Introduction
The insulin (INS) gene is the most important
of the non-human leukocyte antigen (HLA) genes involved in
the pathogenesis of Type 1 Diabetes (T1D), and it may serve as an
immunomodulatory agent preventing or halting β cell stress and/or
cell death (1,2). As the genetic background of T1D has
been extensively studied, there is an increasing focus on
epigenetics, as it is well established that epigenetic mechanisms
contribute to differences in phenotypes via gene hyperexpression or
gene silencing (3-5).
Epigenetics is an important interpretive link connecting the
complex interactions between genetics and environmental factors
that lead to the development of T1D. Epigenetic modifications more
specifically contribute to the pathogenesis of T1D primarily by
regulating the expression of genes that predispose an individual to
T1D. Epigenetic changes may serve as biomarkers of early diagnosis
of a disease in genetically predisposed individuals, and as
potential targets of therapeutic intervention through reversal of
their enzymatic activity (6,7).
DNA methylation, an important epigenetic mechanism
may serve a vital role in the pathogenesis of T1D. It can suppress
gene transcription and occurs at the promoter region of the
INS gene, at specific sites, called cytosine-guanosine
(CpGs) islands (8). These CpGs
islands are frequently observed in close proximity to the
transcriptional start site (TSS) (9). DNMT1, DNMT3A and DNMT3B are members of
the DNA methyltransferases (DNMTs) gene family which catalyze
cytosine methylation. DNMTs are highly expressed during the
progression of diabetes as a result of induced methylation
(10). Following β cell destruction
by cytotoxic T lymphocytes, differentially methylated DNA enters
the circulation (11).
The few studies that have addressed the issue of
INS gene promoter methylation status in T1D patients
reported contradictory results (12-18).
Thus, it still remains questionable whether hypermethylation or
hypomethylation of INS gene promoter is a reliable marker of
T1D appearance or progression.
The aim of the present study was to investigate the
pattern of CpG methylation of the INS gene promoter in
children and adolescents of Greek origin with T1D and compare this
with a control group.
Patients and methods
Determination of the sample size
For the calculation of sample size, we used the
formula [N=2 *σ2 *(Zα +
Z1-β)2/δ2], where N was the number
of patients required in each study group, Zα was a
constant equal to 1.960 at a 5% statistical significance level,
Z1-β corresponded to a value of 0.842 for achieving a
statistical power of 80%, δ was the expected detectable difference
in the INS gene methylation status between intervention and
the control group (that was the primary end point of the study,
estimated to be 5%), while σ was its estimated SD in the healthy
population (assigned a value of 5%) (14). Based on these data, the number of
patients needed in each study group was calculated to be 16.
Participants
A total of 20 children and adolescents (12 males and
8 females) with T1D who met the diagnostic criteria of T1D as
defined by the International Society for Pediatric and Adolescent
Diabetes as well as the American Diabetes Association criteria
(19,20) were recruited from the Pediatric
Diabetes Outpatient Clinic of the Fourth Department of Pediatrics,
at Papageorgiou General Hospital of Thessaloniki between December
2015 and February 2019. Additional inclusion criteria were optimal
glycaemic control with glycated hemoglobin <7.5%, absence of
comorbidities (lipid, thyroid or celiac disease) or other
autoimmune diseases, and a negative infection history for at least
the previous 15 days.
The control group consisted of 20 healthy non-obese
young subjects matched for age, sex and body mass index (BMI)
without personal or family history of autoimmune diseases, and no
history of infection for the last 15 days. These children were
evaluated in the General Pediatric Outpatient Clinic of the same
hospital for any reason other than T1D, such as health
certificates, drug prescription or preventive clinical/laboratory
evaluation. Subjects were clinically examined and their weight
(seca 711 Scale; Seca, GmbH) and height was measured (Harpenden
stadiometer; Veeder-Root), and their BMI was calculated as
kg/m2. Medical history, clinical examination and
laboratory findings were recorded. The age range of both groups was
2-17 years (median age of T1D group 13.6 years, median age of
control group 14 years) and they all had no personal or family
history of other autoimmune diseases. Subgrouping of participants
into prepubertal and pubertal groups was performed after clinical
examination by the same pediatric endocrinologist and application
of the Tanner and Marshal criteria for pubertal development
(prepubertal group, Tanner stage I; pubertal group, Tanner stages
II, III, IV and V) (21,22). Whole blood samples were collected
from both groups after a 12-h fasting period and stored immediately
at -80˚C until further use.
