Introduction
The term aggresome refers to a stress sensitive
subcellular structure, containing misfolded proteins, heat-shock
proteins (HSPs) and proteasomal components, and it is formed when
the protein-degradation system is overwhelmed (1). Aggresome formation requires an intact
microtubule system and the distinct presence of γ-tubulin (2-4), which
ultimately terminates in lysosomal degradation (5,6).
Therefore, the aggresome pathway likely provides a backup system
for delivery of aggregated proteins from the cytoplasm to lysosomes
for degradation (4). Various stress
conditions, including oxidative stress (7), proteasome inhibition (3) and heat shock (8), induce aggresome formation through
different molecular mechanisms. In response to failure of ubiquitin
proteasome system (UPS), p62 assembles the polyubiquitinated
proteins into microaggregates through binding to their ubiquitin
chains with its C-terminal ubiquitin binding domain and
oligomerizing at its N-terminal PB1 domain (9). These microaggregates are then recognized
by histone deacetylase 6 (HDAC6) and retrogradely transported via
the microtubule network to the microtubule-organizing center for
aggresome formation (3,10). Hence, proteasome inhibition-induced
aggresome formation represents a highly organized biological
process to switch polyubiquitinated proteins from the UPS to the
autophagy pathway.
The aggrephagy pathway alleviates proteasome
proteolysis failure-induced cell death under various stress
conditions (5). Loss of aggresome
formation regulators, such as p62, HDAC6 or PTEN-induced kinase 1
(PINK1), causes apoptosis in various cell types during proteasome
inhibition (3,11,12).
Consistently, toxicity of aggregated proteins is strongly enhanced
by inhibition of the microtubule-dependent transport machineries,
which are required for microaggregate transportation (4). More importantly, Parkinson's disease
associated proteins, such as α-synuclein, leucine rich repeat
kinase 2 and mutant DJ1, have been reported to be degraded via
aggrephagy, suggesting that abnormalities in this backup protein
degradation system may cause neurodegeneration (13-15).
However, the mechanism underlying the activation of this
compensatory pathway and the cellular factors that regulate
aggresome formation remain unknown.
Transcriptional control of stress-responsive genes
is a crucial part of the cell response to a wide variety of
stresses (16). In response to
oxidative stress, nuclear factor erythroid 2-related factor 2
(Nrf2), encoded by the NFE2L2 gene, binds to the antioxidant
response elements (ARE) sequence within the promoter of detoxifying
and antioxidant enzymes and activates their transcription (17). Interestingly, the induction of p62 by
oxidative stress is mediated by Nrf2 and, at the same time, p62
competitively binds to Kelch-like ECH-associated protein 1 (Keap1)
to activate Nrf2 (18-20).
Although Nrf2 and p62 are essential for the oxidative stress
response, the function of Nrf2 in aggresome formation during
proteasome inhibition has not been determined.
Here, we showed that the loss of Nrf2 impaired
aggresome formation and caused a hypersensitivity to proteasome
inhibition in stressed cells, which was restored by the
overexpression of p62. This study established Nrf2 as a major
transcriptional mediator for proteasome stress-induced activation
of the aggresome-autophagy pathway.
Materials and methods
Plasmid construction
The human Nrf2 and p62 full-length cDNA was obtained
from Addgene, Inc. Nrf2 cDNA was amplified by Phusion high-fidelity
DNA polymerase (Thermo Fischer Scientific, Inc.). A total of 30
cycles of PCR amplification were performed (98˚C for 5 sec, 55˚C
for 5 sec and 72˚C for 90 sec). PCR products were digested by
BamHI and XbaI. The digested fragment was cloned into
pTango-CFlag vector (BioAtom Technology) linearized with
BamHI/XbaI. The resulting construct is further
referred to as Flag-Nrf2. p62 was amplified by PCR as described
above (30 cycles of 98˚C for 5 sec, 55˚C for 5 sec and 72˚C for 80
sec) and cloned into pTango-NFlag (BioAtom Technology) after
digestion with EcoRI and XbaI. The resulting
construct is further referred to as Flag-p62. The human p62
promoter (-1781/+46) was obtained from genomic DNA purified from
293 cells based using publicly available sequence data (https://www.ncbi.nlm.nih.gov/gene/8878).
Two fragments, namely the p62/SQSTM promoter (PCR, 30 cycles of
98˚C for 5 sec, 55˚C for 5 sec and 72˚C for 40 sec) digested by
BamHI/EcoRI and the enhanced (E) GFP (PCR, 30 cycles
of 98˚C for 5 sec, 55˚C for 5 sec and 72˚C for 30 sec) digested by
EcoRI/XbaI, were cloned into an
BamHI/XbaI-digested pBluescript II vector (Addgene,
Inc.). The resulting construct was a fused EGFP sequence downstream
of the p62 promoter region. All constructs were confirmed by
sequencing. Detailed information for the primers is presented in
Table SI.
Cell lines and reagents
MG132, anisomycin (ANI), actinomycin D (ActD),
TAK715 and N-acetyl-cysteine were obtained from Tocris Bioscience.
293 cells were obtained from the American Type Culture Collection
and were cultured in DMEM (Gibco; Thermo Fischer Scientific, Inc.)
with 10% heat-inactivated fetal bovine serum (FBS; Gibco; Thermo
Fischer Scientific, Inc.) and 1% penicillin/streptomycin (Gibco;
Thermo Fischer Scientific, Inc.) at 37˚C with 5%
CO2.
For the chemical treatment, 50,000 cells were
cultured for 24 h before treatment in 24-well plates at 80%
confluence. Change the medium containing MG132 (2 µM), ANI (5
µg/ml), ActD (2 µg/ml), MG132 (2 µM) + ANI (5 µg/ml), MG132 (2 µM)
+ ActD (2 µg/ml), MG132 (2 µM) + TAK715 (10 µM) or MG132 (2 µM) +
N-acetyl-cysteine (1 mM) and treat for 12 h. In time-dependent
experiments, 293 cells were cultured in MG132 (2 µM)-containing
medium for 12 h and ANI (5 µg/ml) was added at 0, 2, 4 or 6 h. To
evaluate dose dependent effects of MG132 on Nrf2 accumulation, 293
cells were treated with 0.1, 0.5, 1, 2 and 10 µM MG132 for 12
h.
Transient transfection of 293 cells was performed
using Mega-tran 1.0 (OriGene Technologies, Inc.) according to the
manufacturer's instructions. Briefly, 50,000 cells were plated in
24-well plates 24 h prior to transfection (80% confluence). Medium
was replaced with 0.5 ml of complete medium containing FBS and
antibiotics 1 h before transfection. DNA (0.5 µg) was diluted in 50
µl of serum-free DMEM, mixed and 1.5 µl of MegaTran 1.0 was added
and gently mixed. The resulting solution was incubated for 15 min
at room temperature and added to the cells. Medium was replaced 5 h
after adding the transfection solution. Transfection efficiency was
checked 24 h post transfection by western blot.
Generation of Nrf2 knockout
(nrf2-/-) cells
Nrf2 knockout cells were generated from 293 cells
with a CRISPR/Cas9 system designed in our lab. pSpCas9(BB)-2A-Puro
(PX459) V2.0 was a gift (cat. no. 62988; Addgene, Inc.) (21). All single guide (sg) RNA targeting
sequences were predicted as first hits using the CRISPR guide
design website (https://benchling.com). The 293 cells
were first transfected with two PX459 vectors, which targeted
5'-CTTTTTTTGTCTTAAACAT-3' and 5'-GAAAGTTATGGCAGGTTTA-3', to delete
the first exons of the NFE2L2 gene. After 24 h, cells were
diluted and seeded in 96-well plates at 1 cell/well to isolate
monoclonal cells without Nrf2 expression, as determined by
quantitative (q) PCR and western blot analysis. Genomic DNA was
extracted from cells by using PureLink Genomic DNA kit (Invitrogen;
Thermo Fischer Scientific, Inc.). Further PCR analysis (as above;
30 cycles of 98˚C for 5 sec, 55˚C for 5 sec and 72˚C for 30 sec)
was performed to validate the deletion of exon2 of the
NFE2L2 gene in the monoclonal cells; primer sequences are
presented in Table SI.
Generation of stable expression
cells
The coding sequences Flag-p62 and Flag-Nrf2 were
cloned into the plenti6-LVP lentiviral vectors (Thermo Fischer
Scientific, Inc.). Viruses were generated and used to transduce
nrf2-/- cells as described previously (12). Briefly, plenti6-Flag-p62 and
plenti6-Flag-Nrf2 plasmids were co-transfected with lentiviral
packaging mix into 293 cells using Mega-tran 1.0. Supernatant
containing lentivirus was harvested and cellular debris was removed
by centrifugation (500 x g, 10 min, room temperature). A total of
10,000 nrf2-/- cells was added to 24-well plates
and transduced at an MOI of 5. Three days after transduction,
puromycin (2 µg/ml) was added to the culture medium to generate
stable nrf2-/-[Flag-Nrf2] and
nrf2-/-[Flag-p62] cell lines. After puromycin
selection, single cell clones were verified by qPCR analysis to
validate the expression of exogenous Nrf2 and p62. Transfection
controls were prepared with empty vector (plenti6-LVP). To evaluate
the aggresome formation efficiency, nrf2+/+,
nrf2-/-[Vector],
[nrf2-/-[Flag-Nrf2] and
nrf2-/-[Flag-p62] cells were treated with MG132
(2 µM) for 14 h.
Cell death assay
Cell death-inducing effects of proteasomal
inhibition were measured with CF488A-Annexin V (ANXA5) and
propidium iodide (PI) Apoptosis kits (cat. no. 30061; Biotium,
Inc.) as described previously (12).
