Introduction
Familial hypercholesterolemia (FH) inheritable
disorder of abnormal low-density lipoprotein (LDL) metabolism that
is characterized by elevated plasma concentrations of total
cholesterol (TC) and LDL cholesterol (LDL-C), xanthomas
(cholesterol deposits in the skin and tendons) and an increased
risk of premature coronary artery disease (CAD). Homozygous FH
affects 1 in 1,000,000 individuals, and the frequency of
heterozygous FH varies from 1 in 200 to 1 in 500 individuals,
depending on the population (1).
Monogenic FH is caused by defects in several genes
that encode proteins involved in LDL uptake and catabolism
(LDLR, APOB, PCSK9 and LDLRAP1) (1). The majority of patients with autosomal
dominant FH harbor mutations in the LDL receptor gene. To date,
>2,000 FH-causing variants have been reported in LDLR
(2). APOB and PCSK9
mutations account for a smaller percentage of autosomal dominant FH
cases (3). Recessive forms of FH are
associated with variants in LDLRAP1, which encodes for the
LDLR adapter protein 1(1).
Hypercholesterolemia may be associated with other
rare disorders of lipid metabolism, which have very similar
clinical presentations; one example is sitosterolemia, which is
caused by mutations in genes encoding either of two ATP-binding
cassette (ABC) transporters, ABCG5 or ABCG8, which limits
intestinal absorption and promotes biliary excretion of sterols
(4). Next-generation sequencing
(NGS) studies have shown that rare mutations in the ABCG5,
ABCG8, APOE and LIPA genes can cause a FH-like phenotype
(5).
Despite being one of the most common genetic
disorders, FH still remains largely undetected and untreated
worldwide (2,5). Screening during childhood may enhance
the potential identification of individuals with the condition
before establishment of cardiovascular pathologies (6). As lipoprotein metabolism in children is
influenced by fewer environmental factors than it is for adults,
the difference in LDL-C levels between children with and without FH
is more pronounced (7). Pediatric FH
is diagnosed phenotypically by the presence of elevated LDL-C
levels, in addition to a family history of premature CAD, high
baseline TC levels in one parent and/or a FH-causing mutation
(6). It should be noted that there
are no universal criteria for LDL-C cut-offs in the case of
pediatric diagnosis of FH. Although the widely used Simon Broome
criteria proposes an LDL-C cut-off of 4 mmol/l (155 mg/dl) for
individuals under 16(8), another
suggestion is 3.5 mmol/l (140 mg/dl) (9,10). The
younger the child with suspected FH, the lower LDL-C should be
expected to be (11). LDL-C levels
can fluctuate greatly during childhood, and in the case of LDL-C
levels between 2.7-3.5 mmol/l (100-140 mg/dl), it is highly
recommended that the patient is followed-up for at least a few
years by the Japan Pediatric Society and Japan Atherosclerosis
Society (9). As FH can only be
diagnosed definitively in the presence of xanthomas, which are
rarely observed in children and adolescents (7), genetic screening is a fundamental
diagnostic tool. The heterogeneity of FH-causing variants and the
total length of coding regions of FH-associated genes supports the
use of NGS-sequence-based mutation screening as a primary
methodology for diagnostics and improving the overall mutation
detection rate.
The spectrum and prevalence of FH-related mutations
remains to be studied in Russia. Previous studies have used limited
genetic screening methods based on PCR and Sanger sequencing to
study specific relevant genes, such as LDLR and APOB
(12-14).
The aim of the present study was to adapt an NGS-based method for
molecular FH diagnosis in Russian individuals and compare its
efficiency in different age groups. In the present study, mutation
screening in two groups of patients with suspected FH was
presented: The adult group and the children/adolescent group. NGS
was also used to increase the mutation detection rate.
Patients and methods
Subjects
The present study was approved by the Ethics
Committees of Center for Atherosclerosis and Lipid Disorders of
North-Western District Scientific and Clinical Center Named After
L.G. Sokolov, Medical Faculty of Saint-Petersburg State University,
City Hospital No. 40 (St. Petersburg, Russia) and Research Centre
for Medical Genetics (Moscow, Russia), where patients were treated,
and genetic analysis was performed. Written informed consent was
obtained from all patients or the children's legal representatives
prior to the beginning of the study.