Parents or guardians of the participants signed the
written informed consent form, which was designed according to the
Declaration of Helsinki for research involving humans (23). The Bioethics Committee of the School
of Medicine of the Faculty of Health Sciences of the Aristotle
University of Thessaloniki approved the present research protocol
(approval no. 185/30.12.2015).
The protocol was also declared at Clinical
Trials.gov as Methylation of DNA in Children and
Adolescents with Type 1 Diabetes Mellitus (METHYLDIAB; ClinicalTrials.gov Identifier, NCT04139369).
DNA extraction, bisulfite treatment
and PCR
The studied CpGs in close proximity to the
INS promoter were chosen according to similar studies
(12,14). All experimental procedures were
performed at the Laboratory of Biological Chemistry of School of
Medicine of the Faculty of Health Sciences of the Aristotle
University of Thessaloniki.
Isolation of genomic DNA was performed from blood
samples using the DNA extraction kit QIAamp® DNA Blood
Mini kit (Qiagen, Inc.) according to the manufacturer's protocol.
Isolated DNA samples were quantified spectrophotometrically using
the OD ratio 260/280 (1 OD=50 µg/ml) using a BioPhotometer 6131
(Eppendorf AG). Bisulfite-treatment was performed with 300 ng DNA
from each sample using an EZ DNA Methylation-Gold kit (Zymo
Research Corp.) according to the manufacturer's protocol. Treatment
with sodium bisulfate converts unmethylated cytosine residues into
uracil residues, whereas methylated cytosines remain unchanged
under the same conditions (24). For
amplification of the INS gene promoter, the sequences of the
primers were: INS forward, 5'-TATTTTGGAATTTTGAGTTTATT-3' and
INS reverse, 5'-AACAAAAATCTAAAAACAACAA-3'. Additional,
overhang adapter sequence was added to the gene-specific primers
for the regions to be targeted (Nextera Transposase Adaptors;
Illumina, Inc.), for prompt construction of the NGS libraries. The
sequences of the Transposase Adaptors used were: Read1_INSForward
5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3' and Read2_INSReverse
5'-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3'. PCR products were
amplified using a low temperature ramping instrument (9700 Thermal
Cycler; Eppendorf AG; cat. no. 5341) using the preset 9600
emulation mode. The reaction solution consisted of AmpliTaq Gold
DNA Polymerase with Buffer II and MgCl2 (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The total reaction
volume was 25 µl, which consisted of 1.3 µl bisulfite-treated DNA,
2.5 µl 10x Buffer (100 mM Tris-HCl, pH 8.3, 500 mM KCl), 0.2 µM of
each primer, 200 µM dNTPs mix, 2 mM MgCl2 and 1.25 units
AmpliGold Taq Polymerase. PCR conditions were: Initial denaturation
at 95˚C for 3 min; followed by 40 cycles of 95˚C denaturation for
30 sec, 55˚C annealing for 30 sec, and 72˚C extension for 2.5 min;
and subsequently followed by a 1 min final extension step at 72˚C.
After purification of the PCR products using NucleoMag NGS Clean-up
and Size Select (Macherey-Nagel, GmbH; cat. no. 744970.5.), they
were pooled at similar molar quantities and submitted for library
construction according to manufacturer's protocol, (Nextera XT DNA
Library Preparation kit; Illumina, Inc.). For NGS reactions,
paired-end reads were selected at 2x250 base pair read length
formation, on the Illumina MiSeq platform.
