Introduction
Alport Syndrome (AS) is an inherited nephropathy
caused by mutations in one or two of the type IV collagen chains
(α3, α4 and α5) (1-3). AS is characterized by persistent
microscopic hematuria starting during infancy, and eventually
leading to either progressive nephritis or end-stage renal disease
(ESRD), along with extrarenal abnormalities, such as sensorineural
deafness and ocular anomalies (4).
In ~80% of AS cases, patients exhibit X-linked inheritance with
mutations in the COL4A5 gene. Autosomal recessive
inheritance due to biallelic mutations in COL4A3 or
COL4A4 are found in ~15% of patients, and the remaining 5%
of cases are due to autosomal dominant inheritance, due to
heterozygous mutations in COL4A3 or COL4A4 (5).
Patients with AS display a wide array of phenotypic
variability, ranging from progressive renal disease to isolated
hematuria (6). The severity of renal
manifestations in X-linked AS (XLAS) differs between males and
females (6). All male patients with
XLAS develop proteinuria, and eventually progress to renal
insufficiency (7). However, female
patients have milder and more variable clinical presentations,
ranging from isolated hematuria to ESRD, with later onset and
slower disease progression (8).
Electron microscopy analysis of renal biopsies suggested that the
majority of patients exhibit structural alterations in the
glomerular basement membrane (GBM). The earliest change in AS is
diffuse thinning of the GBM (9).
Frequently, the diagnostic ultrastructural changes in the GBM for
adult males are a basket-weave-like change, whereas the predominant
alteration observed in children and female patients with XLAS is
diffuse thinning of the GBM (9).
Therefore, in the absence of a family history of either hematuria
or ESRD, it may be difficult to perform a pathological diagnosis
for female patients with isolated hematuria (4,10,11).
Genetic testing is an effective tool for clinical
diagnosis and prognosis of AS, and to support counseling of
affected patients. In the present study, targeted next-generation
sequencing (NGS) was used to identify four novel and one reported
COL4A5 mutations in Chinese patients suspected of having
AS.
Materials and methods
Patients and ethics
All subjects provided signed informed consent forms
for participation in the present study; consent from the probands
IID2, IID3 and IID4 (all <18 years) was obtained from their
parents. The present study was approved by the Zhengzhou University
Ethics Committee (Zhengzhou, China).
Patients and families
In total, 5 families were included in the present
study who were recruited between June 2018 and May 2019. The age of
patients ranged from 5-26 years old and four male probands and one
female proband were included. Clinical diagnosis of patients was
performed by a nephrologist based on clinical manifestations and
biochemical analysis, such as hematuria, proteinuria and high
creatinine levels. A brief clinical summary of the probands are
presented in Table I.
 | Table IClinical and pathological
characteristics of the patients. |
Table I
Clinical and pathological
characteristics of the patients.
| | | | | Renal biopsy | |
|---|
| IID | Sex | Age, years | Hearing loss | EM | α3/α5 | Family history |
|---|
| IID1 | Male | 26 | Mild | BWC | A/A | P |
| IID2 | Male | 15 | Normal | BWC | M/A | P |
| IID3 | Male | 6 | Normal | BWC | A/A | P |
| IID4 | Male | 10 | Normal | BWC | A/A | P |
| IID5 | Female | 25 | Normal | TBM | Nor/M | N |
Samples and DNA extraction
Genomic DNA was extracted from EDTA peripheral blood
samples using Lab-Aid® 824 DNA extraction kit according
to the manufacturer's protocol (Xiamen Zeesan Biotech Co.,
Ltd.).
Custom panel design
A custom panel was designed for the COL4A3,
COL4A4 and COL4A5 genes using the Ion AmpliSeq™
designer software version 7.4.2 (Thermo Fisher Scientific, Inc.) to
perform mutational screening of patients suspected of having AS.
The coding regions and all the flanking introns up to 50 bp were
targeted. The 3' and 5' untranslated regions were not included in
the panel design. Details of the methodology have been described
previously (12).