A total of 59 unrelated citizens from
Saint-Petersburg and Moscow (29 male/30 female) with suspected FH
were enrolled in the present study. Clinical data were collected,
including the prior lipid levels, family and personal history of
dyslipidemia and the presence of premature atherosclerotic
cardiovascular disease (ASCVD), as well as the presence of
tendon/skin xanthomas and lipoic corneal arcus. Demographic
characteristics and clinical features of the groups are presented
in Table I. The recorded TC and
LDL-C values, independent of pre- or post-treatment, were used in
the present study. Depending on the age, patients were sorted into
two separate groups: The adult group (≥21 years old) and the
children/adolescent group (<21) (15). As children and adolescents were
included, the Simon Broome criteria was used. Exclusion criteria
were the presence of thyroid dysfunction, nephrotic syndrome,
autoimmune disease or primary biliary cirrhosis.
 | Table IClinicopathological and demographic
characteristics of the recruited cohort. |
Table I
Clinicopathological and demographic
characteristics of the recruited cohort.
| Characteristic | Adult, n=31 |
Children/adolescent, n=28 |
|---|
| Age,
yearsa | 47.4±14.1 | 11.0±5.1 |
| Age range,
years | 23-70 | 2-21 |
| Male, n (%) | 12(39) | 17(61) |
| Female, n (%) | 19(61) | 11(39) |
| Family history, n
(%) | 23(74) | 24(86) |
| Maximal total
cholesterol, mmol/la | 11.0±2.2 | 9.3±1.4 |
| LDL cholesterol,
mmol/la | 6.8±2.5 | 6.7±1.8 |
| Tendon xanthomas, n
(%) | 22(71) | 0 (0) |
| Lipoic corneal
arcus, n (%) | 2(6) | 0 (0) |
| Clinical and
instrumental manifestations of ASCVD, n (%) | 17(55) | 0 (0) |
| Increased
intima-media thickness without clinical symptoms, n (%) | 3(10) | 0 (0) |
| Patients on
lipid-lowering therapy, n (%) | 31(100) | 1(4) |
The adult group included 31 patients (12 males and
19 females; median age, 49; age range, 23-70) who fulfilled the
Simon Broome criteria for definite/possible FH (8). The children/adolescent group included
28 children and adolescents (17 males and 11 females; median age,
11; age range, 2-21) who mostly met the strict criteria regarding
lipid levels (TC >7.5 mmol/l or LDL-C >4.9 mmol/l if >16
years; TC >6.7 mmol/l or LDL-C >4.0 mmol/l if <16 years).
Children with LDL-C <4.0 mmol/l were also included in the study
as the diagnosis of FH in children can be based on age and sex
adjusted LDL-C levels, with the 95th percentile being 3.5 mmol/l
for boys and 3.8 mmol/l for girls (16). A previous clinical study stated that
the LDL-C cut-off level may be even lower (3.4 mmol/l) if a
first-degree relative shows increased TC and LDL-C levels, or has
been diagnosed with ASCVD (17). A
total of 86% individuals from the children/adolescent group
represented families with a history of hypercholesterolemia and/or
ASCVD, but never applied for genetic testing. For several patients
included in the study, elevated TC levels were a consequential
finding during routine biochemical examinations due to frequent
respiratory infections.
NGS
NGS was performed as a collaboration between two
genetic laboratories from Saint-Petersburg and Moscow, and the
Illumina MiSeq (Illumina, Inc.) and Ion S5 (Thermo Fisher
Scientific, Inc.) sequencing systems were used, respectively.
