Cells contain specific sensors to monitor DNA repair
and induce apoptotic cell death when the DNA becomes damaged
(1). Tumor suppressors serve
critical roles in the regulation of the genomic integrity and the
DNA repair pathways in various types of cells (2). Increased genomic instability can
promote the development of cancer and neurodegenerative diseases,
such as Alzheimer's disease (AD). The pathogenesis of these
conditions is a multi-step process, accompanied by accumulation of
genetic alterations in the somatic cells. In addition, aging
contributes significantly to the impairment of physiological gene
expression. AD and cancer are both prevalent in the elderly
population (3). Although both are
age-associated diseases, one is degenerative and the other is
proliferative at the cellular level. Various factors that are
upregulated to sustain cell growth in any type of cancer may be
downregulated in AD-neurons contributing to neurodegeneration.
Therefore, it is hypothesized that the inverse relationship between
cancer and AD exhibits a reciprocal inverse association in several
underlying aspects. Epidemiological data suggest that subjects who
develop neurodegenerative diseases due to aging have a decreased
risk of cancer (4). As
neurodegeneration and carcinogenesis share a number of biological
pathways, this inverse association is interesting to explore. The
hypothesis proposes that neurodegeneration and carcinogenesis may
manifest by several distinctive phenomena associated with
senescence (5). The mechanism
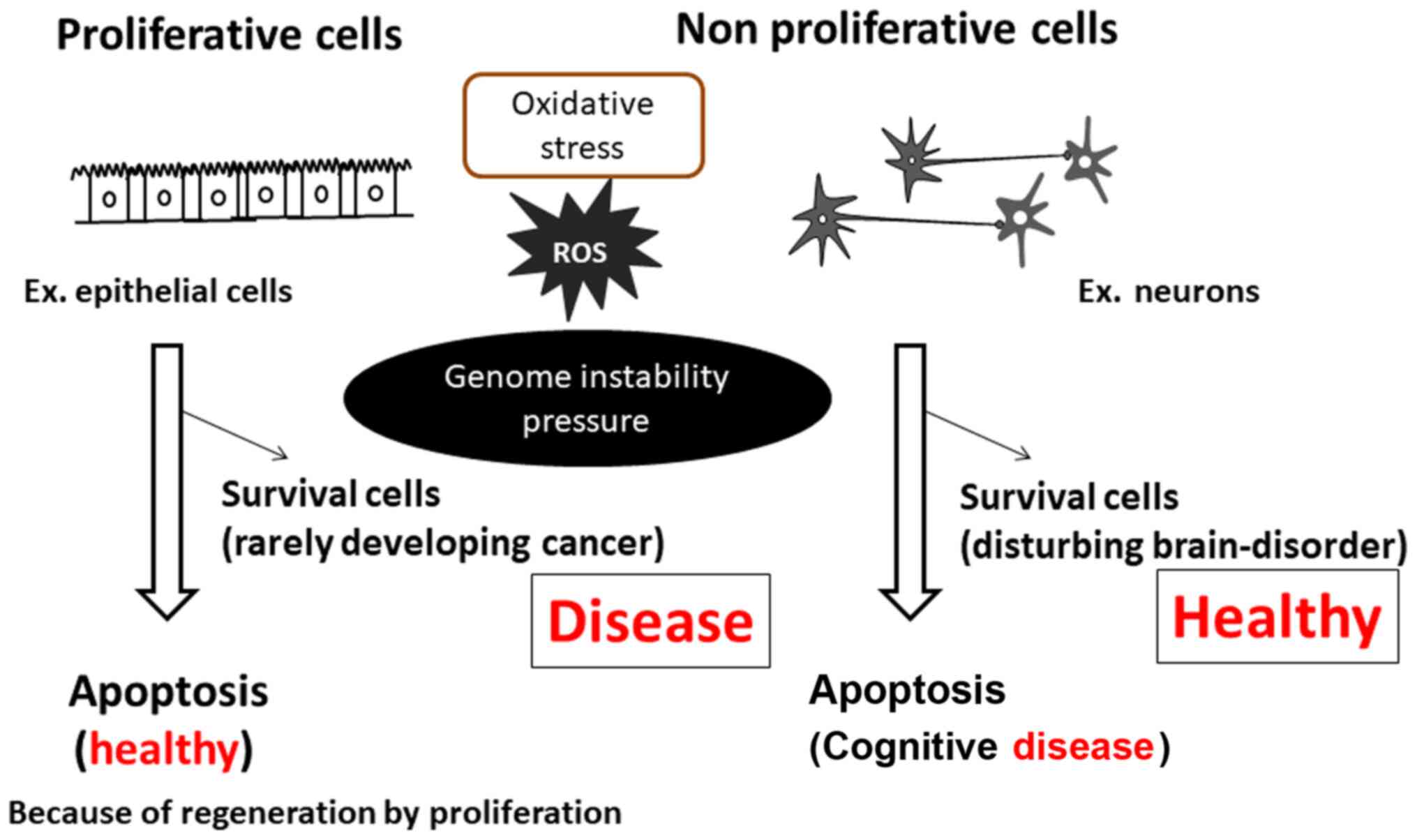
underlying intrinsic susceptibility of the cells towards either
cell proliferative or cell apoptotic outcomes remain unknown
(Fig. 1). A full understanding of
these pathways may aid in the prevention and treatment of cancer or
neurodegeneration (6). At present,
effective treatment strategies for both diseases are lacking. The
identification of the biological mechanisms responsible for the
development of these diseases may provide novel targets for future
therapeutic applications.