Ion torrent personal genome machine
(PGM) sequencing
Library preparation was performed by amplifying 10
ng genomic DNA, using the Ion AmpliSeq™ Library kit 2.0 (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
Libraries were purified using the Agencourt®
AMPure® XP system and quantified using the
Qubit® dsDNA HS assay kit (Invitrogen; Thermo Fisher
Scientific, Inc.) and clonally amplified by emulsion PCR using the
Ion OneTouch™ 2 system (Ion PGM™ Template Hi-Q™ view OT2 200 kit;
Thermo Fisher Scientific, Inc.) all according to the manufacturer's
protocol. The spheres were loaded on to a 316™ v2 chip and
sequenced on the Ion Torrent PGM, using the Ion PGM™ Hi-Q™ view
Sequencing 200 kit v2. Post-run analysis was performed using
Torrent Suite™ version 5.0.4 (Thermo Fisher Scientific, Inc.).
Coverage assessment was performed using the ‘coverage Analysis’
plug-in which provides information regarding the amplicons read
coverage, and variants were called using the ‘variant Caller’
plug-in (12).
Results
In total, 5 probands were included in the present
study. The eye examinations showed there were no abnormalities in
all patients. A total of three missense mutations, one splice site
mutation and one frameshift mutation were identified (Table II). Of the 5 patients, 4 patients
had novel mutations which had not been reported previously, to the
best of our knowledge. These candidate mutations were validated by
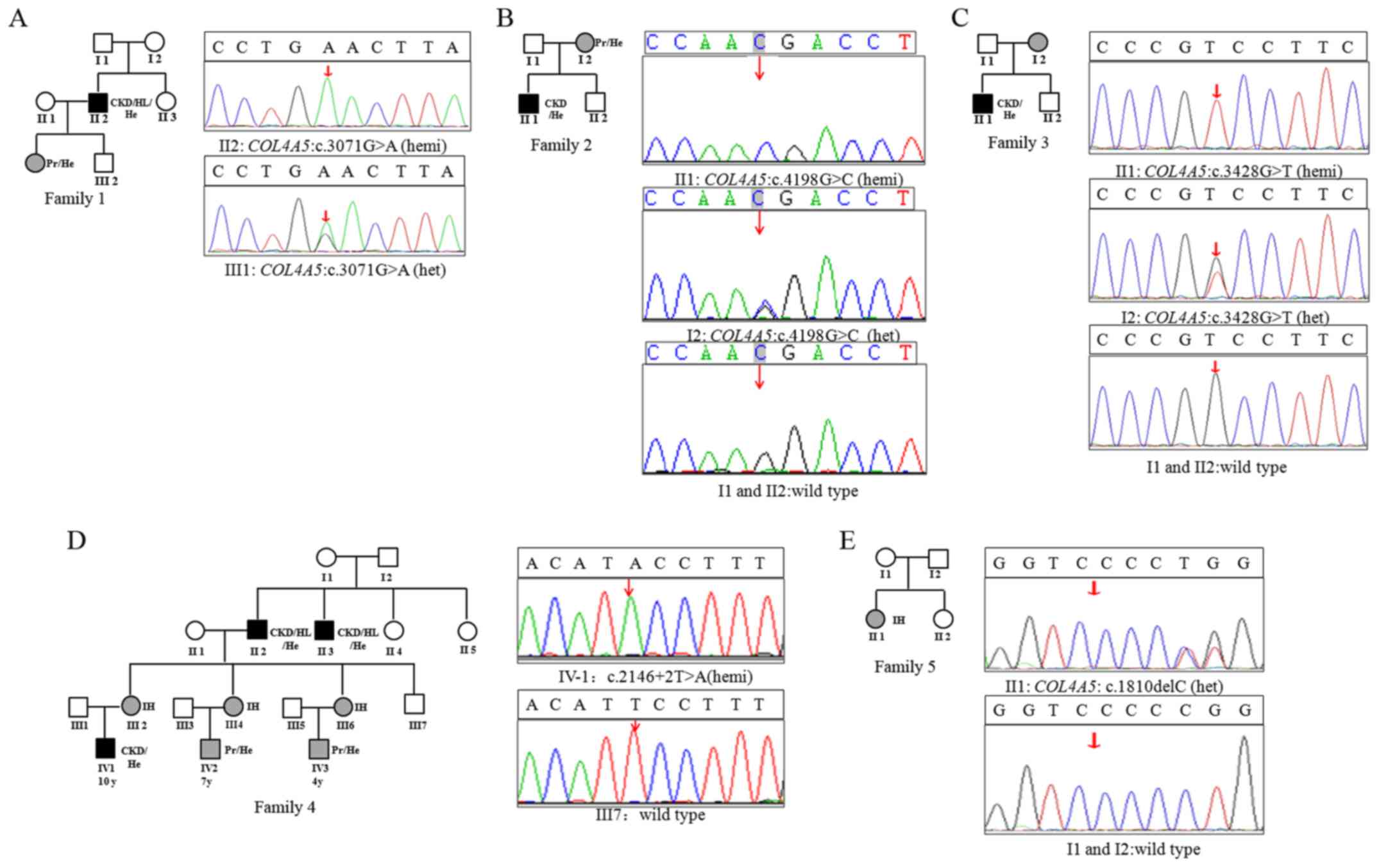
Sanger sequencing (Fig. 1). The
predicted clinical significance of these mutations, which were
assessed using the criteria of clinical significance based on the
American College of Medical Genetics guidelines (13), are listed in Table II.
| Table IIMutations detected in the
COL4A5 gene. |
Table II
Mutations detected in the
COL4A5 gene.
| Sample | Zygosity | Type of mutation | cHGVS | pHGVS | Clinical
significancea | Novelty |
|---|
| IID1 | Hemizygous | Missense | c.3071G>A | p.(Gly1024Glu) | LP | Previously
reported |
| IID2 | Hemizygous | Missense | c.4198G>C | p.(Gly1400Arg) | VUS | Novel |
