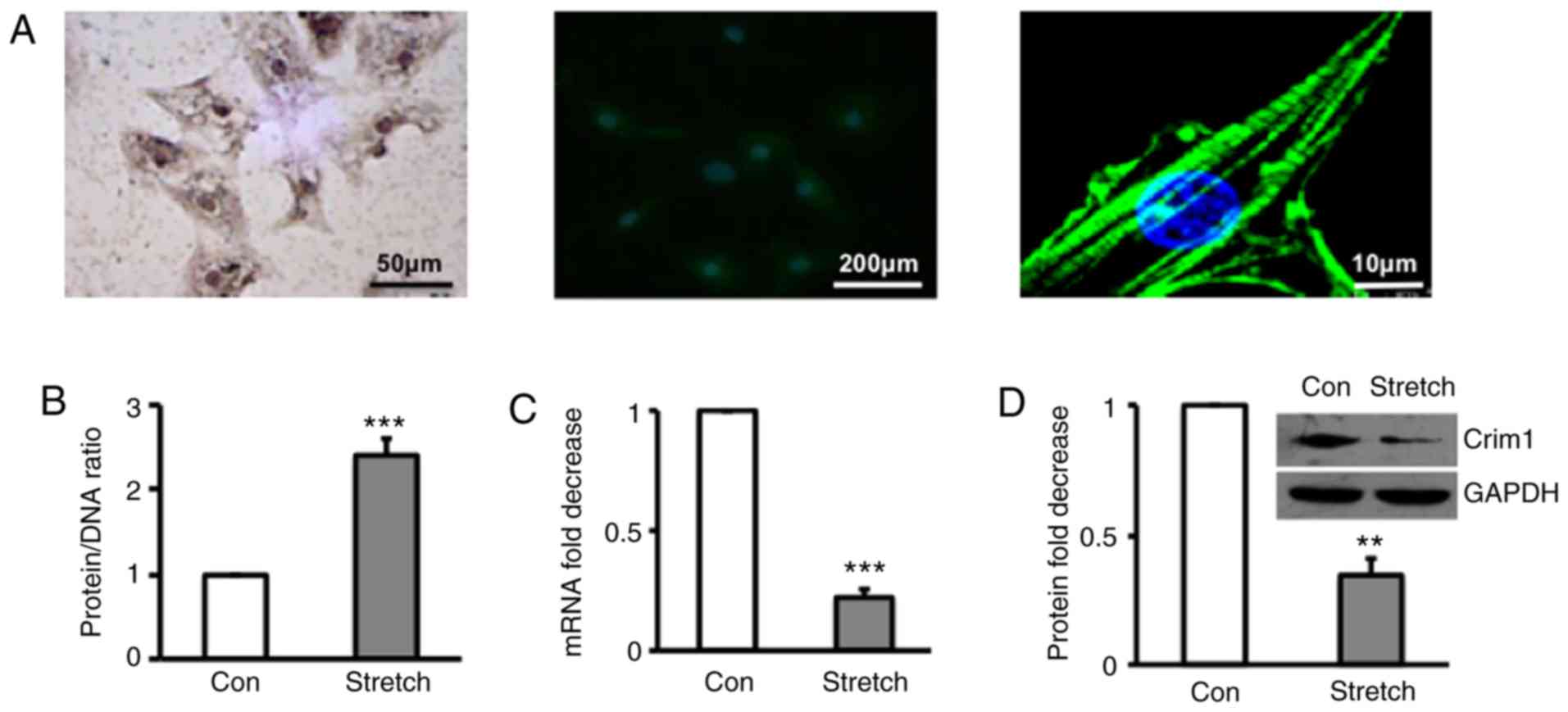 | Figure 1.Expression of Crim1 in rat
hypertrophic ventricular myocytes. (A) Primary myocytes isolated
from left ventricle were identified by immunohistochemical staining
with α-SCA antibodies (left) or immunofluorescent staining with
FITC-conjugated α-striated muscle actin antibodies (middle and
right), with images captured under a confocal microscope
(magnifications, x4 and 200). Primary left ventricular myocytes
were exposed to stretch, with cells without stretch serving as a
control. (B) Protein/DNA ratio in stretched or control cells was
profiled. (C) mRNA and (D) protein expression levels of Crim1 were
examined by reverse transcription-quantitative polymerase chain
reaction analysis and western blotting, respectively. GAPDH served
as internal control. Expression of Crim1 was calculated as the fold
decrease compared with the control. Data were from three
independent experiment, presented as the mean ± SD, and analyzed
using Student's t-test. **P<0.01,
***P<0.001 vs. Con. Con, control. Crim1,
cysteine-rich transmembrane bone morphogenetic protein regulator 1;
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Con, control. |
Increased expression of Crim1
abrogates left ventricular myocyte hypertrophy in vitro
Activating AT1R signaling promotes left ventricular
hypertrophy (20). To determine
whether inhibiting AT1R signaling can have an effect on the
expression of Crim1 in ventricular myocytes, the stretched primary
ventricular myocytes were treated with AT1R antagonist telmisartan.