A total of 100,000 cells/cm2 were grown in 10 cm dishes.
After 24 h, they were treated with DMSO (0.1%) or 2 µM MG132 (in
DMSO) for 20 h. Cells were then harvested by digestion with 0.05%
trypsin-EDTA, washed twice with ANXA5-Binding buffer and
resuspended in 100 µl of this buffer. To each sample, 15 µl
CF488A-ANXA5 and 5 µl PI were added and incubated in the dark for
30 min at room temperature. Unbound dyes were washed off with
binding buffer, cells were mounted onto slides. All and images were
captured using a fluorescence microscope (magnification, x40).
RNA extraction and reverse
transcription-quantitative (RT-q) PCR
Total RNA was extracted from cells using the Qiagen
All-Prep DNA/RNA/miRNA Universal kit (Qiagen, Inc.) according to
manufacturer's protocols. RNA samples were subjected to DNase
digestion using the Turbo DNA-free™ kit (Ambion; Thermo Fischer
Scientific, Inc.). cDNA was synthesized by using ReverTra
Ace® qPCR RT kit (Toyobo Life Science) with a random
primer (30˚C for 10 min, 42˚C for 20 min and 99˚C for 5 min). qPCR
was performed using SYBR Premix Ex Taq (Takara Bio, Inc.) in a
CFX96 real-time PCR system (Bio-Rad Laboratories, Inc.) following
the manufacturer's protocol; thermocycling conditions were as
follows: 95˚C for 30 sec, followed by 40 cycles of 95˚C for 5 sec
and 60˚C for 30 sec. Levels of Nrf2 and p62 were normalized to
GAPDH using the 2-ΔΔCq method (22). The primers used for RT-qPCR were shown
in Table SI.
Immunoblotting and immunocytochemistry
analyses
Proteins were extracted from cells by 1% NP-40 in
Tris buffer (50 mM Tris, 150 mM NaCl, pH 8.0, with protease and
phosphatase inhibitors; Thermo Fischer Scientific, Inc.). The
protein concentration was determined by BCA. Proteins (20 µg) were
separated on 10% SDS-PAGE gels and transferred to PVDF membranes.
Membranes were blocked with 5% non-fat milk in PBST (PBS pH 7.4
with 0.2% Tween-20) for h at room temperature, followed by
incubation at 4˚C overnight with primary antibodies and washes with
PBST (3x10 min at room temperature). Membranes were then incubated
with 0.1 µg/ml Dylight 800 anti-rabbit (cat. no. 5230-0412; KPL,
Inc.) or Dylight 680 anti-mouse (cat. no. 5230-0406; KPL, Inc.)
secondary antibodies in the dark for 2 h at room temperature. After
washing with PBST (3x10 min), images were acquired using a Li-Cor
Odyssey Clx Infrared Imaging System (LI-COR Biosciences).
The following primary antibodies were used:
Anti-Nrf2 (cat. no. ab137550; 1:2,000; Abcam), anti-p62 (cat.no.
18420-1-AP; 1:3,000; ProteinTech Group, Inc.), anti-FlagTag (cat.
no. ANT-146; 1:5,000; Prospec-Tany TechnoGene, Ltd.),
anti-phospho-p38 MAPK (Thr180/Tyr182; cat. no. 9211; 1:1,000; Cell
Signaling Technology, Inc.), anti-p38 MAPK (D13E1) XP (cat. no.
8690; 1:1,000; Cell Signaling Technology, Inc.), anti-β-actin (cat.
no. 60008-1-Ig; 1:5,000; ProteinTech Group, Inc.) and anti-GAPDH
(cat. no. 10494-1-AP; 1:5,000; ProteinTech Group, Inc.).
For the immunocytochemistry, sterilized coverslips
were placed in 24-well plates. Cells (30,000) were seeded and
cultured for 24 h before treatment. After treatment, cover slips
were washed with PBS (3x10 min at room temperature) and fixed with
4% paraformaldehyde for 15 min at room temperature. Cells were then
permeabilized with 0.2% Triton X-100 for 15 min and blocked with 5%
goat serum (Thermo Fischer Scientific, Inc.) for 1 h at room
temperature. Samples were incubated with primary anti-ubiquitin Lys
48 (cat. no. 05-1307; 1:100; EMD Millipore) and anti-FlagTag
antibodies (cat. no. ANT-146; 1:500; Prospec-Tany TechnoGene, Ltd.)
at 4˚C overnight. After washing with PBS (3x10 min at room
temperature), cells were incubated with goat anti-mouse Alexa Flour
488- (cat. no. A32723; 1:300) or goat anti-rabbit Alexa Fluor
568-conjugated secondary antibodies (cat. no. A-11011; 1:300;
Thermo Fischer Scientific, Inc.) for 2 h at room temperature,
stained with DAPI (1 µg/ml) for 5 min at room temperature and
mounted for fluorescence microscopy examination. Fluorescent
microscopy was performed with a confocal microscope (magnification,
x63), with ≥20 observations per experiment.
Statistical analysis
Data are presented as the mean ± SEM. All the
experiments were repeated ≥3 times. Statistical analysis was
performed using one-way ANOVA followed by Tukey's multiple
comparison test using GraphPad Prism 8 (GraphPad Software, Inc.).
Detailed information about statistical analysis for each experiment
are presented in the figure legends. P<0.05 was considered to
indicate a statistically significant difference.
Results
Inhibition of protein synthesis blocks
MG132-induced aggresome formation and Nrf2 accumulation
Previous studies have demonstrated that UbK48-linked
polyubiquitin conjugates are proximal substrates of proteasomal
proteolysis (23). During proteasomal
stress, UbK48-linked polyubiquitinated proteins accumulate and
package to form aggresome, which are recognized by antibodies.
UbK48-positive structures are co-localized with other aggresome
markers, such as p62, HDAC6 and PINK1S (13,24,25). To
examine the role of just synthesized proteins in aggresome
formation, we studied aggresome formation efficiency in
MG132-treated cells after blocking transcription by ActD or
translation by ANI. Immunostaining showed that 58.4% of cells
formed UbK48-positive aggresomes in MG132-treated 293 cells and
4.4% of positive staining was observed in MG132+ANI-treated cells.
ActD+MG132 treatment did not completely inhibit aggresome
formation; however, there was a significant decrease in the number
of aggresome containing cells after treatment with MG132+ActD
compared with the MG132-treated cells (Fig. 1A). ActD treatment resulted in an
accumulation of poly-ubiquitinated proteins in the nucleus, which
may be caused by recruitment of ubiquitin E3 ligase into nuclear,
such as murine double minute protein (26). We further examined aggresome formation
efficiency by treating cells with MG132 and adding ANI at different
incubation times (Fig. 1B).
Inhibition of protein translation by ANI added after 2 h of MG132
pretreatment blocked aggresome formation in 293 cells. Aggresome
formation efficiency increased to 37.9% when translation was
blocked after 4 h of MG132 pretreatment and after 6 h of MG132
pretreatment, ANI had no effect on aggresome formation efficiency
compared with the MG132-terated cells (Fig. 1B). These data indicated that protein
translation in the first hours during proteasomal stress was
critical for aggresome formation.