| IID3 | Hemizygous | Missense | c.3428G>T | p.(Gly1143Val) | LP | Previously
reported |
| IID4 | Hemizygous | Missense | c.2146+2T>A | splicing | LP | Novel |
| IID5 | Heterozygous | Frameshift | c.1810delC |
p.(Thr605Ilefs*13) | LP | Novel |
In AS patient IID 1, systemic edema was first noted
6 years ago without an obvious cause, alongside proteinuria, but
without hematuria or hypertension. The disease was diagnosed as
nephrotic syndrome. Subsequently, 3 years ago, facial edema and
fatigue appeared again, accompanied by intermittent headaches, with
a highest recorded blood pressure of 180/100 mmHg, urinary protein
3+ and blood creatinine levels of 200 µmol/l. This was treated with
hormone therapy. Facial edema and weakness appeared again 1 year
prior to this study, with a highest recorded blood pressure of
200/120 mmHg, hemoglobin levels of 66.0 g/l, urinary protein 3+ and
point albumin levels of 182.56 mg/mmol. The urea level was 49.03
mmol/l, the creatinine levels were 1,115 µmol/l and the uric acid
level was 514 µmol/l. They were diagnosed with CKD-5 with
sensorineural hearing loss, and received regular hemodialysis
treatment for 1 year. The patient progressed to the uremia stage
and received an allograft kidney transplant. Renal biopsy indicated
that there was irregular thickness and wrinkling of the GBM. A
hemizygous spontaneous variation of the COL4A5 gene
[c.3071G>A, p.(Gly1024Glu)] was detected by NGS. The patients
6-year-old daughter presented with intermittent hematuria and
proteinuria, and was found to possess the same heterozygous
mutation [c.3071G>A, p.(Gly1024Glu)] (Fig. 1A).
In AS patient IID 2, urine foaming and hematuria
were observed for >7 months, with blood creatinine levels of 61
µmol/l, heterogeneous small red blood cells, blood pressure of
115/70 mmHg and urinary protein 3+. The renal biopsy showed that
the GBM exhibited irregular thickness and wrinkling. A hemizygous
variation of the COL4A5 gene [c.4198G>C, p.(Gly1400Arg)]
was identified by NGS, which was inherited from the patient's
mother, whom exhibited intermittent microscopic hematuria (Fig. 1B).