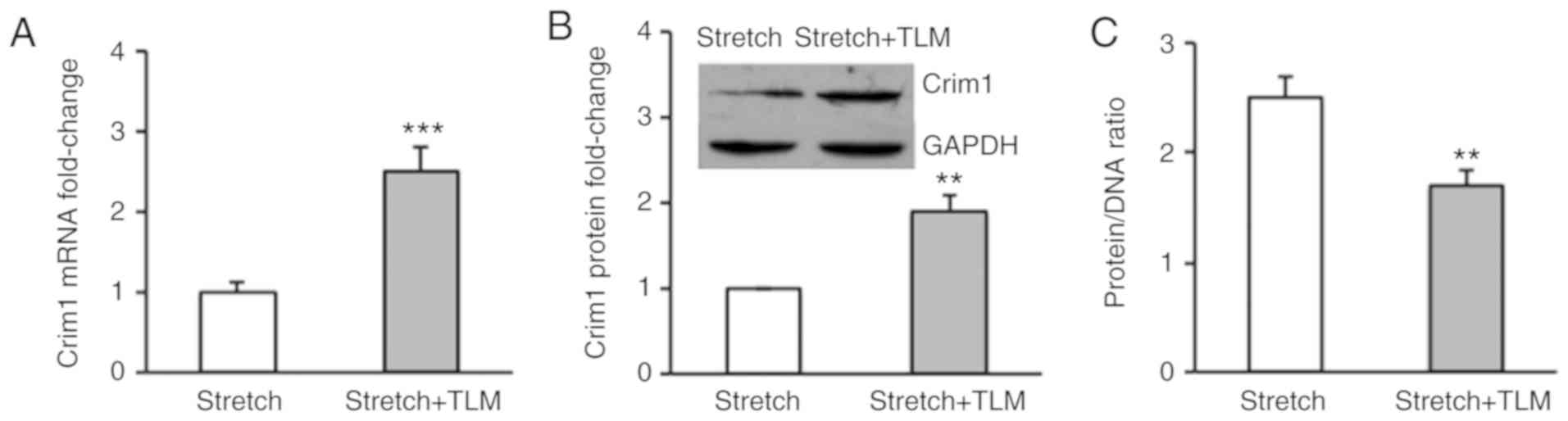
The RT-qPCR assay demonstrated that telmisartan treatment
significantly increased the mRNA expression of Crim1 in the
stretched myocytes compared with that in the saline treatment group
(Fig. 2A). Western blot analysis
further demonstrated that telmisartan treatment significantly
enhanced the protein expression of Crim1 in cells (Fig. 2B). Consequently, telmisartan treatment
abrogated stretch-induced ventricular myocyte hypertrophy, with a
significant decrease in the protein/DNA ratio in the cells. The
average protein/DNA ratio in the control cells and
telmisartan-treated cells were 2.5 and 1.7 respectively (Fig. 2C). To determine whether the
overexpression of Crim1 had an inhibitory effect on ventricular
myocyte hypertrophy, primary left ventricular myocytes were
transfected with Ad-Crim1 or Ad-null. Subsequent western blot
analysis revealed that transfection with Ad-Crim1 enhanced the
expression of Crim1 in the transfected ventricular myocytes,
compared with that in the Ad-null treatment group (Fig. 3A). To examine the suppressive function
of Crim1 on stretch-induced myocyte hypertrophy, crystal violet
staining was performed on the stretched myocytes. Histochemical
analysis demonstrated that Ad-Crim1 transfection (MOI=100) markedly
reduced myocyte hypertrophy, compared with Ad-null treatment or no
viral transfection. Ad-null transfection had no suppressive effect
on myocyte hypertrophy (Fig. 3B).
Analysis of the protein/DNA ratio demonstrated that Ad-Crim1
treatment significantly reduced the ratio in the stretched
myocytes, compared with Ad-null treatment or no viral transfection
(Fig. 3C). The average protein/DNA
ratios in the stretched cells treated with Ad-Crim1, Ad-null, and
without viral transfection were 1.3, 1.9 and 2.1 respectively
(Fig. 3C). The average protein/DNA
ratio in the control myocytes without stretch was 1.15 (Fig. 3C). There was no significant difference
in the protein/DNA ratio between the stretch/Ad-Crim1-treated
myocytes and the control cells (Fig.
3C). Measurements of cell size with Image J software further
confirmed that the overexpression of Crim1 by transfection with
Ad-Crim1 significantly reduced the cell size and inhibited
ventricular myocyte hypertrophy, compared with Ad-null treatment
and no viral transfection (Fig.
3D).