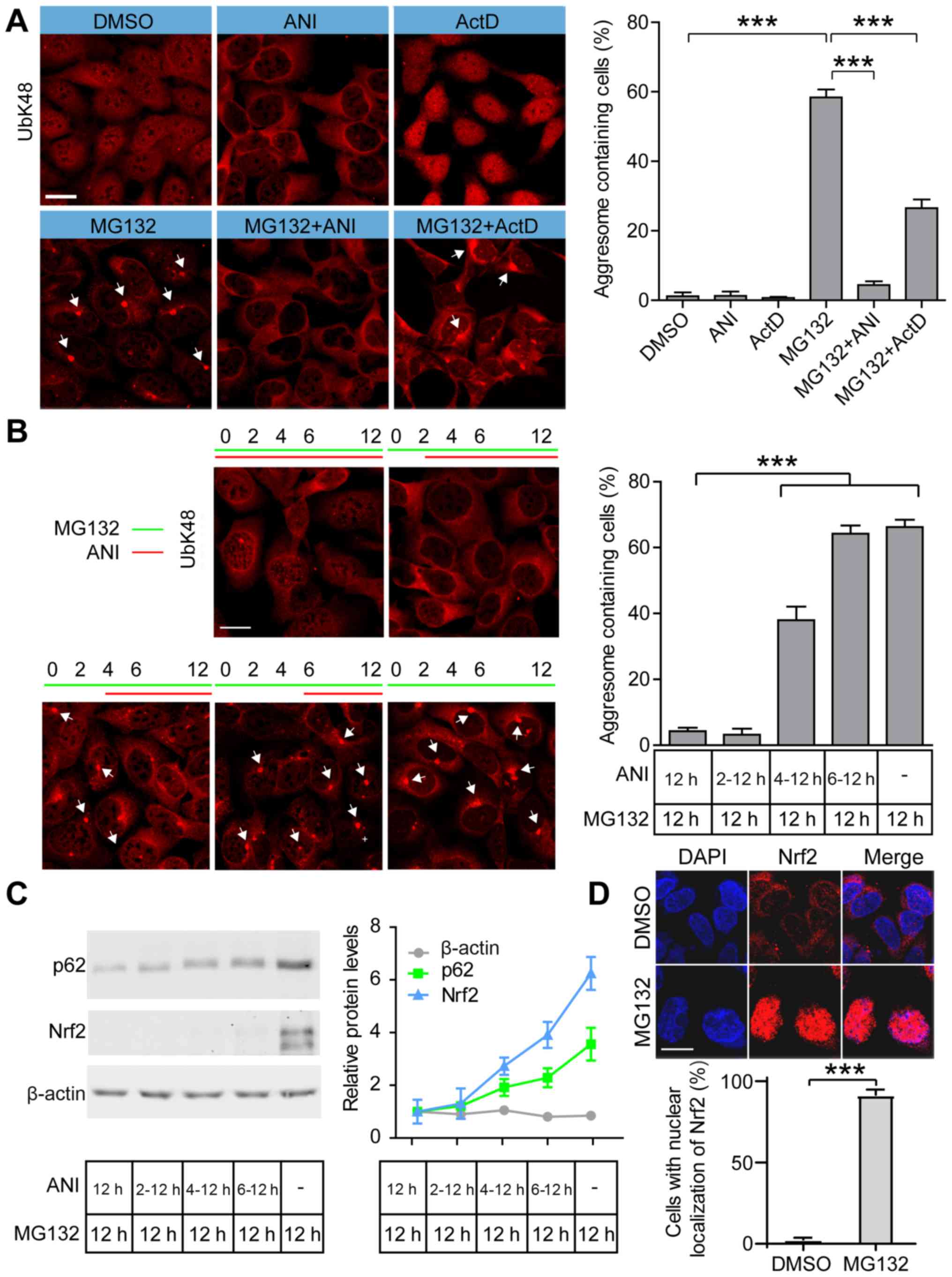 | Figure 1.Inhibition of protein synthesis
blocks proteasome inhibitor-induced aggresome formation. (A)
Representative images and quantification of aggresomes in DMSO,
MG132, MG132+ANI and MG132+ActD treated cells. 293 cells were
treated with ANI (5 µg/ml), ActD (2 µg/ml) and/or MG132 (2 µM) for
12 h and stained with anti-UbK48 to visualize aggresomes
(arrowheads); scale bar, 20 µm. (B) Representative images and
quantification of aggresomes in cells treated with MG132 (2 µM)
and/or ANI (5 µg/ml) for the indicated periods and aggresomes were
stained with anti-UbK48 (arrowheads); scale bar, 20 µm. (C)
Representative immunoblots and quantification of Nrf2 and p62 in
cells treated by MG132 (2 µM) and/or ANI (5 µg/ml). (D)
Representative images and quantification of Nrf2 in DMSO and MG132
treated cells. The 293 cells were treated with MG132 (2 µM) for 12
h and stained with anti-Nrf2 antibody to visualize the localization
of endogenous Nrf2; DAPI was used to detect nuclei; scale bar, 20
µm. Data are presented as the mean ± SEM and were assessed using
one-way ANOVA followed by Tukey's test. ***P<0.001.
Nrf2, nuclear factor erythroid 2-related factor 2; UbK48, ubiquitin
Lys 48; ANI, anisomycin; ActD, actinomycin D. |
Immunoblotting analysis revealed that of protein
translation induced by ANI prevented Nrf2 accumulation at early
stages of stress, compared with the MG132-treated cells (Fig. 1C), and Nrf2 was localized in the
nucleus after MG132 treatment (Fig.
1D). Furthermore, 0.1 to 10 µM MG132 treatment showed that Nrf2
accumulated in a dose-dependent manner (Fig. S1). Levels of p62, critical for
poly-ubiquitinated proteins to form microaggresomes during
proteasome inhibition, were higher in MG132-treated cells compared
with ANI+MG132-treated groups (Fig.
1C). These results suggested that stress-induced protein
synthesis, including the synthesis of Nrf2 and p62, may be
essential for aggresome formation.
Loss of Nrf2 results in failure of
aggresome formation
If Nrf2 was required for MG132-induced aggresome
formation, we hypothesized that knockout of Nrf2 may interfere with
aggresome formation and cell survival after MG132 treatment. To
test this, we generated Nrf2 knockout cell lines using CRISPR/Cas9
(Fig. S2A) (21). Nrf2 is rapidly processed by UPS
(27,28), but protein levels were detected by
western blot in 293 cells following MG132 treatment to induce rapid
accumulation of Nrf2 (Fig. S2B). As
expected, deleting the first exon of NEL2L resulted in loss
of Nrf2 protein expression in nrf2-/- cells
(Fig. S2B). Further PCR analysis of
the monoclonal cell lines without Nrf2 expression showed that the
first exon of NEL2L was successfully deleted (Fig. S2C and D). To determine aggresome formation
efficiency in nrf2+/+ and
nrf2-/- cells, MG132 was used to induce the
aggresome formation. A 14 h treatment with 2 µM MG132 induced
aggresome formation in 68% of nrf2+/+ and 12.5%
of nrf2-/- cells (Fig.
2A). It has been reported that loss of Nrf2 induces increased
reactive oxygen species (ROS) production in cultured cells and
mouse brains (28,29), we wanted to evaluate whether aggresome
formation defects were caused by increased intracellular ROS in
nrf2-/- cells. To reduce intracellular ROS level,
we treated nrf2+/+ and nrf2-/-
cells with N-acetyl-cysteine (NAC), an aminothiol and synthetic
precursor of intracellular cysteine and GSH (30). NAC treatment had no significant effect
on the aggresome formation in nrf2+/+ and
nrf2-/- cells, suggesting that ROS levels may not
influence aggresome formation during proteasome inhibition
(Fig. S2E and F).
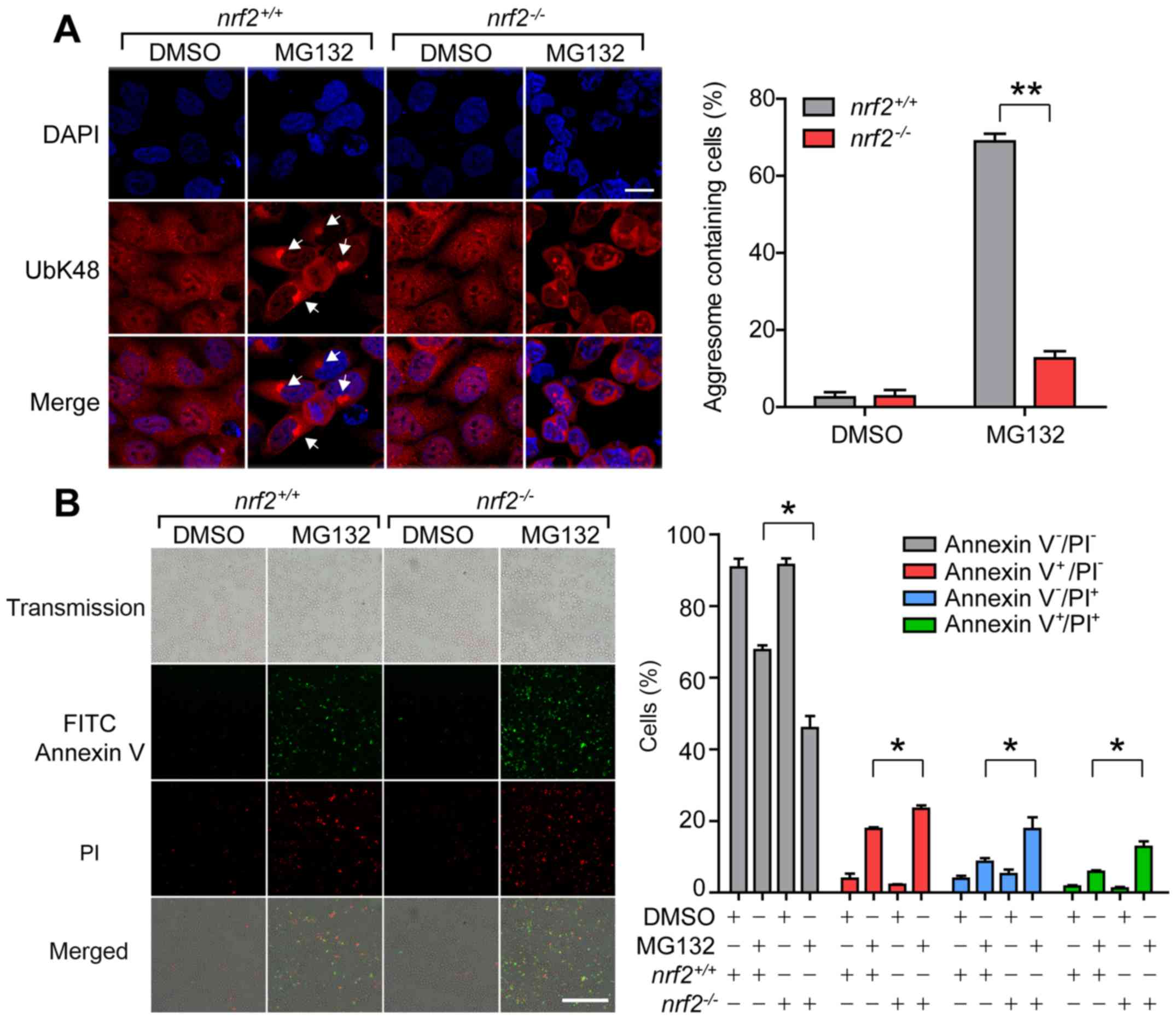 | Figure 2.Loss of Nrf2 results in failure of
proteasome inhibition-induced aggresome formation. (A)
Representative images and quantification of aggresomes in
nrf2+/+ and nrf2-/- cells after
14 h treatment with MG132 (2 µM). Anti-UbK48 was used to visualize
aggresomes (arrowheads) and DAPI staining was used to detect
nuclei; scale bar, 20 µm. (B) Representative images and
quantification of nrf2+/+ and
nrf2-/- cells stained with Annexin V (green) and
PI (red) after 20 h treatment with DMSO or MG132 (2 µM). Live
cells, Annexin V-PI-; early apoptotic cells,
Annexin V+PI-; necrotic cells, Annexin
V-PI+; late apoptotic cells, Annexin
V+PI+; scale bar, 100 µm. Data are presented
as the mean ± SEM representative of three independent experiments
and were assessed using one-way ANOVA followed by Tukey's test.