Analysis of the sequence reads was performed using
FASTQ files and methylation status was estimated using
ampliMethProfiler (25), a
python-based pipeline for targeted deep bisulfite sequenced
amplicons. The methylation status was analyzed at 10 CpG sites of
the INS gene promoter around the TSS site (human genome 11;
INS, NCBI, ref seq NG_007114.1).
Statistical analysis
Data were analyzed using SPSS version 19.0 (IBM,
Corp). The methylation percentage in each of the different examined
loci constitutes a continuous variable that represents the
epigenetic alteration. Thus, every participant in the study was
assigned a percentage of methylation for each examined locus. In
order to determine any differences in the state of methylation
amongst the participants in the study group and the controls,
methylation percentage was compared between the two groups, as a
continuous variable. Regarding any differences in methylation, the
mean methylation status across groups was compared. Continuous
variables were expressed as the mean ± standard deviation (SD) for
normally distributed data and as the median (min-max) for
non-normally distributed data. A Shapiro-Wilk test was used to
examine the normality of distribution. Comparison of variables
between the two groups was performed using either a Student's
t-test or a Mann-Whitney U-test. Categorical variables among groups
were compared using a χ2 test. Correlations between
variables was assessed using a Spearman's Rho correlation
coefficient. P<0.05 was considered to indicate a statistically
significant difference.
Results
The clinicopathological characteristics of the study
population are shown in Table I. The
methylation status as well as the differences between the two
groups analyzed are presented in Table
II. BMI z-scores from both groups was evaluated (Table I). The overall mean methylation
percentage in patients with T1D did not differ compared with the
healthy controls. Methylation was significantly increased at
position -345 in the T1D group of the INS gene (P=0.02) and
a trend towards increased methylation in the T1D group at position
-102 (P=0.06).
 | Table ICharacteristics of the experimental
and control cohorts. |
Table I
Characteristics of the experimental
and control cohorts.
|
Characteristics | T1D patients | Controls |
|---|
| Number | 20 | 20 |
| Sex,
female/male | 8/12 | 11/9 |
| Age,
yearsa | 13.18±3.79 | 13.93±3.80 |
| Age at onset,
yearsa | 7.03±4.00 | - |
| BMI,
kg/m2a | 20.34±2.99 | 19.12±1.66 |
| BMI
z-scorea | 0.51±0.79 | - |
| Diabetes duration,
yearsa | 6.15±4.12 | - |
| HbA1c,
%a | 7.76±0.94 | - |
| Table IIMethylation of the CpG sites in the
INS gene promoter. |
Table II
Methylation of the CpG sites in the
INS gene promoter.
| | Methylation,
%a |
|---|
| CpG
siteb | T1D, n=20 | Control group,
n=20 | P-value |
|---|
| Mean methylation
(range) | 84.13±3.6
(77-92) | 82.28±2.8
(76-87) | 0.084 |
| 1/-357 | 94.00±5.0 | 90.78±7.9 | 0.15 |
| 2/-345 | 96.32±2.2 | 93.28±4.5 | 0.02 |
| 3/-234 | 91.02±6.3 | 89.58±8.4 | 0.65 |
| 4/-206 | 63.16±8.9 | 62.30±9.8 | 0.86 |
| 5/-180 | 85.78±6.6 | 84.35±9.9 | 0.82 |
| 6/-135 | 56.65±9.8 | 52.82±1.0 | 0.25 |
| 7/-102 | 90.01±3.6 | 86,53±6,0 | 0.06 |
| 8/-69 | 80.28±6.2 | 77.72±8.4 | 0.32 |
| 9/-19 | 91.51±5.2 | 89.05±9.2 | 0.67 |
| 10/+60 | 96.37±2.7 | 97.91±1.3 | 0.1 |
Correlation analysis of both age distribution of the
study population and age at diagnosis of T1D was performed. Age of
individuals and age at diagnosis of T1D was not correlated with
methylation status in any of the examined positions. Additionally,
mean methylation levels in all the studied loci of the insulin
promoter was not correlated with the age of participants at
examination or diagnosis of the disease. Finally, when the study
population were sub-grouped into prepubertal children and
adolescents, there was no difference in methylation status observed
between the two age subgroups, at any of the studied insulin
promoter gene positions, in either the diabetic individuals or
controls.