The pediatric AS patient IID 3 had gross hematuria
with no obvious cause 22 days prior to admission. There was
intermittent frequent pain and itchiness when urinating, urinary
protein 2+ and blood creatinine levels of 158 µmol/l. The renal
biopsy showed a GBM with an irregular thickness and wrinkling. A
hemizygous variation of the COL4A5 gene [c.3428G>T,
p.(Gly1143Val)] was detected by NGS, which was inherited from the
patient's mother, who exhibited intermittent microscopic hematuria
(Fig. 1C).
AS patient IID 4 (IV-2 in Fig. 1D) was a 7-year-old male with first
onset of gross hematuria at the age of 4. By the age of 7, he had
already developed proteinuria, and biochemical analysis indicated
high plasma creatinine levels. His mother showed intermittent
microhematuria. IV-1 progressed to chronic kidney disease and the
patient received renal dialysis. IV-3 exhibited hematuria and
proteinuria. III-2 showed intermittent microhematuria and
hypertension, whereas III-6 only presented with microhematuria.
II-2 and II-3 progressed to ESRD at the age 25 and 27 years,
respectively. They both received renal transplants, and both
exhibited sensorineural deafness (Fig.
1D). A novel splicing mutation (c.2146+2T>A) at the splice
donor site at the boundary between exon 27/intron 27 in
COL4A5 in the proband was identified. The candidate mutation
was validated by Sanger sequencing and it was predicted to mutate a
normal splice site according to the Human Splicing Finder
(umd.be/HSF3/index.html). The proband's mother was
heterozygous for the c.2146+2T>A mutation, whereas the healthy
family member, III-7, did not have a mutation at this site
(Fig. 1D).
AS patient IID 5 was a 25-year-old female with gross
hematuria, urinary protein 2+, blood creatinine 100 µmol/l and
point albumin 19.63 mg/mmol. Her urea levels were 69.03 mmol/l, the
creatinine levels were 895 µmol/l and uric acid levels were 324
µmol/l. The renal biopsy showed lesions in the GBM. A novel
spontaneous mutation, c.1810delC (p.Thr605Ilefs*13), was identified
in COL4A5 in the proband (Fig.
1E).
Discussion
The heterotrimer of α3, α4 and α5 chains serve an
important role in the structure and function of the basement
membrane of the glomeruli, cochlea and eyes (14). A collagenous domain of the α5 chain
contains Gly-X-Y triplet sequence repeats (15). A missense mutation replacing the
glycine residue in a Gly-X-Y repeat accounts for ~30% of
COL4A5 mutations in XLAS. Glycine substitution in the
collagenous domain is hypothesized to introduce kinks in the
protein, thus interfering with the proper folding of the collagen
triple helix (14,16). It has been reported that glycine
substitutions in the α5 chain (type IV) result in different
structural changes to the GBM, which are associated with the
clinical phenotype of AS (14). Thus
far, three types of phenotype have been associated with typical
XLAS based on the time taken to progress to ESRD, which is used to
classify the severity (severe type, intermediate type and moderate
type). The severe type progresses to ERSD at ~20 years of age
(early-onset ESRD), with a high incidence of sensorineural deafness
and ocular changes, caused by large rearrangements, premature stop,
frameshift, donor splice and missense mutations in the NC1 domain.
Patients with intermediate type progress to ESRD at ~26 years of
age, due to non-Gly-X-Y missense or Gly-X-Y mutations in exons
21-47. In the moderate type, patients progress to ESRD after ~30
years (late-onset ESRD), which is accompanied by a lower incidence
of sensorineural deafness and ocular changes, due to Gly-X-Y
mutations in exons 1-20 (17-19).