*P<0.05 and **P<0.01. Nrf2, nuclear
factor erythroid 2-related factor 2; PI, propidium iodide;
nrf2-/-, Nrf2 knockout 293 cells; UbK48,
ubiquitin Lys 48. |
To explore the time-dependent manner of MG132
induced cell death, we treated 293 cells with 2 µM MG132 and
analyzed cytotoxicity with FITC-Annexin V and PI assays. After 20 h
treatment with MG132, changes in viability were significant in 293
cells (Fig. S3). We further treated
nrf2+/+ and nrf2-/- cells with
2 µM MG132 for 20 h to induce apoptosis and necrosis. The cell
viability assay demonstrated that the loss of Nrf2 significantly
increased the cells susceptibility towards proteasomal
inhibition-triggered cell death following MG132 treatment (Fig. 2B). MG132 treatment induced apoptosis
in 8.7% and necrosis in 5.8% of nrf2+/+ cells,
and in nrf2-/- cells, apoptosis and necrosis
rates increased to 17.7 and 12.7%, respectively.
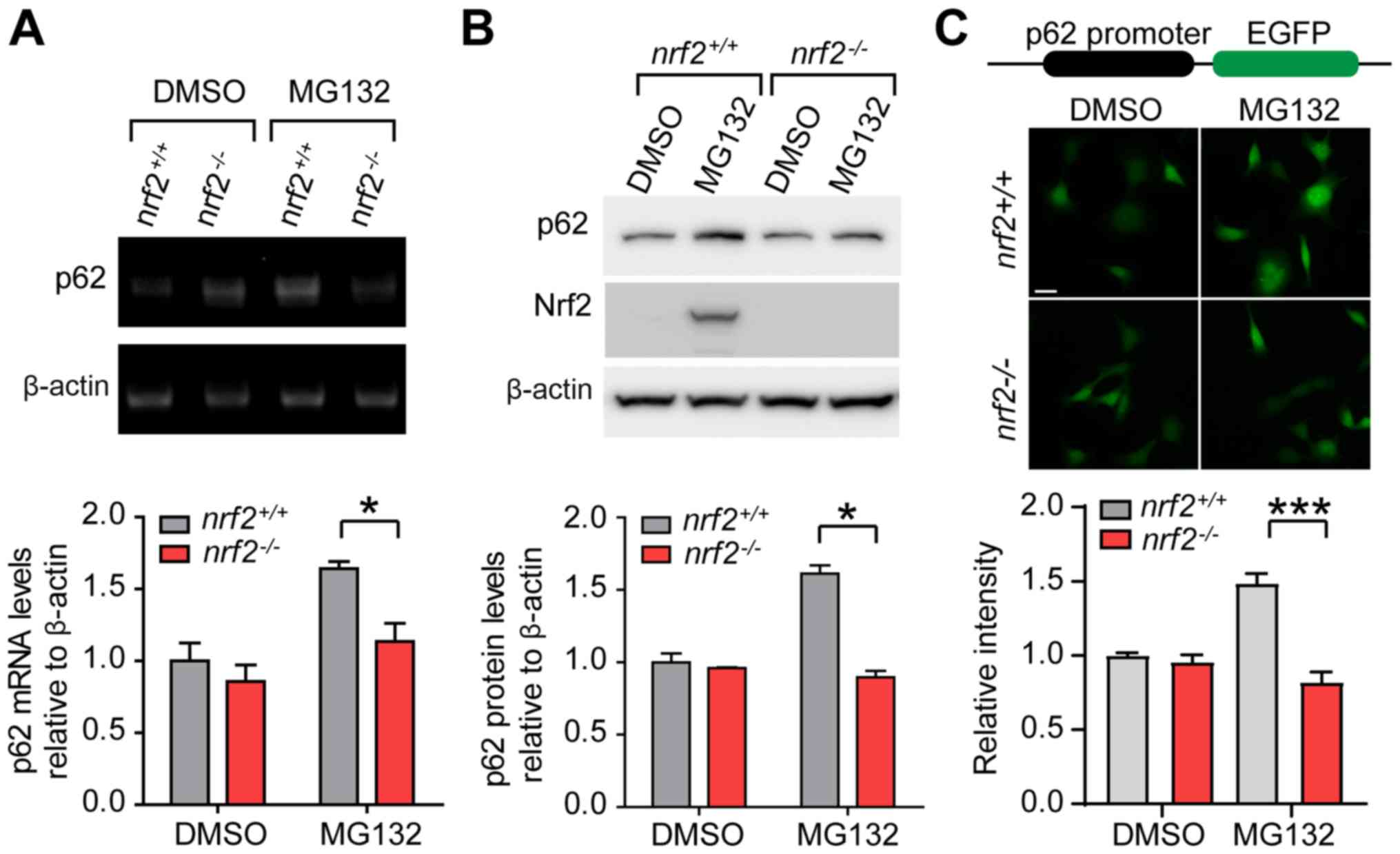
Transcriptional upregulation of p62
during proteasome inhibition is dependent on Nrf2
The presence of the ARE in the p62 gene suggests
that p62 may be transcriptionally activated by Nrf2 during
proteasome inhibition (17). To test
this hypothesis, we examined mRNA and protein levels of p62 in
nrf2+/+ and nrf2-/- cells. mRNA
and protein levels of p62 were significantly increased in
nrf2+/+ cells when treated with MG132 and this
response was inhibited by Nrf2 knockout (Fig. 3A and B).
Similar to p62, heme oxygenase-1, a gene regulated by Nrf2 during
proteasome inhibition (31),
expression decreased in nrf2-/- cells compared
with nrf2+/+ cells following MG132 treatment
(Fig. S4A and B). This further indicated that Nrf2 may be
essential for the transcriptional activation of stress-response
genes. To exclude the possibility that Nrf2 may affect the activity
of other transcriptional factors, we evaluate protein levels of
HSP70, samples in which the transcriptional activation was driven
by HSF1 during MG132 treatment (32).
Loss of Nrf2 had no effect on HSP70 expression compared with
nrf2+/+ cells (Fig.
S4C and D).
To further examine the transcriptional regulation of
p62 by Nrf2, we cloned ~1.8 kb of the human p62 promoter region
upstream of EGFP (33). The reporter
gene was expressed in nrf2+/+ and
nrf2-/- cells, and MG132 treatment significantly
increased EGFP expression in nrf2+/+ but not
nrf2-/- cells (Fig.
3C). The loss of Nrf2 inhibited aggresome formation and p62
transcriptional activation during proteasome inhibition.
Expression of p62 rescues proteasomal
stress response defects in Nrf2 knockout cells
Proteasome inhibition stimulates p62 ubiquitin
binding activity by phosphorylation and assembles ubiquitinated
cargos into microaggregates through self-oligomerization (13,34,35). Loss
of p62 suppresses the formation of aggresomes and clearance of
poly-ubiquitinated proteins during MG132 treatment in 293 cells
(12). If the Nrf2 knockout induced
proteasome formation, defects may be dependent on the loss of p62
in 293 cells, overexpression of p62 in nrf2-/-
cells may rescue these defects. To this end, we generated
nrf2-/-[Flag-Nrf2] and
nrf2-/-[Flag-p62] stable cell lines by
lentivirus-transduction (Fig.
S5).
A 14 h treatment with 2 µM MG132 induced aggresome
formation in 68.8% of nrf2+/+ cells, but
aggresome-containing cells in the nrf2-/-[Vector]
cell line was decreased to 17.1% (Fig.
4A). In nrf2-/- cells, overexpression of
Flag-Nrf2 rescued aggresome formation in 72.5% of cells and
overexpression of Flag-p62 rescued aggresome formation efficiency
to 51%, suggesting that p62 protein levels may be critical for
aggresome formation. The aggresome formation efficiency in
nrf2-/-[Flag-p62] cells lower than that of the
nrf2-/- [Flag-Nrf2] cells, indicating that other
targets regulated by Nrf2 are involved in aggresome formation. A 20
h treatment with 2 µM MG132 induced apoptosis in 19.5% and necrosis
in 11.2% of the nrf2-/-[Vector] cells, and in
nrf2-/-[Flag-Nrf2] cells, apoptosis and necrosis
rates were reduced to 13.2 and 9.7%, respectively (Fig. 4B). Overexpression of Flag-p62 in
nrf2-/- cells decreased apoptosis and necrosis
rates to 8.1 and 5.1%, respectively, suggesting that Nrf2-mediated
transcriptional activation of p62 was required to minimize
proteasomal stress-induced necrosis and apoptosis. Immunoblotting
analysis showed that overexpression of Flag-Nrf2 rescued
MG132-induced transcriptional activation of p62 deficits in
nrf2-/-[Vector] cells (Fig. 4C). Exogenous expressed Flag-Nrf2 in
nrf2-/- cells significantly increased after MG132
treatment compared with the unstressed condition, further
supporting that Nrf2 was sensitive to proteasome inhibition
(Fig. 4C). Together, these results
confirmed that Nrf2-mediated transcriptional activation of p62 was
essential for aggresome formation during proteasome
dysfunction.