A correlation matrix showed the methylation values
(%) at various INS gene CpG sites amongst patients with T1D
(Table III). Fig. 1 shows part of the INS promoter
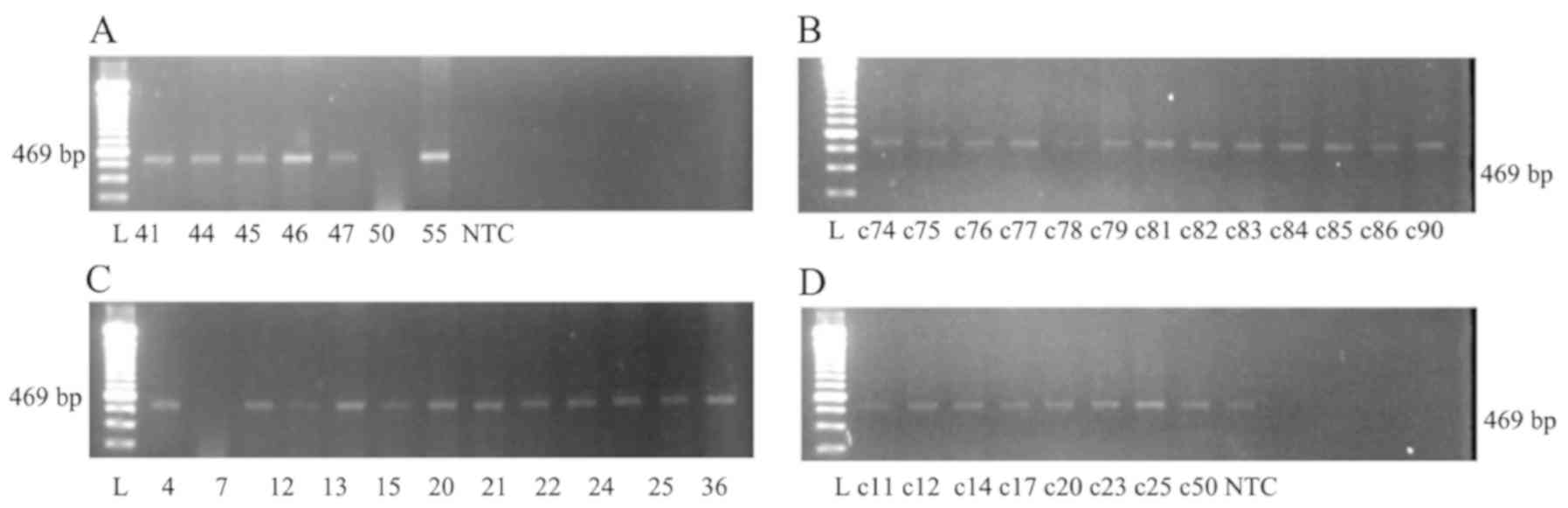
sequence upstream and downstream of the TSS and Fig. 2 shows an agarose gel electrophoresis
analysis of PCR products using modified genomic DNA isolated from
patients with T1D and controls.
| Table IIICorrelation matrix of the methylation
values at various CpG sites of the INS gene promoter among
T1D patients. |
Table III
Correlation matrix of the methylation
values at various CpG sites of the INS gene promoter among
T1D patients.