Cancer cells that originate from the primary site
exhibit altered proliferative activity as a result of gene
mutations responsible for controlling cellular proliferation. The
mutations may result in the upregulation of proto-oncogenes and/or
the downregulation of tumor suppressor genes (7,8). It is
commonly accepted that ROS are the primary trigger of these
carcinogenic mutations (9). In
addition, oxidative stress may contribute to cancer progression by
affecting genomic instability (9).
AD is one of the most notorious neurodegenerative diseases whose
hallmarks include neuronal loss and/or dementia (10). The most significant DNA lesion
affecting the progression of AD is likely caused by oxidative DNA
damage (11). In general, increased
levels of DNA damage and downregulation of cellular DNA repair
capacity have been associated with age-associated diseases.
Accumulation of genomic DNA damage can be caused by increased rates
of oxidative-damage, which may exacerbate cancer and/or AD
progression. Increased DNA oxidation has been reported in the
post-mortem brain tissues of humans with AD (12). It has also been observed that DNA
repair is dysregulated in AD (13).
A stable increase in ROS levels can lead to potent induction of
oxidative stress in cells, which causes genomic instability leading
to the development of both diseases (Fig. 1).
The balance between ROS levels and the reducing
equivalents in the cell determines the redox status and
consequently, cell fate. For example, the therapeutic strategy in
cancer should be based on treatments that increase ROS production
and cause apoptotic cancer cell death (14). In contrast to this hypothesis, the
strategy in treating neurodegenerative diseases should be based on
protecting the activity of the neurons against the development of
oxidative stress (15).
Consequently, the redox status may have prognostic potential for
the development of cancer and neurodegenerative diseases. The
regulation of the redox status may affect the quality of life of
the patients. The production of ROS is regulated by the action of
chemical and enzymatic antioxidant systems, including ascorbate and
the antioxidant enzymes, such as SOD. SOD enzymes possess a
significant antioxidant role characterized by their ability to
scavenge ROS (16). It has been
demonstrated that SOD defects are associated with the development
of several types of disease (17,18).
Cytosolic SOD1 serves an important role in reducing the damage to
the central nervous system (CNS) (19). Loss of SOD1 increases ROS
levels, which triggers the induction of oxidative DNA damage to the
cells. Mutations in the SOD1 gene are responsible for
causing damage to the mitochondria and ultimately leading to the
development of the progressive neurodegenerative diseases, such as
familial amyotrophic lateral sclerosis (ALS) (20). It has also been revealed that
SOD1-null animals develop specific age-related diseases, such as
muscle atrophy (21). SOD2 has been
identified by certain studies as a tumor suppressor since its
expression is reduced in several types of cancer (22). In addition, the roles of SOD2 are
involved in the development of neurodegenerative diseases (23). One of the most significant processes
affected by SOD2 activity is the regulation of energy metabolism.
Furthermore, SOD2 can protect mitochondrial DNA against oxidative
damage. Mitochondrial adenosine triphosphate production activity
has been reported to be impaired in AD (24). Mice containing one deleted copy of
the SOD2 gene exhibit accelerated progression of AD
development (25). SOD2 is localized
within the mitochondrial matrix, which is the crucial site of free
radical production from the electron transport chain (26). SOD3 exists as a secreted form in the
extracellular matrix of several tissues. Downregulation of SOD3 has
been shown to alter DNA copy number and/or to promote the
hyper-methylation of the promoter DNA region (27). SOD3 is secreted to the extracellular
matrix in the CNS tissues. Previous studies have shown that
inhibition of ROS production by SOD activation may reduce neuronal
cell death and glial cell activation, which may have an unusual
effective therapeutic potential compared with conventional
treatments (28,29).
Tumor suppressor gene products are molecules which
may protect a cell from carcinogenesis (30-36).
Accordingly, loss of function in these molecules may be important
in the formation of several types of cancer (30,34).
Well-studied tumor suppressor molecules include TP53, breast cancer
susceptibility gene 1 (BRCA1), phosphatase and tensin homologue
deleted on chromosome 10 (PTEN), APC and the retinoblastoma protein
(Rb), amongst others (31-36).
Mutations in BRCA1 may increase the risk of the development of
breast and ovarian cancer (30).