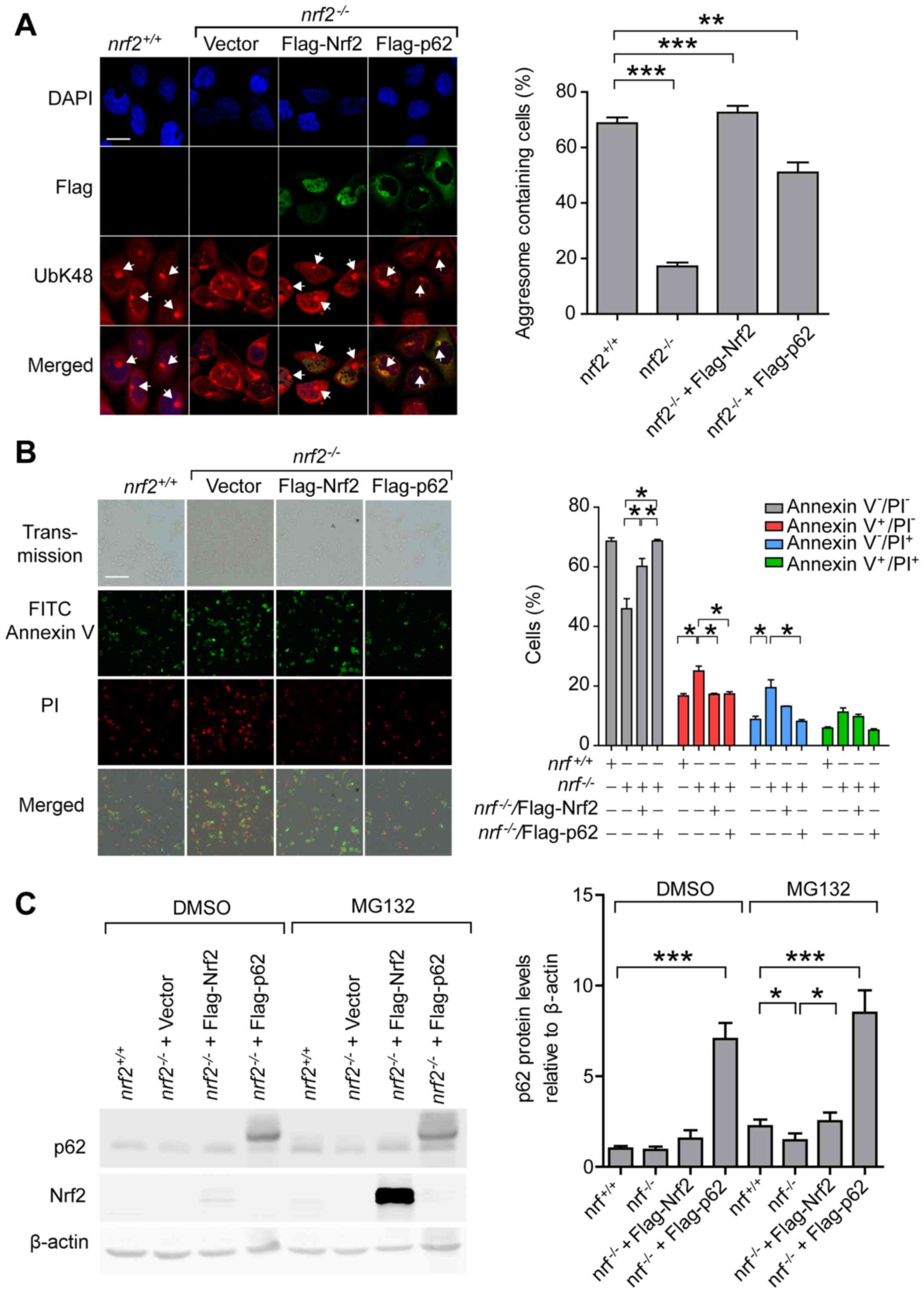 | Figure 4.Expression of p62 rescues the defects
of proteasomal stress response in Nrf2 knockout cells. (A)
Representative images and quantification of aggresomes in
nrf2+/+, nrf2-/-[Vector],
nrf2-/-[Flag-Nrf2] and
nrf2-/-[Flag-p62] cells after 14 h treatment with
MG132 (2 µM). Anti-UbK48 was used to visualize ubiquitin-containing
aggresomes (arrowheads), anti-Flag was used to visualize exogenous
Nrf2 and p62, and DAPI was used to detect nuclei; scale bar, 20 µm.
(B) Representative images and quantification of
nrf2+/+, nrf2-/-[Vector],
nrf2-/-[Flag-Nrf2] and
nrf2-/-[Flag-p62] cells stained with Annexin V
(green) and PI (red) after 20 h treatment with DMSO or MG132. Live
cells, Annexin V-PI-; early apoptotic cells,
Annexin V+PI-; necrotic cells, Annexin
V-PI+; late apoptotic cells, Annexin
V+PI+; scale bar, 100 µm. (C) Representative
immunoblots and quantification of p62 and Nrf2 levels in
nrf2+/+, nrf2-/-[Vector],
nrf2-/-[Flag-Nrf2] and
nrf2-/-[Flag-p62]. Data are presented as the mean
± SEM representative of three independent experiments and were
assessed using one-way ANOVA followed by Tukey's test.
*P<0.05, **P<0.01 and
***P<0.001. Nrf2, nuclear factor erythroid 2-related
factor 2; PI, propidium iodide; nrf2-/-, Nrf2
knockout 293 cells; UbK48, ubiquitin Lys 48; [], transfection
construct. |
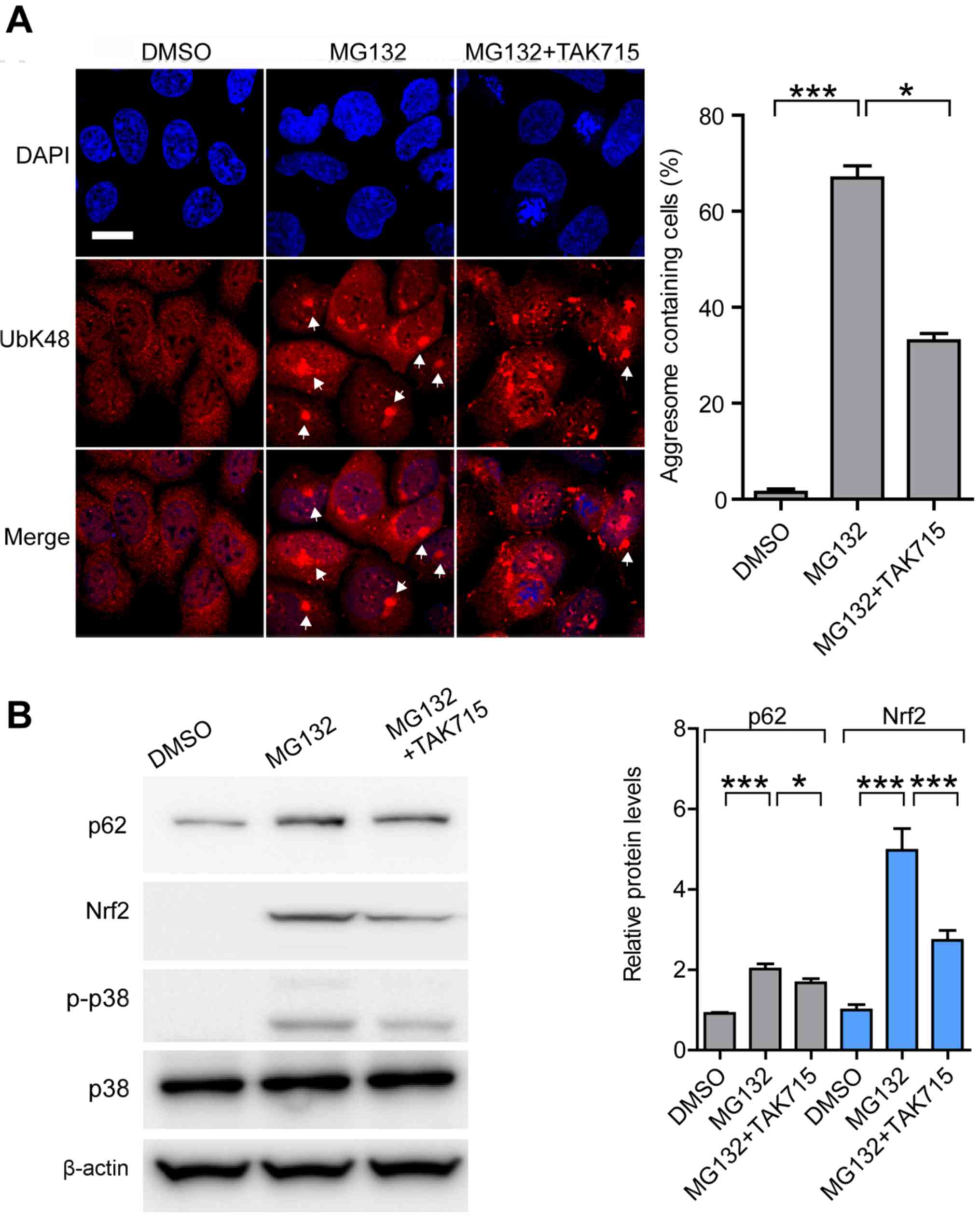
Nrf2 mediated aggresome formation
depends on p38/MAPK kinase activity
It has been reported that Nrf2 activation is
p38/MAPK-dependent, which compromises the cytotoxic effects through
activating transcription of downstream targeting genes during
proteasome inhibition (36). We
blocked p38/MAPK activity using the specific inhibitor TAK715 to
examine, if Nrf2-mediated aggresome formation in MG132-treated
cells was dependent on the p38/MAPK pathway. Consistent with the
previous study, p38 inhibitor TAK715 partially inhibited activation
of p38-induced by proteasomal inhibition; the efficiency of
aggresome formation in MG132+TAK715-treated cells decreased by 50%
compared with the MG132-treated cells (Fig. 5A). Western blot analysis confirmed
that protein levels of Nrf2 and p62 significantly decreased in
MG132+TAK715-treated cells compared with the MG132-treated cells
(Fig. 5B). Taken together, blocking
Nrf2 activation through suppression of the p38/MAPK signaling
pathway may decrease the efficiency of aggresome formation.