Due to the large size of COL4A5 gene, there
are no known mutational hot spots, to the best of our knowledge. To
date, 1,119 mutations in COL4A5 have been recorded according
to the Human Gene Mutation Database (HGMD® Professional
version 2019.2; portal.biobase-international.com/hgmd/pro/start.php),
including missense, nonsense, deletion or splicing mutations, as
well as complex rearrangements. In the present study, three
missense mutations [c.3071G>A p.(Gly1024Glu), c.4198G>C
p.(Gly1400Arg) and c.3428G>T p.(Gly1143Val)] were identified
which resulted in the substitution of glycine located in exons
21-47. The proband with the c.3071G>A p.(Gly1024Glu) mutation in
family 1 progressed to the uremia stage at the age of 26 and
developed sensorineural hearing loss. His daughter (6 years) showed
intermittent hematuria and proteinuria. It has been previously
reported that a patient suffering from AS had the same mutation
(20). These findings suggest that
the mutation may be associated with severe clinical phenotypes and
an earlier age of onset. The probands IID2 (15 years) and IID3 (6
years) both exhibited renal insufficiency due to the mutations
c.4198G>C, p.(Gly1400Arg) and c.3428G>T, p.(Gly1143Val)
inherited from their mothers, who had intermittent microscopic
hematuria. It has been previously reported that a patient suffering
from AS possessed the mutation c.3428G>A, p.(Gly1143Asp) in the
COL4A5 gene (21). Considering their young age,
further follow-up is required to fully assess their clinical
symptoms. Based on a previous report of intermediate type XLAS due
to non-Gly-X-Y missense and Gly-X-Y mutations exons in
21-47(18), it was hypothesized that
the mutations [c.4198G>C, p.(Gly1400Arg) and c.3428G>T,
p.(Gly1143Val)] were also associated with severe clinical
phenotypes.
A novel splicing mutation, c.2146+2T>A was
identified in the COL4A5 gene in the proband IID4. The same
mutation was subsequently found in all 7 affected family members
(II-2, II-3, III-2, III-4, III-6, IV-1 and IV-3), and was absent in
the unaffected members. This variant has not been reported in any
public databases. Based on the phenotypes of affected family
members, it was concluded that the splicing site mutation
c.2146+2T>A was also associated with severe clinical
phenotypes.
Due to the relatively milder phenotypes in female
patients with X-linked AS, it is may be difficult to perform a
pathological diagnosis for females with isolated hematuria. In the
present study, a novel spontaneous mutation [c.1810delC
(p.Thr605Ilefs*13)] in COL4A5 in a 25-year-old female
patient (IID5) was found, who exhibited hematuria and proteinuria,
together with lesions in the GBM, but did not have a family history
of related diseases. This case highlights NGS as an effective
method for obtaining genetic sequencing information in female
patients with XLAS.
In conclusion, four novel mutations of the
COL4A5 gene were identified and were shown to be associated
with AS. The present study broadens the known spectrum of mutations
of the COL4A5 gene and may have implications for genetic
diagnosis, therapy and genetic counseling of affected families.
Acknowledgements
Not applicable.
Funding
This study was supported by the Key Science and
Technology Research Project Fund of Hebei Province of China (grant
no. 19227115D) and the Natural Science Foundation of Hebei Province
of China (grant no. C201920947).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and XK designed the present study. XS, CC and LL
performed the experiments. GZ and JZ collected the data. XS and CW
analyzed the data. XZ and XK wrote the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
All subjects provided signed informed consent forms
for participation in the present study; consent from the probands
IID2, IID3 and IID4 (all <18 years) was obtained from their
parents. The present study was approved by the Zhengzhou University
Ethics Committee (Zhengzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gubler MC: Inherited diseases of the
glomerular basement membrane. Nat Clin Prac Nephrol. 4:24–37.
2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hudson BG, Tryggvason K, Sundaramoorthy M
and Neilson EG: Alport's syndrome, Goodpasture's syndrome, and type
IV collagen. N Engl J Med. 348:2543–2556. 2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Alport AC: Hereditary familial congenital
haemorrhagic nephritis. Br Med J. 1:504–506. 1927.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Han KH, Park JE and Ki CS: De novo
mutations in COL4A5 identified by whole exome sequencing in 2 girls
with Alport syndrome in Korea. Korean J Pediatr. 62:193–197.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Behjati S and Tarpey PS: What is next
generation sequencing? Arch Dis Child Educ Pract Ed. 98:236–238.