Furthermore, it has been shown that the genetic variation in the
BRCA1 gene is associated with prostate cancer development
(31). Activation of the
phosphoinositide 3-kinase (PI3K) enzyme is often associated with
the development of BRCA1-associated breast cancer (32). The functional insufficiency of BRCA1
activates the PI3K/AKT oncogenic pathway, which may also be
associated with the development of neurodegenerative diseases
(33). Reduced levels of BRCA1 have
been found in the brains of patients with AD. In addition,
knockdown of neuronal BRCA1 has been shown to affect synaptic
impairment and memory deficits (34). Accordingly, BRCA1 may support
neuronal integrity and cognitive functions, whereas the reduced
function of the neuronal BRCA1 contributes to cognitive deficits in
AD (35). The changes to DNA repair
as a result of BRCA1 mutations may occur early in the progression
of neurodegeneration. BRCA1 can bind to BRCA2, Rad50, Rad51 and Rb
in order to activate the cell cycle checkpoints (36). Although these protein complexes are
present in neurons of the adult cortex and cerebellum, their
expression is considerably diminished in the neurons of the brain
tissues from AD subjects (37).
These complexes may be involved in DNA repair and the regulation of
the cell cycle checkpoints (38).
The Rb protein is another tumor suppressor involved in cell-cycle
regulation and neural cell apoptosis (39,40). Rb
is used as a potential diagnostic marker for AD (39,40). The
p27Kip1 and p21Waf1 proteins are also
activated by BRCA1 in AD (41). In
addition, mutations of the pathogenic protein presenilin in AD
result in a specific increase in the expression levels of
p21Waf1 and in the expression levels of the proteins
involved in TP53 signaling (42).
The levels of p21Waf1 are increased in AD. The induction
of cell death or cell survival is mediated by the coordinated
action of the BRCA1, p21Waf1 and TP53 proteins dependent
on the type of oxidative damage caused by the cells.
TP53 is a transcription factor ubiquitously
expressed in all cell types, which regulates the cell cycle
checkpoints and the induction of apoptosis following DNA damage
(55). In response to various
cellular stresses, activated TP53 may induce cell cycle arrest
(56). Failure of the DNA repair
machinery to correct extensive DNA damage activates the induction
of apoptotic cell death mediated by the TP53 protein (56). The genomic integrity is maintained by
the cells decision to induce DNA repair or to activate the
apoptotic cascade (57). The
TP53 gene is frequently mutated in numerous types of cancer
cells, suggesting that it serves a critical role in preventing
normal cells from malignant transformation (58). The importance of TP53 as an
inherited cancer susceptibility gene product has been demonstrated
in Li-Fraumeni syndrome, and is associated with a high risk of
developing cancer in multiple types of malignancies (59). The functions of TP53 are supported by
different downstream targets and several effectors. Among these,
cyclin-dependent kinase (CDK) inhibitors such as p21Waf1
are important mediators of TP53(60). As mentioned earlier,
p21Waf1 is activated by BRCA1 in AD. Furthermore, the
specific severity of the clinical progression of AD is associated
with mutation of the presenilin protein into a pathogenic isoform
that is frequently observed in patients with AD (42). This mutation may be mediated by
p21Waf1 and TP53 signaling proteins (42). Accumulation of DNA damage
irrespective of the TP53 function may also lead to neuronal cell
death following AD.
The comprehensive roles of these tumor suppressor
signaling molecules can be used to explore the potential inverse
associations between AD and cancer. Cell survival or apoptosis may
be governed by the balance between DNA damage and repair, which has
received increasing attention as a major pathway used in the
treatment of cancer and neurodegenerative diseases (61,62).