| CpG site | -357 | -345 | -234 | -206 | -180 | -135 | -102 | -69 | -19 | 60 |
|---|
| INS-357 | | 0.241 | 0.453a | 0.081 | 0.356 | 0.480a | 0.420 | 0.191 | 0.430 | -0.020 |
| | | 0.307 | 0.045 | 0.734 | 0.123 | 0.032 | 0.066 | 0.420 | 0.058 | 0.935 |
| INS-345 | | | 0.346 | 0.083 | 0.030 | 0.011 | -0.186 | 0.298 | 0.195 | 0.090 |
| | | | 0.135 | 0.729 | 0.900 | 0.965 | 0.431 | 0.202 | 0.409 | 0.705 |
| INS-234 | | | | 0.451a | 0.487a | 0.215 | 0.105 | 0.259 | -0.092 | 0.105 |
| | | | | 0.046 | 0.029 | 0.363 | 0.659 | 0.271 | 0.701 | 0.659 |
| INS-206 | | | | | 0.400 | 0.223 | 0.314 | 0.271 | -0.117 | 0.223 |
| | | | | | 0.081 | 0.346 | 0.177 | 0.248 | 0.622 | 0.346 |
| INS-180 | | | | | | 0.328 | 0.153 | 0.021 | -0.012 | 0.408 |
| | | | | | | 0.158 | 0.519 | 0.930 | 0.960 | 0.075 |
| INS-135 | | | | | | | 0.395 | 0.138 | 0.323 | 0.069 |
| | | | | | | | 0.084 | 0.561 | 0.164 | 0.772 |
| INS-102 | | | | | | | | 0.095 | 0.141 | 0.029 |
| | | | | | | | | 0.691 | 0.552 | 0.905 |
| INS-69 | | | | | | | | | 0.068 | 0.245 |
| | | | | | | | | | 0.777 | 0.298 |
| INS-19 | | | | | | | | | | -0.176 |
| | | | | | | | | | | 0.458 |
Discussion
Human studies showed that, most notably,
unmethylated DNA of the INS gene is a valuable marker of
silent β cell stress and death, while that was not the case with
other biomarkers such as proinsulin/C-peptide ratio and various
non-coding RNAs (12-14,16-18,26).
The observed increase of unmethylated INS gene in patients
that developed T1D was correlated with a decrease in insulin
secretion, indicative of active β cell loss. Unmethylated
INS DNA may thus be considered a novel biomarker of ongoing
β cell death used for the evaluation and prediction of progression
of T1D progression (18).
In the present study, ten CpG sites in a close
proximity with the TSS of INS gene were analyzed in order to
evaluate the methylation status in patients with T1D of Greek
origin, with a mean diabetes duration of ~6 years. Despite the fact
that the CpG position sites were mostly the same compared with
other studies (12,14,15,17), in
the present study, position -345 was found to exhibit increased
methylation compared with the controls. Furthermore, position -102
showed a tendency for methylation.
Hypermethylation of the INS gene promoter CpG
at position -345 observed in the present study is in partial
agreement with the tissue-specific pattern of methylation proposed
by Kuroda et al (17). They
found that in insulin-expressing β cells, this site was completely
demethylated, whereas in non-insulin expressing human pancreatic
exocrine tissue, it was methylated. In the present study, promoter
methylation of the INS gene was evaluated in genomic DNA
extracted from whole blood cells.
Regarding INS gene promoter CpG methylation
at position -102, the tendency for increased methylation was in
contrast to the results of previous studies (12,14),
which reported either hypomethylation or comparable methylation
levels. It is worth mentioning that the methodology of the study of
Fradin et al (14) that
failed to detect any difference in methylation is comparable to the
one used in the present study, as DNA was extracted from
non-immortalised human whole blood cells, using pyrosequencing, 7.5
years after T1D diagnosis. Nevertheless, based on the inconsistency
of the results, this trend may be an incidental finding and
requires further verification.