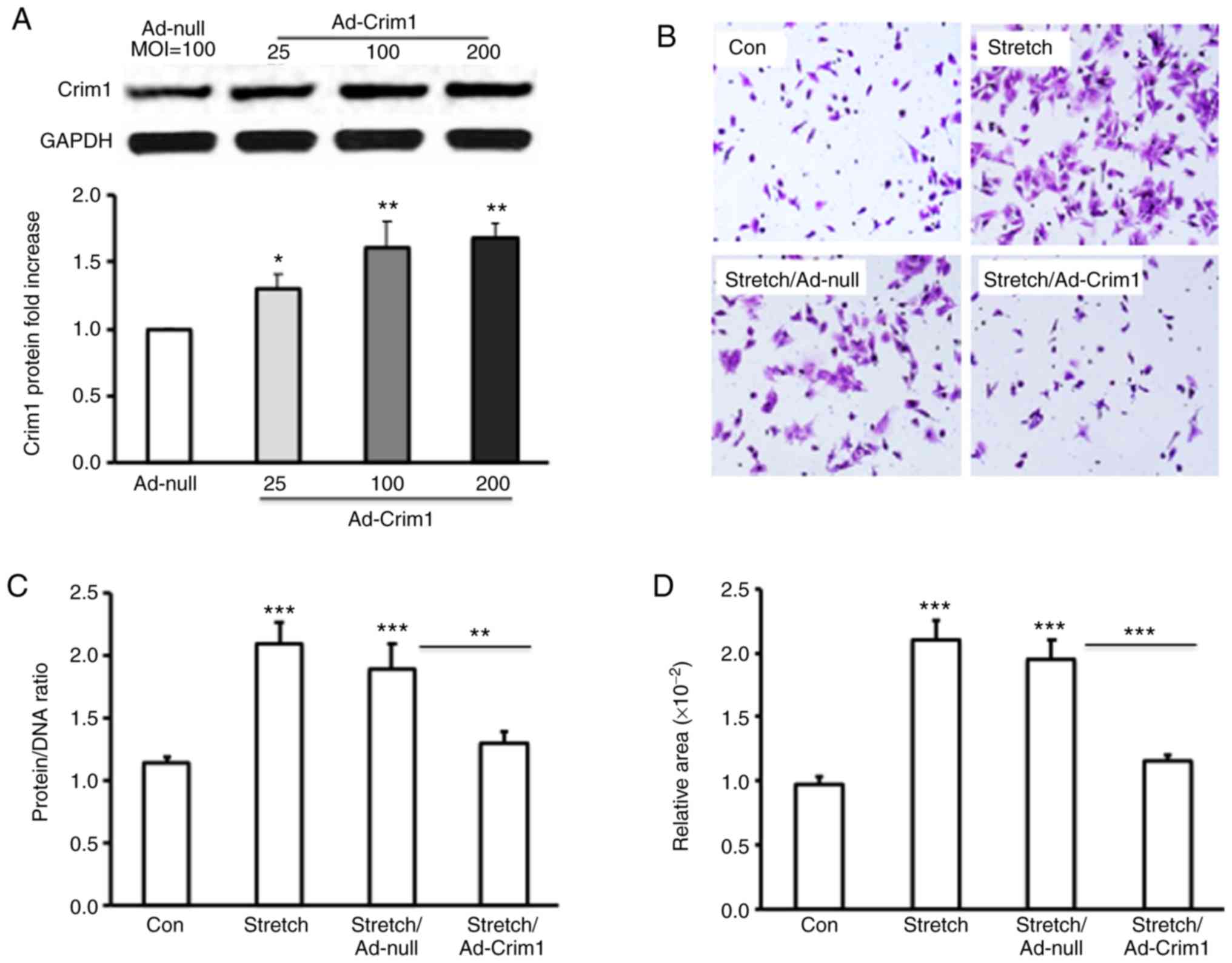 | Figure 3.Effect of the overexpression of Crim1
on left ventricular myocyte hypertrophy. (A) Primary rat left
ventricular myocytes were infected with Ad-Crim1 or Ad-null.
Proteins were isolated from the treated cells and subjected to
western blotting with anti-Crim1 or GAPDH specific antibodies.
Protein expression levels of Crim1 in Ad-Crim1-treated cells are
presented as the fold increase compared with Ad-null-treated cells.
(B) Primary left ventricular myocytes were subjected to stretch and
transfected with Ad-Crim1, Ad-null, or without transfection.
Ventricular myocytes without treatment served as a control. The
cells were then stained with crystal violet and images were
captured under a microscope (magnification, x20). (C) Protein/DNA
ratio in treated or control ventricular myocytes. (D)
Cross-sectional area of ventricular myocytes was analyzed using
ImageJ software. Data are from three independent experiments,
presented as the mean ± SD and analyzed by one-way ANOVA.
*P<0.05, **P<0.01,
***P<0.001. Individual treatment was compared with
the control; stretch/Ad-Crim1 treatment was also compared with
stretch/Ad-null treatment. Crim1, cysteine-rich transmembrane bone
morphogenetic protein regulator 1; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; MOI, multiplicity of
infection; Con, control. |
Enhancement of Crim1 by telmisartan
inhibits left ventricular myocyte hypertrophy in vivo
To examine whether enhancement of the protein
expression of Crim1 can suppress AAC-induced left ventricular
myocyte hypertrophy, the AAC-treated rats were subjected to
telmisartan gavage and the protein expression of Crim1 in the left
ventricle was analyzed. Western blot analysis demonstrated that the
protein expression of Crim1 was significantly decreased in the left
ventricle of the AAC-treated rats, compared with that in the sham
group (Fig. 4A). However, protein
expression of Crim1 in the left ventricle of the AAC-treated rats
with telmisartan gavage was significantly increased compared with
that in the group without telmisartan treatment (Fig. 4A). H&E staining further
demonstrated that telmisartan treatment abrogated AAC-induced left
ventricular myocyte hypertrophy compared with that in the absence
of telmisartan treatment (Fig. 4B).