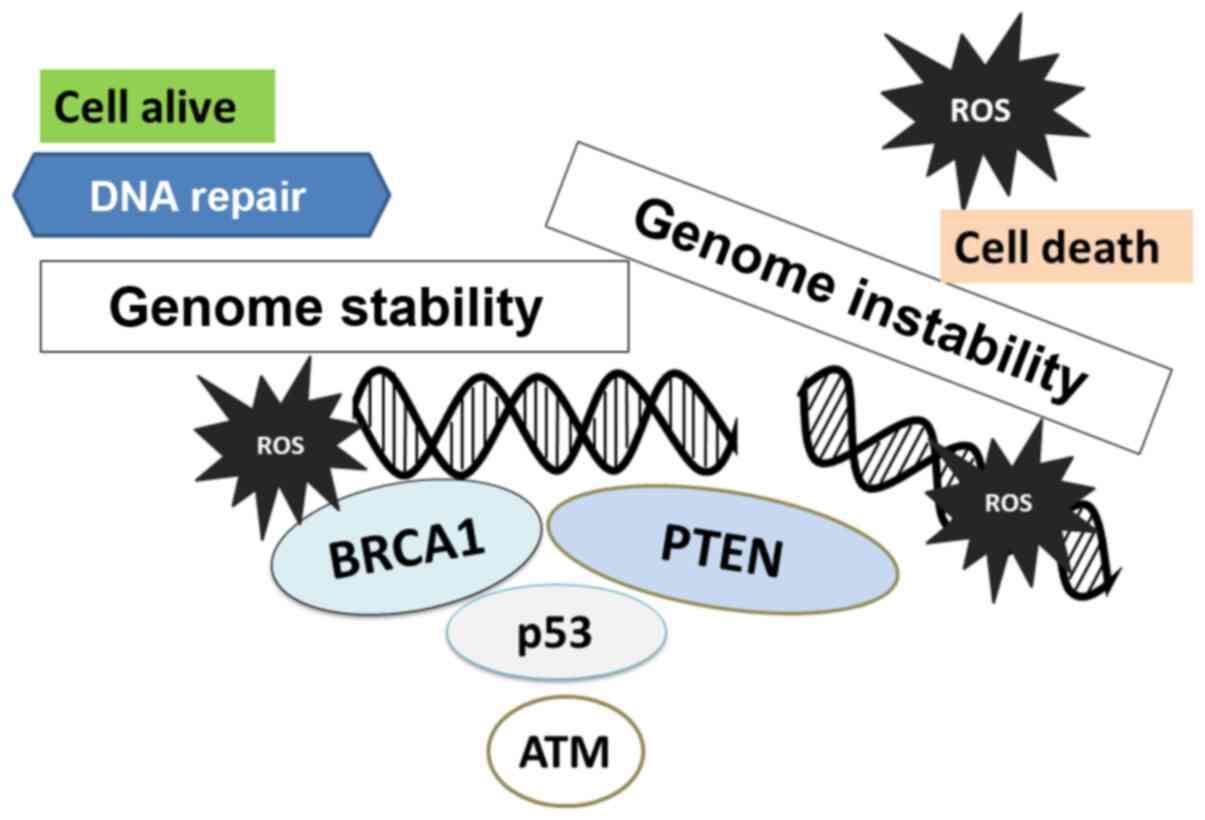
Genomic integrity may be sustained through the
function of several tumor suppressor genes, which reduces
pathologic alterations, such as the development of carcinogenesis
and neurodegeneration. Loss of the function of the tumor suppressor
genes may reduce DNA repair and induce genomic instability, which
can lead to cell apoptosis. Furthermore, it may enhance the
sensitivity of cancer cells to irradiation. With regards to
therapeutic applications, this treatment may be beneficial for
patients with cancer. Neurodegenerative diseases can be caused by
neuronal cell apoptosis of non-proliferative neuronal cells
(Fig. 1). The use of alkylating
agents and/or irradiation for cancer therapy may promote
neurodegeneration due to the genomic instability caused from excess
production of ROS (73). In
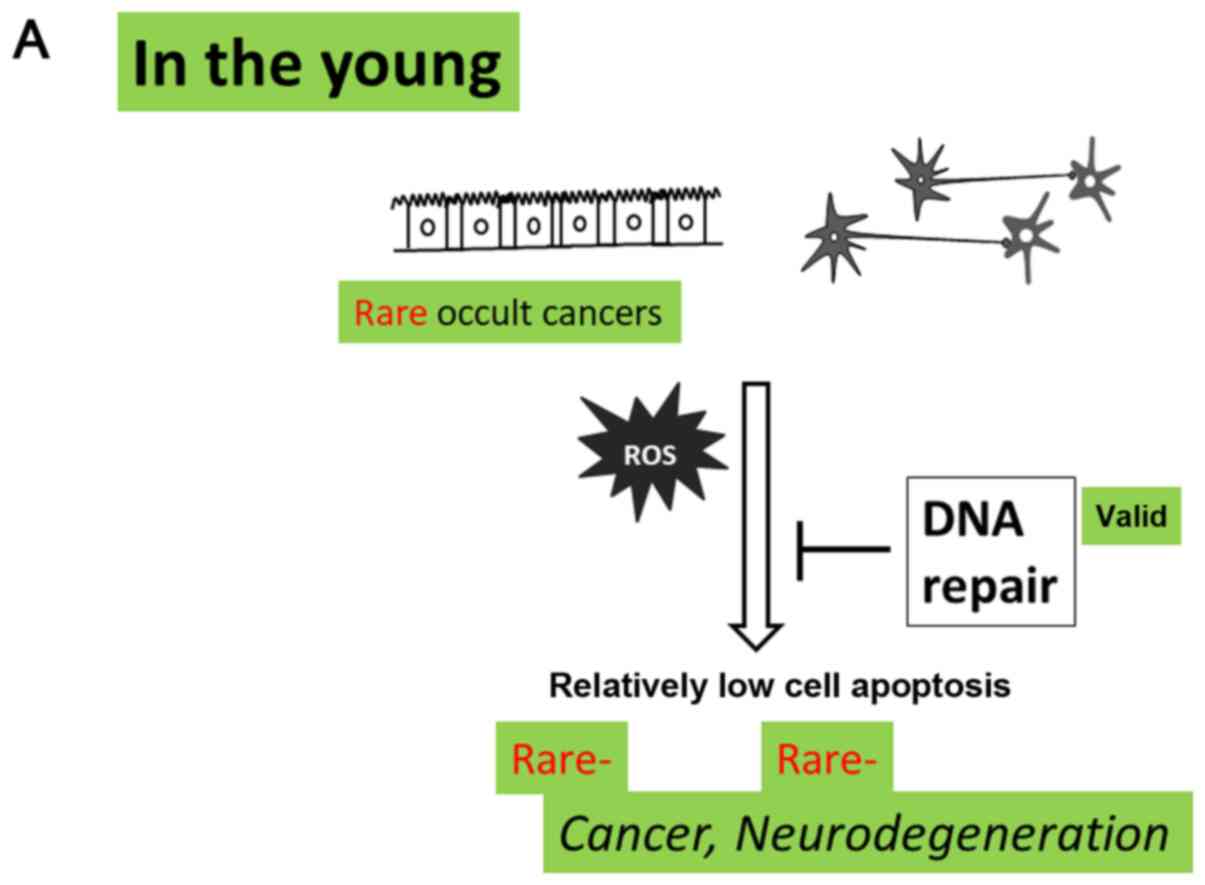
conclusion, the present review reports on the increased number of
age-related occult cancer and the reduced potential of
anti-oxidative defense mechanisms that would determine the bipolar
fates of patients with regard to the development of cancer or of
neurodegenerative diseases in elderly individuals (Fig. 2). This may explain why the inverse
association occurs in the elderly in contrast to younger patients
(Fig. 2). In the absence of the
excess ROS production, bacterial inflammation and/or
life-style-associated fatty diseases may increase ROS levels in the
elderly, which may in turn increase cancer incidence and the
induction of neuronal apoptosis. Consequently, the extent of DNA
repair capacity would determine the tendency to develop either
cancer or neurodegenerative diseases (Fig. 2).
Tumor suppressor genes can act in a cooperative
manner to maintain genomic integrity (Fig. 3). The understanding of the complete
assembly of the tumor suppressor proteins can aid in the
development of effective treatment approaches for cancer and
neurodegenerative diseases. Further mechanistic studies are
required in order to understand the precise assembly among tumor
suppressor proteins and the molecular signaling mechanisms
responsible for facilitating the development of effective
treatments, which can regulate genomic stability and improve
therapeutic efficacy. These factors may influence the development
of neurodegeneration and/or regulate cognitive dysfunction in the
elderly. Further studies are required to elucidate the biological
mechanisms underlying the inverse association between cancer and AD
with the goal of identifying preventative molecular targets.