Discussion
The aggresome has emerged as a key stress-response
subcellular structure in the clearance of misfolded protein
aggregates during many stress conditions, which are often linked
with cell death in neurodegenerative diseases (37). Despite the potential importance of the
aggresome in managing the stress response and its association with
disease, few proteins critical for aggresome formation have been
identified (38-40).
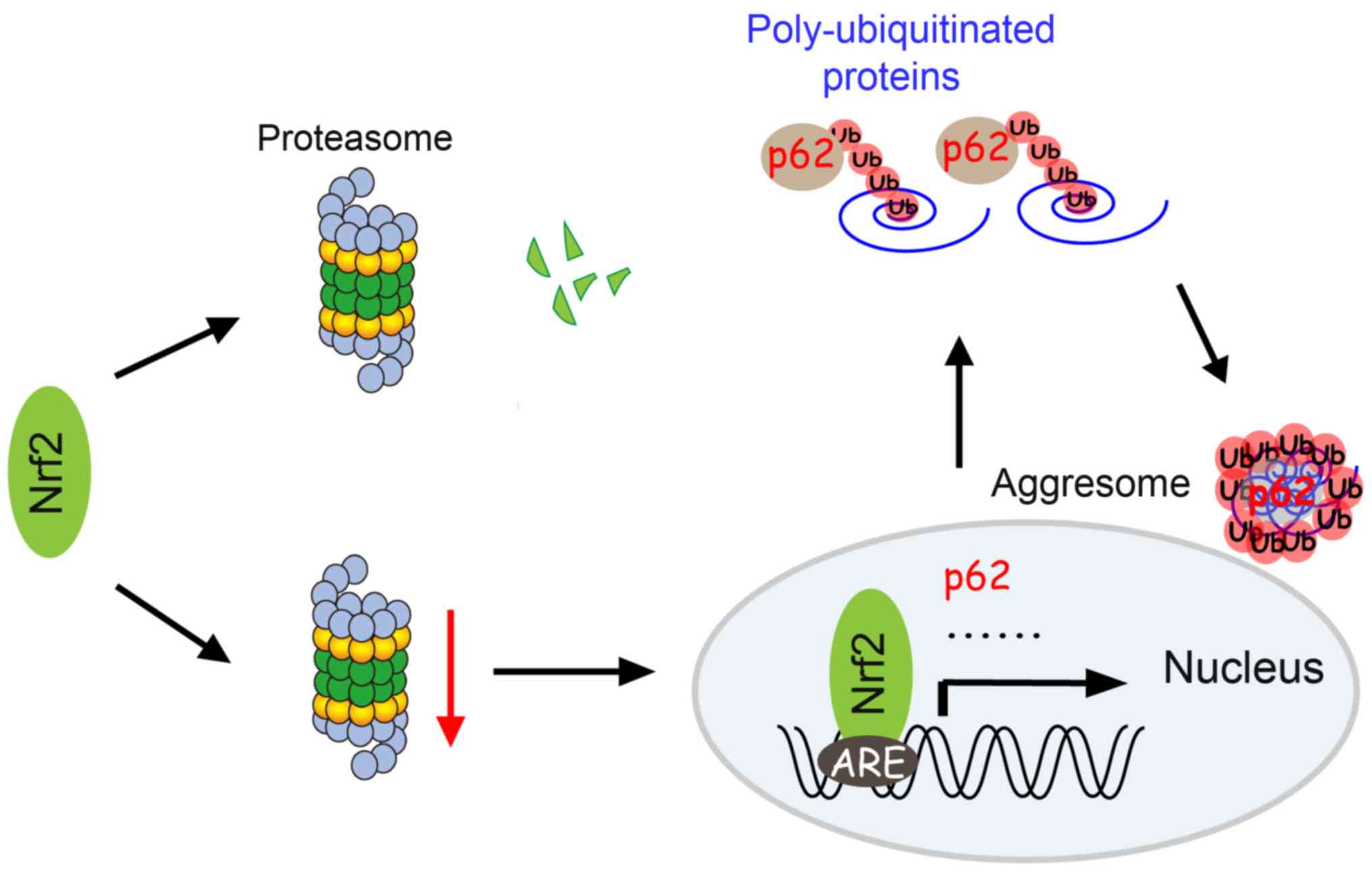
In this study, we reported that Nrf2 was crucial for aggresome
formation and cell survival in response to proteasome
inhibition-induced stress. Based on our results, we proposed a
model, which described that Nrf2-mediated mRNA transcription may be
induced through cellular stress and mediated by proteasome
dysfunction (Fig. 6).
During mRNA translation, the synthesized polypeptide
chain is elongated by the ribosome with the linear chain folding
into its three dimensional structure and then being targeted to a
precise intracellular location. If misfolded, the polypeptides are
rapidly degraded by UPS (41,42). However, when UPS is impaired or
overwhelmed, misfolded polypeptides are sequestered into
microaggregates and then retrogradely transported to the
microtubule-organizing center to form aggresomes (43,44). How
does the UPS dysfunction activate downstream signals to form the
aggresome? As shown in previous studies, during proteasome
inhibition, newly synthesized proteins play a special regulatory
role in the initiation of aggresome formation (40,45).
Therefore, we hypothesized that inhibitors of transcription and
translation may suppress aggresome formation. Inhibition of protein
translation by ANI completely inhibited aggresome formation,
whereas transcriptional inhibitor ActD, which prevents mRNA
synthesis, only partially reduced the efficiency of aggresome
formation. These data supported the notion that translational
regulation provided immediate and effective changes in protein
levels involved in aggresome formation. However, transcriptional
regulation is essential in mediating the strength of stress
response (46).
Cells activate mechanisms to regulate gene
expression after exposure to environmental stress, largely at the
transcriptional level. These stress responses enable cells to cope
with and adapt to stressful situations, such as starvation,
oxidative and DNA damage (47). A
central mechanism for the regulation of stress-induced
transcription is mediated by activation of the specific
transcriptional factors (48).
Transcriptional factor Nrf2 is the major regulator of
cytoprotective responses to oxidative and electrophilic stress
(49). Nrf2 is rapidly turned over by
UPS, with a short half-life under physiological conditions (7-15
min) that is increased to 30-100 min in the presence of a stress
inducer (50,51). Proteasome inhibitors, such as
clasto-lactacystin, β-lactone, MG132 and MG115, lead to the rapid
accumulation of Nrf2 in the nuclei (52,53). Here,
we showed that MG132, at low concentration, such as 0.5 µM and 1
µM, induced Nrf2 accumulation, indicating that Nrf2 was extremely
sensitive to proteasome inhibition making it a potential candidate
to serve as an upstream modulator in the proteasomal stress
response. Nrf2 knockout cells displayed reduced efficiency in
aggresome formation, as well as increased cell death. This
phenotype was suppressed by the overexpression of Nrf2 and p62,
suggesting that the reduced aggresome formation efficiency in
nrf2-/- cells may be due to their lack of p62.
These results supported claims of a major role of Nrf2 in the
transduction of proteasome impairment signaling to the downstream
aggresome-autophagy machinery.
Loss the Nrf2 expression caused decreases in
aggresome formation efficiency (68.7 and 17.1% for
nrf2+/+ and nrf2-/-,
respectively), while transcriptional inhibitor ActD only reduced
the efficiency to 37.9%. The data suggested that Nrf2 regulated
aggresome formation not only through transcriptional upregulation
of targeting mRNA, such as p62, but also through an unknown
pathway. Loss of Nrf2 leads to mitochondrial depolarization,
decreased ATP levels and impaired respiration (54). Microaggregate transport, through the
microtubule mediated by HDAC6, is an energy-consuming process
(3), and decreased ATP levels may
contribute to aggresome formation defects in
nrf2-/- cells, a process which is independent of
Nrf2-regulated transcriptional reprogramming. Although
overexpression of p62 restored aggresome formation efficiency to
51%, levels were still different compared with
nrf2-/-[Flag-Nrf2] cells (72.5%), indicating that
Nrf2 influenced aggresome formation through multiple pathways.
Genetic activation of Nrf2 by deletion of Keap1 increases
the mitochondrial membrane potential and ATP levels, the rate of
respiration and the efficiency of oxidative phosphorylation
(54). This may explain the small
increase of aggresome formation efficiency in Nrf2 overexpressing
in nrf2-/- cells compared with the
nrf2+/+[Vector] cells. Further studies are needed
to evaluate the association between Nrf2-mediated mitochondrial
function and aggresome formation.
Proteasome inhibitors have been shown to induce
aggresome formation in many cell types, including 293, MEF, as well
as neuronal-like cells PC12 and N2A, in a time- and dose-dependent
manner (26,55-57).