Quantification of myocyte cross-sectional area further confirmed
that the size of ventricular myocytes in AAC-treated rats with
telmisartan gavage was significantly reduced compared with that in
the rats without telmisartan treatment (Fig. 4C). The expression of Crim1 and myocyte
size in the left ventricle of AAC/telmisartan-treated rats did not
differ significantly from those in the sham group or in the rats
without AAC but with telmisartan treatment (Fig. 4A-C).
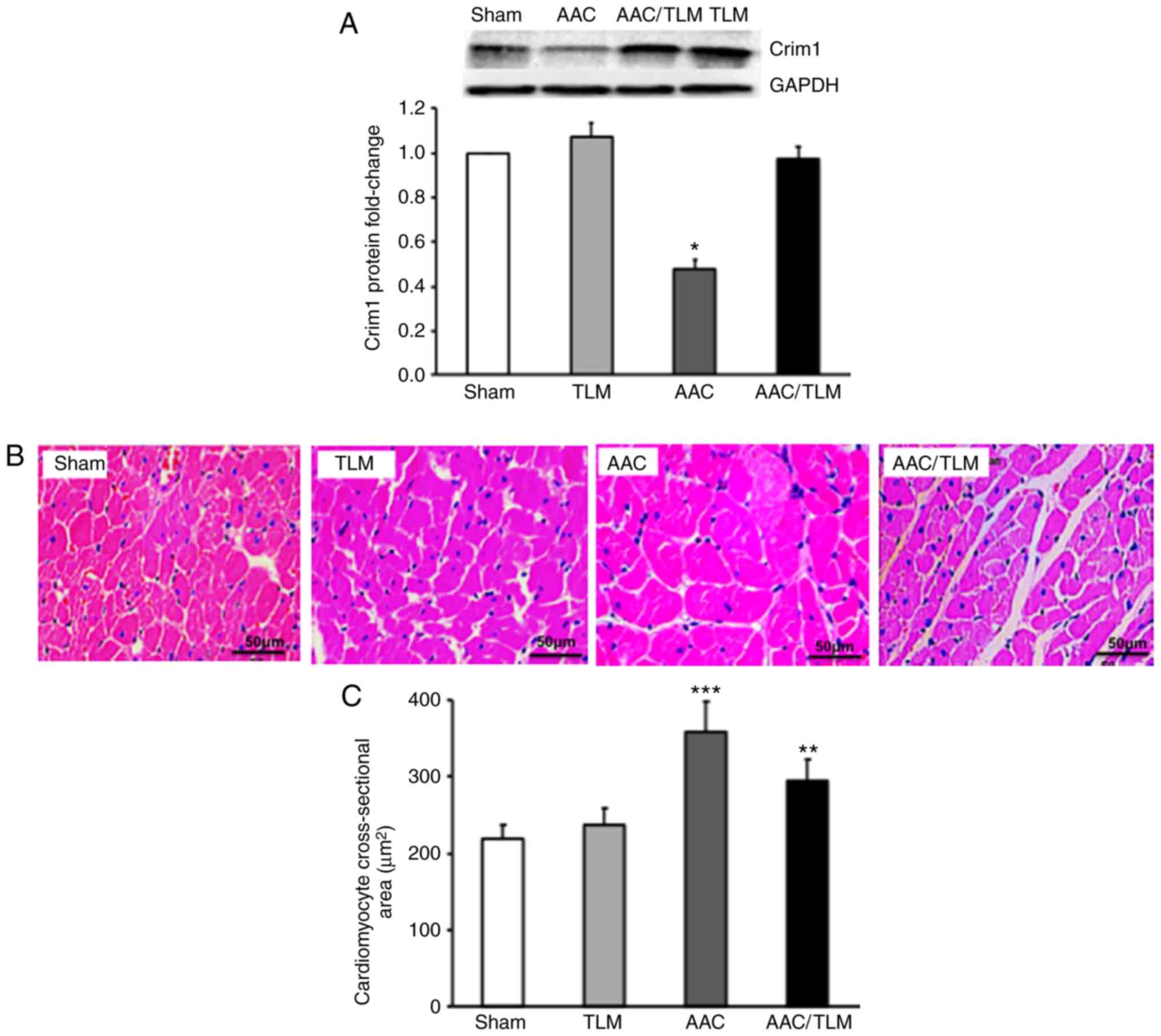 | Figure 4.Effects of TLM on the expression of
Crim1 and ventricular hypertrophy in vivo. Rats were
subjected to AAC treatment with (AAC/TLM) or without (ACC) TLM
gavage, or TLM gavage without AAC (TLM), or sham treatment. (A)
Proteins were extracted from left ventricle of the four groups of
rats and subjected to western blotting with anti-Crim1 or
anti-GAPDH antibodies. GAPDH served as the internal protein
control. The fold decrease or increase in Crim1 protein in the
TLM-, AAC-or AAC/TLM-treated groups were compared with the sham
group. (B) Paraffin-fixed left ventricular sections from the four
groups of rats were subjected to hematoxylin and eosin staining and
images were captured under a confocal microscope (magnification,
x400, scale bar=50 µm). (C) Cross-sectional area of left
ventricular myocytes was analyzed using ImageJ software. Data were
from three independent experiments, presented as the mean ± SD, and
analyzed using one-way ANOVA. *P<0.05,
**P<0.01, ***P<0.001. Individual
treatment was compared with sham; AAC/TLM treatment was also
compared with AAC treatment. Crim1, cysteine-rich transmembrane
bone morphogenetic protein regulator 1; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; TLM, telmisartan; AAC,
abdominal aortic coarctation. |
Injection of Ad-Crim1 suppresses
AAC-induced left ventricular hypertrophy in vivo
To examine whether the administration of Ad-Crim1
suppresses AAC-induced left ventricular hypertrophy in the rat
model, as Ad-Crim1 did on primary ventricular myocytes in
vitro (Fig. 3B-D), the
AAC-treated rats were subjected to intramuscular injection of
Ad-Crim1, Ad-null or saline. Echocardiography (Fig. 5A) demonstrated that the left
ventricular end-diastolic anterior wall (LAVWd) in the AAC-treated
rats was significantly thicker compared with that in the sham group
(Fig. 5B). There were no significant