Regarding the remaining INS gene promoter CpG
sites; -357, -234, -206, -180, -135, -69 and -19, there were no
statistically significant differences in the methylation levels
between patients and healthy subjects observed in the present
study. This is in agreement with the findings of Fradin et
al (14) only for CpG sites at
-69 and -206. A study quantitating the hyper- or hypomethylation at
the CpG position -69 in pediatric patients with new-onset T1D, by
droplet digital PCR, found elevated levels of unmethylated and
methylated DNA sequences of the INS gene compared with the
control group, and this elevation may arise from β cells and other
cell types related with T1D autoimmunity. The levels of methylated
DNA remained high and the levels of unmethylated DNA dropped after
eight weeks of onset of the disease, and they both returned to
control levels 1-year post-onset (15). In the present study, comparable
methylation patterns were observed at CpG position -69 between
patients and controls. However, it cannot be ruled out that at
onset, there may have been a difference in the methylation levels
at this CpG site, or indeed, any other CpG site. Thus, additional
studies are required, examining the levels of methylation at the
CpG sites in the INS promoter gene at the onset of the
disease, and assessing the changes in the methylation levels over
time.
In animal studies, analysis of the mouse Ins1
and Ins2 promoters revealed unique demethylation of
INS gene in β cells suggesting it to be a strong biomarker
of β cell death (12,17,27-30).
DNA methylation was found to regulate insulin gene expression in
the Ins1 and Ins2 genes in non-obese diabetic (NOD)
mice, whereas increases in cytokine transcription as NOD mice age
can induce methylation of markers by activating methyltransferases
(10).
In the present study, NGS instead of Sanger
sequencing was used, as NGS has the ability to sequence, in
parallel, thousands to millions of DNA fragments, with the ability
to detect mutations or variants in the DNA sequence (31-34).
In addition, NGS has high sensitivity and specificity, even when
sequencing small genomes, and provides specific information of the
methylation status of each CpG. To the best of our knowledge, only
one study in the literature reported the use of a similar DNA
sequencing method in circulating free DNA in newly diagnosed T1D
patients (16).
According to the results of the present study,
methylated DNA leading to INS gene silencing remains
elevated at positions -345 and -102 long after the onset of
diabetes. However, there was no evidence of SNPs at the positions
-345 and -102 in T1D samples (data not shown). A previous study
performed at the onset of T1D detected unmethylated INS DNA
(16). To the best of our knowledge,
the present study is the first to address the methylation status of
the INS gene a long time after the initial diagnosis with
NGS, thus highlighting potential future research opportunities to
investigate the effect of hyperglycemia on alteration of CpG
methylation status. As methylation can vary over time and metabolic
and immunological factors may serve a role in the alteration of
methylation dependent on the duration of diabetes, glycemic
control, BMI and low-grade inflammation, further studies are
required to clarify if the epigenetic changes identified are
associated with T1D or are incidental findings.
In the present study, children and adolescents with
T1D exhibited good glycemic control, a normal BMI, and no signs and
symptoms of low-grade inflammation, thus reducing the potential
impact of these factors on the level of methylation over time.
As it is difficult to measure the methylation status
directly in the pancreatic islets, whole blood DNA was used as an
acceptable alternative to detect the methylation status, serving as
a liquid biopsy. Furthermore, these data indicate that methylated
compared with unmethylated DNA at certain CpG sites may possibly
serve as an even more specific and reliable methylation pattern,
and it is may be worth investigating the influence of long term
glycemic control of T1D.
The present study has some limitations: The
participants were only of Greek origin and their number was
limited. Larger scale studies are required to confirm the findings
pf the present study. In the future, patients recently diagnosed
with T1D diabetes will also be assessed.
There are no concrete explanations which account for
the discrepancies between our results and those of previous
studies. They may possibly be attributed to the different time
frames after disease onset to examination, the ethnicity of the
groups assessed, differences in the PCR methods used or the
different times of blood sample collection compared with the other
studies mentioned above.
In conclusion, according to our data the methylation
level in the promoter region of the INS gene quantitated
using NGS, remains elevated in certain CpG sites long after the
onset of diabetes. However, the DNA methylation status in other CpG
sites did not differ between patients with T1D and healthy
subjects. Previous studies performed at the onset of T1D detected
unmethylated INS DNA. The present study is the first study
to assess the hypermethylation status of the INS gene long
after the initial diagnosis using NGS, to the best of our
knowledge.