However, some misfolded proteins form dispersed aggregates rather
than aggresomes during proteasome inhibition, such as HeLa, Huh7
and Cos7 (55,58). In contrast to aggresomes formed around
MTOC, dispersed aggregates are distributed in the cells and do not
result in cytoskeleton rearrangements, suggesting that dispersed
aggregates may represent intermediate particles during aggresome
assembly (24,58). The defects of aggresome formation in
these cells may be caused by the lack of critical regulators of
aggresome assembly, such as the subtype of p38 kinase (24). Due to their biochemical and
morphological similarities to the protein inclusion bodies in
neurodegenerative diseases, aggresomes have received much attention
in the past twenty years (41,59).
Aggresomes have been thought to play a critical role in protein
surveillance and the pathogenesis of neurodegenerative diseases
(60). Induction of aggresome
formation can increase cell survival when overexpressing a mutant
huntingtin fragment in primary cultured neuronal cells (61). Reducing the load of aggresomes by
genetically inhibiting ubiquitination enhances polyQ-mediated
neuronal death in a mouse model (62). An Nrf2-mediated aggresome formation
mechanism may not only explain the activation of autophagy by
proteasome dysfunction, but also implicate the inefficient switch
between different protein degradation pathways, as one pathological
mechanism of neurodegenerative diseases.
In summary, our study revealed transcriptional
events associated with Nrf2 as critical signaling modules for the
perinuclear aggresome formation during proteasomal stress (Fig. 6). These findings supported claims that
the stress-induced aggresome-autophagy pathway may be critical for
cell survival during proteasomal failure. Given that protein
aggregation and accumulation is related to many diseases (63), our findings suggested the p38/Nrf2/p62
pathway as an important therapeutic target.
Supplementary Material
Low levels of MG132 induce Nrf2
accumulation. (A) Representative immunoblots and (B) quantification
of Nrf2 in cells treated with varying amounts of MG132. Data are
presented as the mean ± SEM representative of three independent
experiments and were assessed using one-way ANOVA followed by
Tukey's test. **P<0.01 and ***P<0.001. Nrf2, nuclear factor
erythroid 2-related factor 2.
Generation of Nrf2 knockout cells. (A)
Schematic diagram of CRISPR/Cas9 genome editing of NEF2L2.
(B) Representative immunoblots of Nrf2 in nrf2+/+
and nrf2-/- cells treated with MG132 (2 μM) for
12 h. (C) Representative images of NEF2L2 gene fragments in
nrf2+/+ and nrf2-/- cells;
amplified genomic DNA covered the sgRNA-targeting sequence. (D) DNA
sequences of PCR fragments in created by PCR. (E) Representative
images and (F) quantification of aggresomes in MG132 or MG132+NAC
(1 mM) treated cells; anti-ubiquitin Lys 48 visualizes aggresomes;
scale bar, 20 μm. Data are presented as the mean± SEM
representative of three independent experiments and were assessed
using one-way ANOVA followed by Tukey's test.
***P<0.001. Nrf2, nuclear factor erythroid 2-related
factor 2; NS, not significant; sgRNA, single guide RNA;
nrf2-/-, Nrf2 knockout 293 cells; NAC,
N-acetyl-cysteine.
MG132 induces cell death in 293 cells.
Representative quantification of viable 293 cells stained with
Annexin V and PI at different time points after MG132 (2 μM)
treatment; live cells, Annexin V-PI-. Data
are presented as the mean ± SEM representative of three independent
experiments and were assessed using one-way ANOVA followed by
Tukey's test. *P<0.05, ***P<0.001 and
****P<0.0001. Nrf2, nuclear factor erythroid
2-related factor 2; PI, propidium iodide.
Loss of Nrf2 reduces HO1 expression
without affecting other stress-induced genes during proteasomal
inhibition. (A) Representative immunoblots and (B) quantification
of HO1 in cells treated with DMSO or MG132 (2 μM) for 12 h. (C)
Representative immunoblots and (D) quantification of HSP70 in cells
treated with DMSO or MG132 (2 μM) for 12 h. Data are presented as
the mean ± SEM representative of three independent experiments and
were assessed using one-way ANOVA followed by Tukey's test.
**P<0.01. Nrf2, nuclear factor erythroid 2-related
factor 2; nrf2-/-, Nrf2 knockout 293 cells; HO1,
heme oxygenase-1; HSP70, 70-kDa heat shock protein.
Generation of
nrf2-/-[Flag-p62] and
nrf2-/-[Flag-Nrf2] cells. (A) Nrf2 mRNA levels in
nrf2+/+, nrf2-/-,
nrf2-/-[Flag-Nrf2],
nrf2+/+[Vector] and
nrf2-/-[Vector] cells. (B) p62 mRNA levels in
nrf2+/+, nrf2-/-,
nrf2-/-[Flag-p62],
nrf2+/+[Vector] and
nrf2-/-[Vector] cells. Data are presented as the
mean ± SEM representative of three independent experiments and were
assessed using one-way ANOVA followed by Tukey's test.
***P<0.001, ****P<0.0001. Nrf2, nuclear
factor erythroid 2-related factor 2; nrf2-/-,
Nrf2 knockout 293 cells; [], transfection construct.
Low levels of MG132 induce Nrf2
accumulation. (A) Representative immunoblots and (B) quantification
of Nrf2 in cells treated with varying amounts of MG132. Data are
presented as the mean ± SEM representative of three independent
experiments and were assessed using one-way ANOVA followed by
Tukey's test. **P<0.01 and ***P<0.001. Nrf2, nuclear factor
erythroid 2-related factor 2.
Primers used in this study.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 30871032 and 31600616), the
Natural Science Foundation of Fujian Province (grant no.
2017J05142), the Scientific Foundation of Fuzhou Health Department
(grant no. 2016-S-124-10) and the Scientific Foundation of Fujian
Provincial Health Commission (grant no. 2016-1-87).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
CJ and JG conceived and designed the experiments.
SQ and JG performed experiments and analyzed the data. JG wrote the
paper. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Corboy MJ, Thomas PJ and Wigley WC:
Aggresome formation. Methods Mol Biol. 301:305–327. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Johnston JA, Ward CL and Kopito RR:
Aggresomes: A cellular response to misfolded proteins. J Cell Biol.
143:1883–1898. 1998.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance
JM, Ito A and Yao TP: The deacetylase HDAC6 regulates aggresome
formation and cell viability in response to misfolded protein
stress. Cell. 115:727–738. 2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Webb JL, Ravikumar B and Rubinsztein DC:
Microtubule disruption inhibits autophagosome-lysosome fusion:
Implications for studying the roles of aggresomes in polyglutamine
diseases. Int J Biochem Cell Biol. 36:2541–2550. 2004.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo
T and Mouradian MM: Aggresomes formed by alpha-synuclein and
synphilin-1 are cytoprotective. J Biol Chem. 279:4625–4631.
2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rubinsztein DC, Ravikumar B,
Acevedo-Arozena A, Imarisio S, O'Kane CJ and Brown SD: Dyneins,
autophagy, aggregation and neurodegeneration. Autophagy. 1:177–178.
2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Marambio P, Toro B, Sanhueza C, Troncoso
R, Parra V, Verdejo H, García L, Quiroga C, Munafo D, Díaz-Elizondo
J, et al: Glucose deprivation causes oxidative stress and
stimulates aggresome formation and autophagy in cultured cardiac
myocytes. Biochim Biophys Acta. 1802:509–518. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kovacs I, Lentini KM, Ingano LM and Kovacs
DM: Presenilin 1 forms aggresomal deposits in response to heat
shock. Journal of molecular neuroscience. J Mol Neurosci. 29:9–19.
2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ang E, Pavlos NJ, Rea SL, Qi M, Chai T,
Walsh JP, Ratajczak T, Zheng MH and Xu J: Proteasome inhibitors
impair RANKL-induced NF-kappaB activity in osteoclast-like cells
via disruption of p62, TRAF6, CYLD, and IkappaBalpha signaling
cascades. J Cell Physiol. 220:450–459. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kirkin V, McEwan DG, Novak I and Dikic I:
A role for ubiquitin in selective autophagy. Mol Cell. 34:259–269.
2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gao J, Li M, Qin S, Zhang T, Jiang S, Hu
Y, Deng Y, Zhang C, You D, Li H, et al: Cytosolic PINK1 promotes
the targeting of ubiquitinated proteins to the aggresome-autophagy
pathway during proteasomal stress. Autophagy. 12:632–647.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Olzmann JA, Li L, Chudaev MV, Chen J,
Perez FA, Palmiter RD and Chin LS: Parkin-mediated K63-linked
polyubiquitination targets misfolded DJ-1 to aggresomes via binding
to HDAC6. J Cell Biol. 178:1025–1038. 2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Waxman EA, Covy JP, Bukh I, Li X, Dawson
TM and Giasson BI: Leucine-rich repeat kinase 2 expression leads to
aggresome formation that is not associated with alpha-synuclein
inclusions. J Neuropathol Exp Neurol. 68:785–796. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
McLean PJ, Ribich S and Hyman BT:
Subcellular localization of alpha-synuclein in primary neuronal
cultures: Effect of missense mutations. J Neural Transm Suppl.
58:53–63. 2000.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Harding HP, Novoa I, Zhang Y, Zeng H, Wek
R, Schapira M and Ron D: Regulated translation initiation controls
stress-induced gene expression in mammalian cells. Mol Cell.
6:1099–1108. 2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jain A, Lamark T, Sjøttem E, Larsen KB,
Awuh JA, Øvervatn A, McMahon M, Hayes JD and Johansen T: p62/SQSTM1
is a target gene for transcription factor NRF2 and creates a
positive feedback loop by inducing antioxidant response
element-driven gene transcription. J Biol Chem. 285:22576–22591.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lau A, Zheng Y, Tao S, Wang H, Whitman SA,
White E and Zhang DD: Arsenic inhibits autophagic flux, activating
the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol.