differences in LAVWd among the three groups of rats treated with
Ad-Crim1, Ad-null or without viral injection (Fig. 5B). Measurement of the left ventricular
mass revealed that AAC treatment significantly increased left
ventricular weight compared with that following sham treatment
(Fig. 5C). However, rats treated with
Ad-Crim1 had significantly lower left ventricular weight compared
with the those treated with Ad-null or without viral injection
(Fig. 5C). Quantification of myocyte
cross-sectional area further confirmed that AAC treatment markedly
increased the size of ventricular myocytes compared with that in
the sham treatment group (Fig. 5D);
Ad-Crim1 treatment significantly reduced the size of left
ventricular myocytes compared with that in the Ad-null treatment or
without viral injection groups (Fig.
5D). There was no significant difference in myocyte size
between rats treated with Ad-null and without viral injection
(Fig. 5D). H&E staining of the
left ventricle confirmed that AAC treatment induced ventricular
myocyte hypertrophy compared with that in the sham treatment group
(Fig. 5E). Intramuscular injection of
Ad-Crim1 markedly suppressed AAC-induced myocyte hypertrophy
compared with that in the Ad-null treatment group (Fig. 5E).
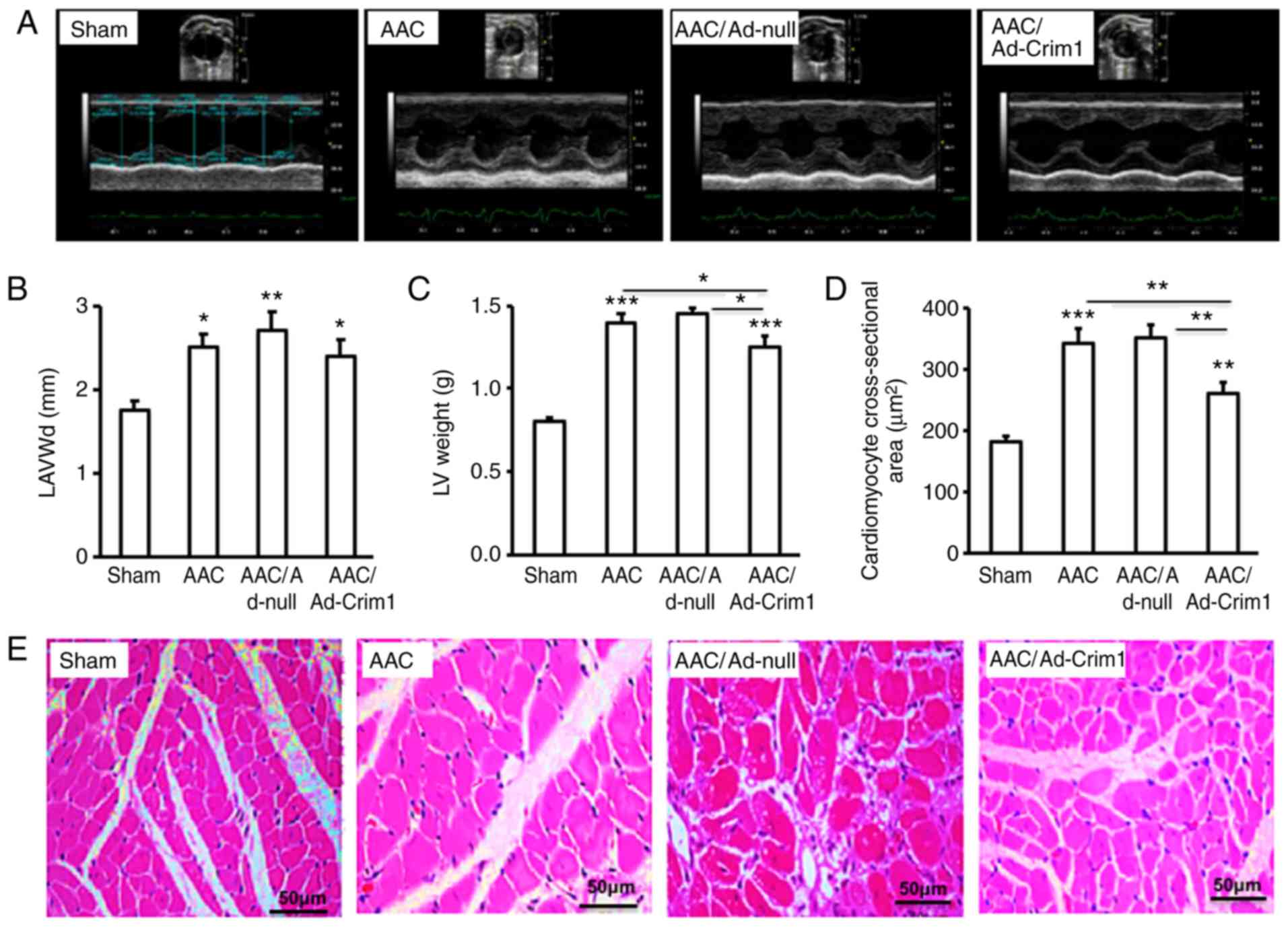 | Figure 5.Effects of Ad-Crim1 on left
ventricular hypertrophy in vivo. (A) AAC-treated rats were
subjected to thoracic intramuscular injection of Ad-Crim1, Ad-null,
or saline. Sham group rats received no treatment. Rats in the four
groups were examined by echocardiography. (B) LAVWd and (C) LV
weight were calculated and presented. (D) Cross-sectional areas of
left ventricular myocytes were analyzed using ImageJ software. (E)
Paraffin-fixed left ventricular sections were subjected to
hematoxylin and eosin staining and images were captured under a
confocal microscope (magnification, x400, scale bar=50 µm). Data
are from three independent experiments, presented as the mean ± SD,
and analyzed using one-way ANOVA. *P<0.05,
**P<0.01, ***P<0.001. Individual
treatment was compared with sham; AAC/Ad-Crim1 treatment was also
compared with AAC or AAC/Ad-null treatment. Crim1, cysteine-rich
transmembrane bone morphogenetic protein regulator 1; AAC,
abdominal aortic coarctation; LAVWd, left ventricular end-diastolic
anterior wall; LV, left ventricle. |