Not applicable.
The present work was supported in part by JSPS
KAKENHI (grant no. JP18K17964). In addition, funding was received
in part by a grant from the Nara Women's University in Japan.
Not applicable.
SM has contributed to the conception and design of
the study. MM, YI, YN, AT, YK and SM participated in drafting and
revising the article. All authors have read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Awasthi P, Foiani M and Kumar A: ATM and
ATR signaling at a glance. J Cell Sci. 128:4255–4262.
2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Van Haaften G, Plasterk RH and Tijsterman
M: Genomic instability and cancer: Scanning the Caenorhabditis
elegans genome for tumor suppressors. Oncogene. 23:8366–8375.
2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ghezzi EM and Ship JA: Systemic diseases
and their treatments in the elderly: Impact on oral health. J
Public Health Dent. 60:289–296. 2000.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhang Q, Guo S, Zhang X, Tang S, Shao W,
Han X, Wang L and Du Y: Inverse relationship between cancer and
Alzheimer's disease: A systemic review meta-analysis. Neurol Sci.
36:1987–1994. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Musicco M, Adorni F, Di Santo S, Prinelli
F, Pettenati C, Caltagirone C, Palmer K and Russo A: Inverse
occurrence of cancer and Alzheimer disease: A population-based
incidence study. Neurology. 81:322–328. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Nudelman KN, Risacher SL, West JD,
McDonald BC, Gao S and Saykin AJ: Alzheimer's disease neuroimaging
initiative. Association of cancer history with Alzheimer's disease
onset and structural brain changes. Front Physiol.
5(423)2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu Y, Chen Q and Zhang JT: Tumor
suppressor gene 14-3-3sigma is down-regulated whereas the
proto-oncogene translation elongation factor 1delta is up-regulated
in non-small cell lung cancers as identified by proteomic
profiling. J Proteome Res. 3:728–735. 2004.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Boiko AD, Porteous S, Razorenova OV,
Krivokrysenko VI, Williams BR and Gudkov AV: A systematic search
for downstream mediators of tumor suppressor function of p53
reveals a major role of BTG2 in suppression of Ras-induced
transformation. Genes Dev. 20:236–252. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Payne CM, Bernstein C, Dvorak K and
Bernstein H: Hydrophobic bile acids, genomic instability, Darwinian
selection, and colon carcinogenesis. Clin Exp Gastroenterol.