33:2436–2446. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ichimura Y, Waguri S, Sou YS, Kageyama S,
Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et
al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during
selective autophagy. Mol Cell. 51:618–631. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Pickart CM and Fushman D: Polyubiquitin
chains: Polymeric protein signals. Curr Opin Chem Biol. 8:610–616.
2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang C, Gao J, Li M, Deng Y and Jiang C:
p38δ MAPK regulates aggresome biogenesis by phosphorylating SQSTM1
in response to proteasomal stress. J Cell Sci.
131(131)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hao R, Nanduri P, Rao Y, Panichelli RS,
Ito A, Yoshida M and Yao TP: Proteasomes activate aggresome
disassembly and clearance by producing unanchored ubiquitin chains.
Mol Cell. 51:819–828. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mekhail K, Khacho M, Carrigan A, Hache RR,
Gunaratnam L and Lee S: Regulation of ubiquitin ligase dynamics by
the nucleolus. J Cell Biol. 170:733–744. 2005.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cui T, Lai Y, Janicki JS and Wang X:
Nuclear factor erythroid-2 related factor 2 (Nrf2)-mediated protein
quality control in cardiomyocytes. Front Biosci. 21:192–202.
2016.PubMed/NCBI
|
|
28
|
Hochmuth CE, Biteau B, Bohmann D and
Jasper H: Redox regulation by Keap1 and Nrf2 controls intestinal
stem cell proliferation in Drosophila. Cell Stem Cell. 8:188–199.
2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kovac S, Angelova PR, Holmström KM, Zhang
Y, Dinkova-Kostova AT and Abramov AY: Nrf2 regulates ROS production
by mitochondria and NADPH oxidase. Biochim Biophys Acta.
1850:794–801. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Arakawa M and Ito Y: N-acetylcysteine and
neurodegenerative diseases: Basic and clinical pharmacology.
Cerebellum. 6:308–314. 2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sheng XJ, Tu HJ, Chien WL, Kang KH, Lu DH,
Liou HH, Lee MJ and Fu WM: Antagonism of proteasome
inhibitor-induced heme oxygenase-1 expression by PINK1 mutation.
PLoS One. 12(e0183076)2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kim D, Kim SH and Li GC: Proteasome
inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce
hsp70 and hsp27 expression. Biochem Biophys Res Commun.
254:264–268. 1999.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Vadlamudi RK and Shin J: Genomic structure
and promoter analysis of the p62 gene encoding a non-proteasomal
multiubiquitin chain binding protein. FEBS Lett. 435:138–142.
1998.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lim J, Lachenmayer ML, Wu S, Liu W, Kundu
M, Wang R, Komatsu M, Oh YJ, Zhao Y and Yue Z: Proteotoxic stress
induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective
autophagic clearance of protein aggregates. PLoS Genet.
11(e1004987)2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Matsumoto G, Wada K, Okuno M, Kurosawa M
and Nukina N: Serine 403 phosphorylation of p62/SQSTM1 regulates
selective autophagic clearance of ubiquitinated proteins. Mol Cell.
44:279–289. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Du ZX, Yan Y, Zhang HY, Liu BQ, Gao YY,
Niu XF, Meng X and Wang HQ: Proteasome inhibition induces a p38
MAPK pathway-dependent antiapoptotic program via Nrf2 in thyroid
cancer cells. J Clin Endocrinol Metab. 96:E763–E771.
2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kopito RR: Aggresomes, inclusion bodies
and protein aggregation. Trends Cell Biol. 10:524–530.
2000.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Szebenyi G, Wigley WC, Hall B, Didier A,
Yu M, Thomas P and Krämer H: Hook2 contributes to aggresome
formation. BMC Cell Biol. 8(19)2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bolhuis S and Richter-Landsberg C: Effect
of proteasome inhibition by MG-132 on HSP27 oligomerization,
phosphorylation, and aggresome formation in the OLN-93
oligodendroglia cell line. J Neurochem. 114:960–971.
2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Meriin AB, Zaarur N and Sherman MY:
Association of translation factor eEF1A with defective ribosomal
products generates a signal for aggresome formation. J Cell Sci.
125:2665–2674. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hyttinen JM, Amadio M, Viiri J, Pascale A,
Salminen A and Kaarniranta K: Clearance of misfolded and aggregated
proteins by aggrephagy and implications for aggregation diseases.
Ageing Res Rev. 18:16–28. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Lamark T and Johansen T: Aggrephagy:
Selective disposal of protein aggregates by macroautophagy. Int J
Cell Biol. 2012(736905)2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Takalo M, Salminen A, Soininen H, Hiltunen
M and Haapasalo A: Protein aggregation and degradation mechanisms
in neurodegenerative diseases. Am J Neurodegener Dis. 2:1–14.
2013.PubMed/NCBI
|
|
44
|
Brüning A and Jückstock J: Misfolded
proteins: From little villains to little helpers in the fight
against cancer. Front Oncol. 5(47)2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Park J, Park Y, Ryu I, Choi MH, Lee HJ, Oh
N, Kim K, Kim KM, Choe J, Lee C, et al: Misfolded polypeptides are
selectively recognized and transported toward aggresomes by a CED
complex. Nat Commun. 8(15730)2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Liu B and Qian SB: Translational
reprogramming in cellular stress response. Wiley Interdiscip Rev
RNA. 5:301–315. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sonenberg N and Hinnebusch AG: Regulation
of translation initiation in eukaryotes: Mechanisms and biological
targets. Cell. 136:731–745. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Espinosa-Diez C, Miguel V, Mennerich D,
Kietzmann T, Sánchez-Pérez P, Cadenas S and Lamas S: Antioxidant
responses and cellular adjustments to oxidative stress. Redox Biol.
6:183–197. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Baird L and Dinkova-Kostova AT: The
cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol.
85:241–272. 2011.PubMed/NCBI View Article : Google Scholar
|
|
50
|
McMahon M, Thomas N, Itoh K, Yamamoto M
and Hayes JD: Redox-regulated turnover of Nrf2 is determined by at
least two separate protein domains, the redox-sensitive Neh2 degron
and the redox-insensitive Neh6 degron. J Biol Chem.
279:31556–31567. 2004.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Nguyen T, Sherratt PJ, Huang HC, Yang CS
and Pickett CB: Increased protein stability as a mechanism that
enhances Nrf2-mediated transcriptional activation of the
antioxidant response element. Degradation of Nrf2 by the 26 S
proteasome. J Biol Chem. 278:4536–4541. 2003.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Itoh K, Wakabayashi N, Katoh Y, Ishii T,
O'Connor T and Yamamoto M: Keap1 regulates both cytoplasmic-nuclear
shuttling and degradation of Nrf2 in response to electrophiles.
Genes Cells. 8:379–391. 2003.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Furukawa M and Xiong Y: BTB protein Keap1
targets antioxidant transcription factor Nrf2 for ubiquitination by
the Cullin 3-Roc1 ligase. Mol Cell Biol. 25:162–171.
2005.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Holmström KM, Baird L, Zhang Y, Hargreaves
I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova-Kostova AT
and Abramov AY: Nrf2 impacts cellular bioenergetics by controlling
substrate availability for mitochondrial respiration. Biol Open.
2:761–770. 2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Pilecka I, Sadowski L, Kalaidzidis Y and
Miaczynska M: Recruitment of APPL1 to ubiquitin-rich aggresomes in
response to proteasomal impairment. Exp Cell Res. 317:1093–1107.
2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Salemi LM, Almawi AW, Lefebvre KJ and
Schild-Poulter C: Aggresome formation is regulated by RanBPM
through an interaction with HDAC6. Biol Open. 3:418–430.
2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Dron M, Dandoy-Dron F, Farooq Salamat MK
and Laude H: Proteasome inhibitors promote the sequestration of
PrPSc into aggresomes within the cytosol of prion-infected CAD
neuronal cells. J Gen Virol. 90:2050–2060. 2009.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Beaudoin S, Goggin K, Bissonnette C,
Grenier C and Roucou X: Aggresomes do not represent a general
cellular response to protein misfolding in mammalian cells. BMC
Cell Biol. 9(59)2008.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Dantuma NP and Bott LC: The
ubiquitin-proteasome system in neurodegenerative diseases:
Precipitating factor, yet part of the solution. Front Mol Neurosci.
7(70)2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Cummings CJ, Mancini MA, Antalffy B,
DeFranco DB, Orr HT and Zoghbi HY: Chaperone suppression of
aggregation and altered subcellular proteasome localization imply
protein misfolding in SCA1. Nat Genet. 19:148–154. 1998.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Arrasate M, Mitra S, Schweitzer ES, Segal
MR and Finkbeiner S: Inclusion body formation reduces levels of
mutant huntingtin and the risk of neuronal death. Nature.
431:805–810. 2004.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Cummings CJ, Reinstein E, Sun Y, Antalffy
B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL and Zoghbi HY:
Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion
frequency while accelerating polyglutamine-induced pathology in
SCA1 mice. Neuron. 24:879–892. 1999.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Gandhi J, Antonelli AC, Afridi A, Vatsia
S, Joshi G, Romanov V, Murray IV and Khan SA: Protein misfolding
and aggregation in neurodegenerative diseases: A review of
pathogeneses, novel detection strategies, and potential
therapeutics. Rev Neurosci. 30:339–358. 2019.PubMed/NCBI View Article : Google Scholar
|