Acknowledgements
Parts of the study have been presented at the 57 and
58th ESPE Annual Meeting (2018 and 2019) and at the 56th
PanHellenic Paediatric Congress (2018).
Funding
This study was funded by the Hellenic Association
for the Study and Education of Diabetes Mellitus (grant no.
2015).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
KM and AGT conceived the study. AGT, KM and GT
designed the study. KM, AK, AF, IK, AS, VRT and SG collected the
data. EPK, KM, AGT and GT analyzed and interpreted the data. KM,
AK, VRT AS, SG, AGT, GT, IK, EPK, assisted in writing or revising
the manuscript for important intellectual content. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Parents or guardians provided written informed
consent for the inclusion of their children in the present in
accordance with the guidelines stated in the Declaration of
Helsinki for research involving human subjects. The study protocol
was approved by the Bioethics Committee of the Medical Department,
Faculty of Health Sciences, Aristotle University of Thessaloniki
(approval no. 185/30.12.2015) (Thessaloniki, Greece).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pugliese A: The insulin gene in type 1
diabetes. IUBMB Life. 57:463–468. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Steck AK and Rewers MJ: Genetics of type 1
diabetes. Clin Chem. 57:176–185. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bansal A and Pinney SE: DNA methylation
and its role in the pathogenesis of diabetes. Pediatr Diabetes.
18:167–177. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Handy DE, Castro R and Loscalzo J:
Epigenetic modifications: Basic mechanism and role in
cardiovascular disease. Circulation. 17:2145–2156. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cano-Rodriguez D and Rots MG: Epigenetic
editing: On the verge of reprogramming gene expression at will.
Curr Genet Med Rep. 1:170–179. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Dang MN, Buzzetti R and Pozzilli P:
Epigenetics in autoimmune diseases with focus on type 1 diabetes.
Diabetes Metab Res Rev. 29:8–18. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang Z, Xie Z, Lu Q, Chang C and Zhou Z:
Beyond genetics: What causes type 1 diabetes. Clin Rev Allergy
Immunol. 52:273–286. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Moore LD, Le T and Fan G: DNA methylation
and its basic function. Neuropsychopharmacology. 38:23–38.
2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 15:1010–1022.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Riu J, Deng S, Lebastchi J, Clark PL,
Usmani-Brawn S and Herold KC: Methylation of insulin DNA in
response to proinflammatory cytokines during the progression of
autoimmune diabetes in NOD mice. Diabetologia. 59:1021–1029.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang K, Lin G, Han Y, Xie J and Li J:
Circulating unmethylated insulin DNA as a potential non-invasive
biomarker of beta cell death in type 1 diabetes: A review and
future prospect. Clin Epigenetics. 26(44)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Husseiny MI, Kaye A, Zebabua E, Kandeel F
and Ferreri K: Tissue-specific methylation of human insulin gene
and PCR assay for monitoring beta cell death. PLoS One.
9(e94591)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Neiman D, Moss J, Hecht M, Magenheim J,
Piyanzin S, Shapiro AM, de Koning EJ, Razin A, Cedar H, Shemer R
and Dor Y: Islet cells share promoter hypomethylation independently
of expression, but exhibit cell-type-specific methylation in
enhancers. Proc Natl Acad Sci USA. 19:13525–13230. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Fradin D, Le Fur S, Mille C, Naoui N,
Groves C, Zelenika D, McCarthy MI, Lathrop M and Bougnères P:
Association of the CpG methylation pattern of the proximal insulin
gene promoter with type 1 diabetes. PLoS One.
7(336278)2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fisher MM, Watkins RA, Blum J,
Evans-Molina C, Chalasani N, DiMeglio LA, Mather KJ, Tersey SA and
Mirmira RG: Elevations in circulating methylated and unmethylated
preproinsulin DNA in new-onset type 1 diabetes. Diabetes.