Discussion
In the present study, it was demonstrated that Crim1
had an inhibitory effect on left ventricular hypertrophy.
Enhancement of the expression of Crim1 in myocytes abrogated left
ventricular hypertrophy.
Crim1 protein has six conserved domains of
chordin-like von Willebrand C-type cysteine-rich repeats (CRR)
(5), existing in the Golgi,
endoplasmic reticulum and on the cell surface (6,7). Crim1 CRR
domains can intracellularly bind several cystine knot-containing
growth factors, including vascular endothelial growth factor α
(VEGFα), bone morphogenetic protein (BMP)2 and BMP4, and these
molecules are antagonized through reducing its expression,
processing and secretion (7,21,22).
The activation of BMP2 or BMP4 promotes the development of cardiac
hypertrophy (23-25).
Triggering VEGFα signal transduction facilitates tissue growth and
angiogenesis in cardiac hypertrophy (26,27). It is
known that Crim1 inhibits BMP signaling by either limiting BMP
precursors on the cell surface or preventing the maturation or
secretion of BMP (7). Data from the
present study demonstrated that the increased expression of Crim1
in cardiomyocytes by infection with Ad-Crim1 suppressed left
ventricular hypertrophy. One of the mechanistic actions of Crim1 in
suppressing ventricular/cardiac hypertrophy may be through
inhibiting the activation of BMP2 or BMP4 signaling. Crim1 can also
maintain retinal vascular and renal microvascular stability by
regulating VEGFα signal transduction in vascular endothelial cells
(22,28). It is possible that the inhibitory
effect of Crim1 on ventricular hypertrophy is mediated by affecting
the delivery and function of VEGFα (21,22). Crim1
can also bind to β-integrin and regulate its signaling (29). Cardiomyocytes can undergo
biomechanical change through receptors, including integrins
(30). β-integrin, a mechanical
sensor, can transduce mechanical force into biological information
in collapsible muscle cells (31).
β-integrin is also important in heart self-protection and the
compensatory reaction in response to stress. The data obtained in
the present study demonstrated that Crim1 prevented stretch-induced
left ventricular hypertrophy. It is possible that the inhibitory
function of Crim1 on myocardial hypertrophy may be through
regulating biological stress-induced β-integrin signal
transduction.