1:19–47. 2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fujikake N, Shin M and Shimizu S:
Association between autophagy and neurodegenerative diseases. Front
Neurosci. 12(255)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Madabhushi R, Pan L and Tsai LH: DNA
damage and its links to neurodegeneration. Neuron. 83:266–282.
2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bradley-Whitman MA, Timmons MD, Beckett
TL, Murphy MP, Lynn BC and Lovell MA: Nucleic acid oxidation: An
early feature of Alzheimer's disease. J Neurochem. 128:294–304.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Canugovi C, Shamanna RA, Croteau DL and
Bohr VA: Base excision DNA repair levels in mitochondrial lysates
of Alzheimer's disease. Neurobiol Aging. 35:1293–1300.
2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chen W, Zou P, Zhao Z, Chen X, Fan X,
Vinothkumar R, Cui R, Wu F, Zhang Q, Liang G and Ji J: Synergistic
antitumor activity of rapamycin and EF24 via increasing ROS for the
treatment of gastric cancer. Redox Biol. 10:78–89. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhao L, Wang JL, Wang YR and Fa XZ:
Apigenin attenuates copper-mediated β-amyloid neurotoxicity through
antioxidation, mitochondrion protection and MAPK signal
inactivation in an AD cell model. Brain Res. 1492:33–45.
2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang Y, Branicky R, Noë A and Hekimi S:
Superoxide dismutases: Dual roles in controlling ROS damage and
regulating ROS signaling. J Cell Biol. 217:1915–1928.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pong K: Oxidative stress in
neurodegenerative diseases: Therapeutic implications for superoxide
dismutase mimetics. Expert Opin Biol Ther. 3:127–139.
2003.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nazıroğlu M, Muhamad S and Pecze L:
Nanoparticles as potential clinical therapeutic agents in
Alzheimer's disease: Focus on selenium nanoparticles. Expert Rev
Clin Pharmacol. 10:773–782. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cudkowicz ME, Pastusza KA, Sapp PC,
Mathews RK, Leahy J, Pasinelli P, Francis JW, Jiang D, Andersen JK
and Brown RH Jr: Survival in transgenic ALS mice does not vary with
CNS glutathione peroxidase activity. Neurology. 59:729–734.
2002.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sangwan S and Eisenberg DS: Perspective on
SOD1 mediated toxicity in Amyotrophic Lateral Sclerosis. Postepy
Biochem. 62:362–369. 2016.PubMed/NCBI
|
|
21
|
Kostrominova TY: Advanced age-related
denervation and fiber-type grouping in skeletal muscle of SOD1
knockout mice. Free Radic Biol Med. 49:1582–1593. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kang SW: Superoxide dismutase 2 gene and
cancer risk: Evidence from an updated meta-analysis. Int J Clin Exp
Med. 8:14647–14655. 2015.PubMed/NCBI
|
|
23
|
Flynn JM and Melov S: SOD2 in
mitochondrial dysfunction and neurodegeneration. Free Radic Biol
Med. 62:4–12. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hroudová J, Singh N and Fišar Z:
Mitochondrial dysfunctions in neurodegenerative diseases: Relevance
to Alzheimer's disease. Biomed Res Int. 2014(175062)2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hinerfeld D, Traini MD, Weinberger RP,
Cochran B, Doctrow SR, Harry J and Melov S: Endogenous
mitochondrial oxidative stress: Neurodegeneration, proteomic
analysis, specific respiratory chain defects, and efficacious
antioxidant therapy in superoxide dismutase 2 null mice. J
Neurochem. 88:657–667. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Papa L, Hahn M, Marsh EL, Evans BS and
Germain D: SOD2 to SOD1 switch in breast cancer. Biol Chem.
289:5412–5416. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Teoh-Fitzgerald ML, Fitzgerald MP, Jensen
TJ, Futscher BW and Domann FE: Genetic and epigenetic inactivation
of extracellular superoxide dismutase promotes an invasive
phenotype in human lung cancer by disrupting ECM homeostasis. Mol
Cancer Res. 10:40–51. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Baluchnejadmojarad T, Mansouri M, Ghalami
J, Mokhtari Z and Roghani M: Sesamin imparts neuroprotection
against intrastriatal 6-hydroxydopamine toxicity by inhibition of
astroglial activation, apoptosis, and oxidative stress. Biomed
Pharmacother. 88:754–761. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Petro M, Jaffer H, Yang J, Kabu S, Morris
VB and Labhasetwar V: Tissue plasminogen activator followed by
antioxidant-loaded nanoparticle delivery promotes
activation/mobilization of progenitor cells in infarcted rat brain.
Biomaterials. 81:169–180. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Dilawari A, Cangiarella J, Smith J, Huang
A, Downey A and Muggia F: Co-existence of breast and ovarian
cancers in BRCA germ-line mutation carriers. Ecancermedicalscience.