64:3867–3872. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lehmann-Werman R, Neiman D, Zemmour H,
Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgård B,
Blennow K, Zetterberg H, et al: Identification of tissue-specific
cell death using methylation patterns of circulating DNA. Proc Natl
Acad Sci USA. 113:E1826–E1834. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kuroda A, Rauch TA, Todorov I, Ku HT,
Al-Abdullah I, Kandeel F, Mullen Y, Pfeifer GP and Ferreri K:
Insulin gene expression is regulated by DNA methylation. PLoS One.
9(e6953)2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Herold KC, Usmani-Brown S, Ghazi T,
Lebastchi J, Beam CA, Bellin MD, Ledizet M, Sosenko JM, Krischer JP
and Palmer JP: Type 1 Diabetes TrialNet Study Group. β cell death
and dysfunction during type 1 diabetes development in at-risk
individuals. J Clin Invest. 125:1163–1173. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Mayer-Davis EJ, Kahkoska AR, Jefferies C,
Dabelea D, Balde N, Gong CX, Aschner P and Craig ME: ISPAD Clinical
practice consensus guidelines 2018: Definition, epidemiology, and
classification of diabetes in children and adolescents. Pediatr
Diabetes. 19:7–19. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
American Diabetes Association.
Classification and diagnosis of diabetes: Standards of medical care
in diabetes-2018. Diabetes Care. 41 (Suppl 1):S13–S27.
2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Marshall WA and Tanner JM: Variations in
the pattern of pubertal changes in boys. Arch Dis Child. 45:13–23.
1970.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Marshall WA and Tanner JM: Variations in
pattern of pubertal changes in girls. Arch Dis Child. 44:291–303.
1969.PubMed/NCBI View Article : Google Scholar
|
|
23
|
World Medical Association. World medical
association declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194.
2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li Y and Tollefsbol TO: DNA methylation
detection: Bisulfite genomic sequencing analysis. Methods Mol Biol.
791:11–21. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Scala G, Affinito O, Palumbo D, Florio E,
Monticelli A, Miele G, Chiariotti L and Cocozza S:
AmpliMethProfiler: A pipeline for the analysis of CpG methylation
profiles of targeted deep bisulfite sequenced amplicons. BMC
Bioinformatics. 25(484)2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mirmira RG, SimsEK Syed F and Evans-Morina
C: Biomarkers of β-cell stress and death in type 1 diabetes. Curr
Diab Rep. 16(95)2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Husseiny MI, Kuroda A, Kaye AN, Nair I,
Kandeel F and Ferreri K: Development of a quantitative
methylation-specific polymerase chain reaction method for
monitoring beta cell death in type 1 diabetes. PLoS One.
7(e47942)2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fisher MM, Chumbiauca CN, Mather KJ,
Mirmira RG and Tersey SA: Detection of islet β-cell death in vivo
by multiplex PCR analysis of differentially methylated DNA.
Endocrinology. 154:3476–3481. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Usmani-Brown S, Lebastchi J, Steck AK,
Beam S, Herold KC and Ledizet M: Analysis of β-cell death in type 1
diabetes by droplet digital PCR. Endocrinol. 155:3694–3698.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Akirav EM, Lebastchi J, Galvan EM,
Henegariu O, Akirav M, Ablamunits V, Lizardi PM and Herold KC:
Detection of β cell death in diabetes using differentially
methylated circulating DNA. Proc Natl Acad Sci USA.
108:19018–19023. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Grada A and Weinbrecht K: Next generation
sequencing: Methodology and application. J Invest Dermatol.
133(e11)2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Behjati S and Tarpey PS: What is next
generation sequencing? Arch Dis Child Educ Pract Ed. 98:236–238.
2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin
D, Lu L and Law M: Comparison of next generation sequencing
systems. J Biomed Biotechnol. 2012(251364)2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Buermans HPJ and den Dunnen JT: Next
generation sequencing technology: Advantages and applications.
Biochim Biophys Acta. 1842:1932–1941. 2014.PubMed/NCBI View Article : Google Scholar
|