AT1R is a major pathogenic molecule contributing to
cardiac damage (32). The
overstimulation of AT1R causes hypertension and cardiac hypertrophy
through either the AngII-dependent or AngII-independent pathway
(19,32-34).
The inhibition of AT1R attenuates or reverses myocardial
hypertrophy (20,35). In the present study, data showed that
AT1R antagonist telmisartan inhibited the cardiomyocyte hypertrophy
induced by stretch or AAC. Of note, stretch or AAC treatment
significantly reduced the expression of Crim1 in hypertrophic
ventricular myocytes. By contrast, inhibiting AT1R signaling by
telmisartan significantly increased the expression of Crim1 in
cardiomyocytes, suggesting a potential suppressive role of AT1R
signal on the expression of Crim1 in cardiomyocytes.
In conclusion, the present study showed that the
expression of Crim1 was downregulated in hypertrophic ventricular
myocytes. The increased expression of Crim1 by inhibiting AT1R or
the intramuscular injection of Crim1-expressing recombinant
adenovirus prevented left ventricular hypertrophy. The effect of
Crim1 on left ventricular hypertrophy supports the view that Crim1
can be utilized as a novel therapeutic target for the treatment of
cardiac hypertrophy.
Acknowledgements
The authors would like to thank Dr Ryan Jajosky
(Department of Pathology and Laboratory Medicine, Emory University
School of Medicine) for editing the manuscript.
Funding
This study was supported by grants from the Guizhou
Provincial Science and Technology Fund [grant no. (2019) 1209], the
Chinese National Natural Science Foundation to YL (grant no.
81260040), the Chinese National Clinical Key Specialty Construction
Project to YL [grant no. (2013) 544] and the Clinical Research
Center Project of Department of Science & Technology of Guizhou
Province, China to YL [grant no. (2017) 5405].
Availability of data and materials
All data obtained in this study are included in this
published article.
Authors' contributions
LY and JH designed the experiments, LY, JH, GX, JY,
QT and YY performed the experiments, LY, JH, JY and JD analyzed and
interpreted data, LY and JD wrote the manuscript. All authors read
and approved the final manuscript.
Ethical approval and consent to
participate
The Animal Care Committee of Guizhou Provincial
People's Hospital approved the animal experimental protocol.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Soliman EZ and Prineas RJ:
Antihypertensive therapies and left ventricular hypertrophy. Curr
Hypertens Rep. 19(79)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ikeda Y, Kumagai H, Motozawa Y, Suzuki J
and Komuro I: Biased agonism of the angiotensin II type I receptor.
Int Heart J. 56:485–488. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang S, Gong H, Jiang G, Ye Y, Wu J, You
J, Zhang G, Sun A, Komuro I, Ge J and Zou Y: Src is required for
mechanical stretch-induced cardiomyocyte hypertrophy through
angiotensin II type 1 receptor-dependent beta-arrestin2 pathways.
PLoS One. 9(e92926)2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kolle G, Georgas K, Holmes GP, Little MH
and Yamada T: CRIM1, a novel gene encoding a cysteine-rich repeat
protein, is developmentally regulated and implicated in vertebrate
CNS development and organogenesis. Mech Dev. 90:181–193.
2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Glienke J, Sturz A, Menrad A and Thierauch
KH: CRIM1 is involved in endothelial cell capillary formation in
vitro and is expressed in blood vessels in vivo. Mec Dev.
119:165–175. 2002.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wilkinson L, Kolle G, Wen D, Piper M,
Scott J and Little M: CRIM1 regulates the rate of processing and
delivery of bone morphogenetic proteins to the cell surface. J Biol
Chem. 278:34181–34188. 2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nakashima Y and Takahashi S: Induction of
cysteine-rich motor neuron 1 mRNA expression in vascular
endothelial cells. Biochem Biophys Res Commun. 451:235–238.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Iyer S, Pennisi DJ and Piper M: Crim1-, a
regulator of developmental organogenesis. Histol Histopathol.
31:1049–1057. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Iyer S, Chou FY, Wang R, Chiu HS, Raju VK,
Little MH, Thomas WG, Piper M and Pennisi DJ: Crim1 has
cell-autonomous and paracrine roles during embryonic heart
development. Sci Rep. 6(19832)2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pennisi DJ, Wilkinson L, Kolle G, Sohaskey
ML, Gillinder K, Piper MJ, McAvoy JW, Lovicu FJ and Little MH:
Crim1KST264/KST264 mice display a disruption of the Crim1 gene
resulting in perinatal lethality with defects in multiple organ
systems. Dev Dyn. 236:502–511. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chiu HS, York JP, Wilkinson L, Zhang P,
Little MH and Pennisi DJ: Production of a mouse line with a
conditional Crim1 mutant allele. Genesis. 50:711–716.
2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Golden HB, Gollapudi D, Gerilechaogetu F,
Li J, Cristales RJ, Peng X and Dostal DE: Isolation of cardiac
myocytes and fibroblasts from neonatal rat pups. Methods Mol Biol.