2(109)2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Modena A, Iacovelli R, Scarpa A, Brunelli
M, Ciccarese C, Fantinel E, Bimbatti D, Massari F, Martignoni G and
Tortora G: Investigating BRCA mutations: A breakthrough in
precision medicine of castration-resistant prostate cancer. Target
Oncol. 11:569–577. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Guirouilh-Barbat JK, Wilhelm T and Lopez
BS: AKT1/BRCA1 in the control of homologous recombination and
genetic stability: The missing link between hereditary and sporadic
breast cancers. Oncotarget. 1:691–699. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ogino M, Ichimura M, Nakano N, Minami A,
Kitagishi Y and Matsuda S: Roles of PTEN with DNA repair in
Parkinson's disease. Int J Mol Sci. 17(954)2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Gonzalez ME, Li X, Toy K, DuPrie M,
Ventura AC, Banerjee M, Ljungman M, Merajver SD and Kleer CG:
Downregulation of EZH2 decreases growth of estrogen
receptor-negative invasive breast carcinoma and requires BRCA1.
Oncogene. 28:843–853. 2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Suberbielle E, Djukic B, Evans M, Kim DH,
Taneja P, Wang X, Finucane M, Knox J, Ho K, Devidze N, et al: DNA
repair factor BRCA1 depletion occurs in Alzheimer brains and
impairs cognitive function in mice. Nat Commun.
6(8897)2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Brown EJ: Analysis of cell cycle
progression and genomic integrity in early lethal knockouts.
Methods Mol Biol. 280:201–212. 2004.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jacobsen E, Beach T, Shen Y, Li R and
Chang Y: Deficiency of the Mre11 DNA repair complex in Alzheimer's
disease brains. Brain Res Mol Brain Res. 128:1–7. 2004.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jhanwar-Uniyal M: BRCA1 in cancer, cell
cycle and genomic stability. Front Biosci. 8 (Suppl):S1107–S1117.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
39
|
Hradek AC, Lee HP, Siedlak SL, Torres SL,
Jung W, Han AH and Lee HG: Distinct chronology of neuronal cell
cycle re-entry and tau pathology in the 3xTg-AD mouse model and
Alzheimer's disease patients. J Alzheimers Dis. 4:57–65.
2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Padmanabhan J, Brown K and Shelanski ML:
Cell cycle inhibition and retinoblastoma protein overexpression
prevent Purkinje cell death in organotypic slice cultures. Dev
Neurobiol. 67:818–826. 2007.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Delobel P, Lavenir I, Ghetti B, Holzer M
and Goedert M: Cell-cycle markers in a transgenic mouse model of
human tauopathy: Increased levels of cyclin-dependent kinase
inhibitors p21Cip1 and p27Kip1. Am J Pathol. 168:878–887.
2006.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bialopiotrowicz E, Szybinska A, Kuzniewska
B, Buizza L, Uberti D, Kuznicki J and Wojda U: Highly pathogenic
Alzheimer's disease presenilin 1 P117R mutation causes a specific
increase in p53 and p21 protein levels and cell cycle dysregulation
in human lymphocytes. J Alzheimers Dis. 32:397–415. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yin Y and Shen WH: PTEN: A new guardian of
the genome. Oncogene. 27:5443–5453. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wise HM, Hermida MA and Leslie NR:
Prostate cancer, PI3K, PTEN and prognosis. Clin Sci (Lond).
131:197–210. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yakubov E, Ghoochani A, Buslei R,
Buchfelder M, Eyüpoglu IY and Savaskan N: Hidden association of
Cowden syndrome, PTEN mutation and meningioma frequency.
Oncoscience. 3:149–155. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hou SQ, Ouyang M, Brandmaier A, Hao H and
Shen WH: PTEN in the maintenance of genome integrity: From DNA
replication to chromosome segregation. Bioessays:.
39(1700082)2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ming M and He YY: PTEN in DNA damage
repair. Cancer Lett. 319:125–129. 2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Fiandalo MV and Kyprianou N: Caspase
control: Protagonists of cancer cell apoptosis. Exp Oncol.
34:165–175. 2012.PubMed/NCBI
|
|
49
|
Krishnan A and Zochodne DW: Is cytoplasmic
PTEN a specific target for neuronal survival? Mol Neurobiol.
52:1758–1764. 2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Asua D, Bougamra G, Calleja-Felipe M,
Morales M and Knafo S: Peptides acting as cognitive enhancers.
Neuroscience. 370:81–87. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Frere S and Slutsky I: Targeting PTEN
interactions for Alzheimer's disease. Nat Neurosci. 19:416–418.
2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Knafo S, Sánchez-Puelles C, Palomer E,
Delgado I, Draffin JE, Mingo J, Wahle T, Kaleka K, Mou L,
Pereda-Perez I, et al: PTEN recruitment controls synaptic and
cognitive function in Alzheimer's models. Nat Neurosci. 19:443–453.