2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wei G, Zhihong L, Huiping C, Caihong Z,
Zhaohong C and Leishi L: Spectrum of clinical features and type IV
collagen alpha-chain distribution in Chinese patients with Alport
syndrome. Nephrology, dialysis, transplantation: Official
publication of the European Dialysis and Transplant Association.
European Renal Association. 21:3146–3154. 2006.
|
|
7
|
Barker DF, Hostikka SL, Zhou J, Chow LT,
Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL and
Tryggvason K: Identification of mutations in the COL4A5 collagen
gene in Alport syndrome. Science. 248:1224–1227. 1990.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kashtan CE: Alport syndromes: Phenotypic
heterogeneity of progressive hereditary nephritis. Pediatr Nephrol.
14:502–512. 2000.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Meleg-Smith S, Magliato S, Cheles M,
Garola RE and Kashtan CE: X-linked Alport syndrome in females. Hum
Pathol. 29:404–408. 1998.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jais JP, Knebelmann B, Giatras I, De
Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO,
Flinter F, et al: X-linked Alport syndrome: Natural history and
genotype-phenotype correlations in girls and women belonging to 195
families: A ‘European community Alport syndrome concerted action’
study. J Am Soc Nephrol. 14:2603–2610. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tsiakkis D, Pieri M, Koupepidou P,
Demosthenous P, Panayidou K and Deltas C: Genotype-phenotype
correlation in X-linked Alport syndrome patients carrying missense
mutations in the collagenous domain of COL4A5. Clin Genet.
82:297–299. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhao X, Chen C, Wei Y, Zhao G, Liu L, Wang
C Zhang J and Kong X: Novel mutations of COL4A3, COL4A4, and COL4A5
genes in Chinese patients with Alport syndrome using next
generation sequence technique. Mol Genet Genomic Med.
7(e653)2019.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang YF, Ding J, Wang F and Bu DF: Effect
of glycine substitutions on alpha5(IV) chain structure and
structure-phenotype correlations in Alport syndrome. Biochem
Biophys Res Commun. 316:1143–1149. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hudson BG: The molecular basis of
Goodpasture and Alport syndromes: Beacons for the discovery of the
collagen IV family. J Am Soc Nephrol. 15:2514–2527. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kashtan CE: Alport Syndrome and thin
basement membrane nephropathy. In: Adam MP, Ardinger HH, Pagon RA,
Wallace SE, Bean LJH, Stephens K, et al, editors.
GeneReviews((R)). Seattle (WA) 1993.
|
|
17
|
Slajpah M, Gorinsek B, Berginc G, Vizjak
A, Ferluga D, Hvala A, Meglic A, Jaksa I, Furlan P, Gregoric A, et
al: Sixteen novel mutations identified in COL4A3, COL4A4, and
COL4A5 genes in Slovenian families with Alport syndrome and benign
familial hematuria. Kidney Int. 71:1287–1295. 2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Demosthenous P, Voskarides K, Stylianou K,
Hadjigavriel M, Arsali M, Patsias C, Georgaki E, Zirogiannis P,
Stavrou C, Daphnis E, et al: X-linked Alport syndrome in Hellenic
families: Phenotypic heterogeneity and mutations near interruptions
of the collagen domain in COL4A5. Clin Genet. 81:240–248.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xiu X, Yuan J, Deng X, Xiao J, Xu H, Zeng
Z, Guan L, Xu F and Deng S: A novel COL4A5 mutation identified in a
Chinese Han family using exome sequencing. Biomed Res Int.
2014(186048)2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Morinière V, Dahan K, Hilbert P, Lison M,
Lebbah S, Topa A, Bole-Feysot C, Pruvost S, Nitschke P, Plaisier E,
et al: Improving mutation screening in familial hematuric
nephropathies through next generation sequencing. J Am Soc Nephrol.
25:2740–2751. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhou J, Hertz JM and Tryggvason K:
Mutation in the alpha 5(IV) collagen chain in juvenile-onset Alport
syndrome without hearing loss or ocular lesions: Detection by
denaturing gradient gel electrophoresis of a PCR product. Am J Hum
Genet. 50:1291–1300. 1992.PubMed/NCBI
|