843:205–214. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang GJ, Yao YS and Wang HX: Comparing
effects of U50488H, prazosin and/or propranolol on cardiac
hypertrophy induced by NE in rat. Zhongguo Ying Yong Sheng Li Xue
Za Zhi. 26:82–85. 2010.(In Chinese). PubMed/NCBI
|
|
15
|
Yang Y, Zhang H, Li X, Yang T and Jiang Q:
Effects of PPARα/PGC-1α on the energy metabolism remodeling and
apoptosis in the doxorubicin induced mice cardiomyocytes in vitro.
Int J Clin Exp Pathol. 8:12216–12224. 2015.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Deng J, Pennati A, Cohen JB, Wu Y, Ng S,
Wu JH, Flowers CR and Galipeau J: GIFT4 fusokine converts leukemic
B cells into immune helper cells. J Transl Med.
14(106)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Huang J, Wang D, Zheng J, Huang X and Jin
H: Hydrogen sulfide attenuates cardiac hypertrophy and fibrosis
induced by abdominal aortic coarctation in rats. Mol Med Rep.
5:923–928. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sadoshima J, Xu Y, Slayter HS and Izumo S:
Autocrine release of angiotensin II mediates stretch-induced
hypertrophy of cardiac myocytes in vitro. Cell. 75:977–984.
1993.PubMed/NCBI
|
|
20
|
Arumugam S, Sreedhar R, Thandavarayan RA,
Karuppagounder V, Krishnamurthy P, Suzuki K, Nakamura M and
Watanabe K: Angiotensin receptor blockers: Focus on cardiac and
renal injury. Trends Cardiovasc Med. 26:221–228. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wilkinson L, Gilbert T, Kinna G, Ruta LA,
Pennisi D, Kett M and Little MH: Crim1KST264/KST264 mice implicate
Crim1 in the regulation of vascular endothelial growth factor-A
activity during glomerular vascular development. J Am Soc Nephrol.
18:1697–1708. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Fan J, Ponferrada VG, Sato T, Vemaraju S,
Fruttiger M, Gerhardt H, Ferrara N and Lang RA: Crim1 maintains
retinal vascular stability during development by regulating
endothelial cell Vegfa autocrine signaling. Development.
141:448–459. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen
C, Liu HB, Li N, Li CB, Guo WT, et al: Bone morphogenetic protein-4
mediates cardiac hypertrophy, apoptosis, and fibrosis in
experimentally pathological cardiac hypertrophy. Hypertension.
61:352–360. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sun B, Sheng Y, Huo R, Hu CW, Lu J, Li SL,
Liu X, Wang YC and Dong DL: Bone morphogenetic protein-4
contributes to the down-regulation of Kv4.3 K+ channels in
pathological cardiac hypertrophy. Biochem Biophys Res Commun.
436:591–594. 2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shahid M, Spagnolli E, Ernande L, Thoonen
R, Kolodziej SA, Leyton PA, Cheng J, Tainsh RE, Mayeur C, Rhee DK,
et al: BMP type I receptor ALK2 is required for angiotensin
II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol.
310:H984–H994. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Izumiya Y, Shiojima I, Sato K, Sawyer DB,
Colucci WS and Walsh K: Vascular endothelial growth factor blockade
promotes the transition from compensatory cardiac hypertrophy to
failure in response to pressure overload. Hypertension. 47:887–893.
2006.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ferrara N: Vascular endothelial growth
factor. Arterioscler Thromb Vasc Biol. 29:789–791. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wilkinson L, Gilbert T, Sipos A, Toma I,
Pennisi DJ, Peti-Peterdi J and Little MH: Loss of renal
microvascular integrity in postnatal Crim1 hypomorphic transgenic
mice. Kidney Int. 76:1161–1171. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang Y, Fan J, Ho JW, Hu T, Kneeland SC,
Fan X, Xi Q, Sellarole MA, de Vries WN, Lu W, et al: Crim1
regulates integrin signaling in murine lens development.
Development. 143:356–366. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ross RS and Borg TK: Integrins and the
myocardium. Circ Res. 88:1112–1119. 2001.PubMed/NCBI
|
|
31
|
Israeli-Rosenberg S, Manso AM, Okada H and
Ross RS: Integrins and integrin-associated proteins in the cardiac
myocyte. Circ Res. 114:572–586. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Takezako T, Unal H, Karnik SS and Node K:
Current topics in angiotensin II type 1 receptor research: Focus on
inverse agonism, receptor dimerization and biased agonism.
Pharmacol Res. 123:40–50. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zou Y, Akazawa H, Qin Y, Sano M, Takano H,
Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, et al: Mechanical
stress activates angiotensin II type 1 receptor without the
involvement of angiotensin II. Nat Cell Biol. 6:499–506.
2004.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Akazawa H and Komuro I: Mechanisms
underlying angiotensin II-independent activation of angiotensin II
type 1 receptor. Nihon Rinsho. 70:1492–1498. 2012.(In Japanese).
PubMed/NCBI
|
|
35
|
Dargad RR, Parekh JD, Dargad RR and
Kukrety S: Azilsartan: Novel angiotensin receptor blocker. J Assoc
Physicians India. 64:96–98. 2016.PubMed/NCBI
|