2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhang YY, Huang J, Yang M, Gu LJ, Ji JY,
Wang LJ and Yuan WJ: Effect of a low-protein diet supplemented with
Keto-acids on autophagy and inflammation in 5/6 nephrectomized
rats. Biosci Rep. 35(e00263)2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Estevez AO, Morgan KL, Szewczyk NJ, Gems D
and Estevez M: The neurodegenerative effects of selenium are
inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like
growth factor signaling in Caenorhabditis elegans. Neurotoxicology.
41:28–43. 2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Machado-Silva A, Perrier S and Bourdon JC:
p53 family members in cancer diagnosis and treatment. Semin Cancer
Biol. 20:57–62. 2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Stewart-Ornstein J and Lahav G: p53
dynamics in response to DNA damage vary across cell lines and are
shaped by efficiency of DNA repair and activity of the kinase ATM.
Sci Signal. 10(eaah6671)2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zhang XP, Liu F, Cheng Z and Wang W: Cell
fate decision mediated by p53 pulses. Version 2. Proc Natl Acad Sci
USA. 106:12245–50. 2009.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Bénard J, Douc-Rasy S and Ahomadegbe JC:
TP53 family members and human cancers. Hum Mutat. 21:182–191.
2003.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Varley JM: Germline TP53 mutations and
Li-Fraumeni syndrome. Hum Mutat. 21:313–320. 2003.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhao J, Lammers P, Torrance CJ and Bader
AG: TP53-independent function of miR-34a via HDAC1 and
p21(CIP1/WAF1.). Mol Ther. 21:1678–1686. 2013.PubMed/NCBI View Article : Google Scholar
|
|
61
|
O'Neil N and Rose A: DNA repair. WormBook.
Jan, 13:1–12. 2006.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Golubnitschaja O: Cell cycle checkpoints:
The role and evaluation for early diagnosis of senescence,
cardiovascular, cancer, and neurodegenerative diseases. Amino
Acids. 32:359–371. 2007.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Hazan I, Hofmann TG and Aqeilan RI: Tumor
suppressor genes within common fragile sites are active players in
the DNA damage response. PLoS Genet. 12(e1006436)2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Thurn KT, Thomas S, Raha P, Qureshi I and
Munster PN: Histone deacetylase regulation of ATM-mediated DNA
damage signaling. Mol Cancer Ther. 12:2078–2087. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Le Corre L, Fustier P, Chalabi N, Bignon
YJ and Bernard-Gallon D: Effects of resveratrol on the expression
of a panel of genes interacting with the BRCA1 oncosuppressor in
human breast cell lines. Clin Chim Acta. 344:115–121.
2004.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Matsuda S, Nakagawa Y, Kitagishi Y,
Nakanishi A and Murai T: Reactive oxygen species, superoxide
dimutases, and PTEN-p53-AKT-MDM2 signaling loop network in
mesenchymal Stem/Stromal cells regulation. Cells.
7(36)2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Hair JM, Terzoudi GI, Hatzi VI, Lehockey
KA, Srivastava D, Wang W, Pantelias GE and Georgakilas AG: BRCA1
role in the mitigation of radiotoxicity and chromosomal instability
through repair of clustered DNA lesions. Chem Biol Interact.
188:350–358. 2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Liu W, Zhou Y, Reske SN and Shen C: PTEN
mutation: Many birds with one stone in tumorigenesis. Anticancer
Res. 28:3613–3619. 2008.PubMed/NCBI
|
|
69
|
Kobayashi H, Ogawa K, Kawahara N, Iwai K,
Niiro E, Morioka S and Yamada Y: Sequential molecular changes and
dynamic oxidative stress in high-grade serous ovarian
carcinogenesis. Free Radic Res. 51:755–764. 2017.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Bankoglu EE, Tschopp O, Schmitt J, Burkard
P, Jahn D, Geier A and Stopper H: Role of PTEN in oxidative stress
and DNA damage in the liver of whole-body Pten Haplodeficient mice.
PLoS One. 11(e0166956)2016.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Armstrong CW, Maxwell PJ, Ong CW, Redmond
KM, McCann C, Neisen J, Ward GA, Chessari G, Johnson C, Crawford
NT, et al: PTEN deficiency promotes macrophage infiltration and
hypersensitivity of prostate cancer to IAP antagonist/radiation
combination therapy. Oncotarget. 7:7885–7898. 2016.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Zhang R, Zhu L, Zhang L, Xu A, Li Z, Xu Y,
He P, Wu M, Wei F and Wang C: PTEN enhances G2/M arrest in
Etoposide-treated MCF-7 cells through activation of the ATM
pathway. Oncol Rep. 35:2707–2714. 2016.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Yin H, Zhou Y, Wen C, Zhou C, Zhang W, Hu
X, Wang L, You C and Shao J: Curcumin sensitizes glioblastoma to
temozolomide by simultaneously generating ROS and disrupting
AKT/mTOR signaling. Oncol Rep. 32:1610–1616. 2014.PubMed/NCBI View Article : Google Scholar
|