Genomic DNA (gDNA) was extracted from whole blood using the Magna
Pure system (Roche Diagnostics) or with the use of a Diatom DNA
Prep reagent kit (Biocom) according to the manufacturer's
protocols. Concentration of gDNA as well as DNA concentration of
the libraries afterwards was determined using a Quantus
Fluorometer™ (Promega Corporation) or Qubit™ Fluorometer (Thermo
Fisher Scientific, Inc.). gDNA was subjected to electrophoresis in
1% agarose gel and the optical density ratio was used to confirm
its integrity and purity.
DNA samples were prepared for the targeted NGS
covering all of the coding exons of the LDLR (NM_000527),
APOB (NM_000384), PCSK9 (NM_174936), LDLRAP1
(NM_015627), ABCG5 (NM_022436) and ABCG8 (NM_022437)
genes. The panel used for mutation screening in the
children/adolescent group additionally included LIPA
(NM_001127605), as LIPA disorders manifest with FH-like
clinical features at a young age.
For sequencing on the Illumina platform, DNA
libraries were prepared from 200 ng using a KAPA LTP Library
Preparation kit with a custom designed SeqCap® EZ Choice
Library Enrichment kit [Roche Diagnostics; cat. no. KK8232
(07961880001) and 170911_HG19_gb40_cardio_EZ_HX3, respectively].
Validation of the libraries was performed on the Agilent 4200 Tape
Station (Agilent Technologies, Inc.). Concentration in nmol was
calculated based on the size of the libraries and concentration in
ng/µl. Libraries were normalized to 4 nmol before pooling and
denaturation to get a final loading concentration of 12.5 pmol.
Paired-end sequencing of the 150 bp libraries was performed on an
Illumina MiSeq Sequencer (Illumina, Inc.) using a MiSeq Reagent kit
v2 (300 cycles) (Illumina, Inc., cat. MS-102-2002) to obtain the
FastQ data.
For sequencing on the Ion S5 system, DNA libraries
were constructed with the Ion AmpliSeq™ custom panel and Ion
AmpliSeq™ Library kit 2.0 (Thermo Fisher Scientific, Inc.; cat.
nos. 04779971_Dyslipidemia_IAD175748_182 and 4480442,
respectively). DNA quality was confirmed by the final stage of
library preparation using test PCR performed with the included
manufacturer's primers to adaptors sequences. The thermocycling
conditions used were as follows: 95˚C for 40 sec, 68˚C for 35 sec
and 72˚C for 75 sec; the number of cycles used was dependent on the
library concentration. PCR results were visualized on
silver-stained 8% acrylamide gel (staining time 10 min at 4˚C).
Massive parallel sequencing of pooled libraries with loading
concentration of 75 pmol was performed using an Ion 540™ Chip kit
(Thermo Fisher Scientific, Inc., cat. A27766) and Ion 540™ Kit-Chef
(Thermo Fisher Scientific, Inc., cat. mo. A30011) with an average
amplicon length of 175 bps.
Bioinformatics analysis
The 1000 Genomes human reference genome assembly
(b37) was used for data analysis (18). All samples were analyzed using a
bioinformatics pipeline based on the BWA-MEM version 0.7.15-r1140,
PicardTools version 2.2.2 (broadinstitute.github.io/picard/) and Genome Analysis
Tool kit (GATK) version 3.5 (github.com/broadinstitute/gatk/releases) software
according to the GATK Best Practices workflow (software.broadinstitute.org/gatk/best-practices/)
(19,20). Target enrichment metrics were
collected using the Picard CalculateHsMetrics tool. All samples
included in the dataset were jointly genotyped using the GATK
GenotypeGVCFs tool. Variants were hard filtered using threshold
values recommended by GATK. Variant annotation was performed using
SnpEff and SnpSift packages. The following resources and databases
were used for variant annotation: dbSNP build 146, 1000 Genomes
phase 3(21); Exome Aggregation
Consortium r. 0.3.1(22); ClinVar
version 2018-04-01 and dbNSFP version 2.9(23). To determine splicing alterations, the
NetGene2 Server (cbs.dtu.dk/services/NetGene2) was used. The frequency
of the identified variants was additionally assessed following the
Northwest Russia variant compendium (24). Novel variants were defined based on
the following criteria: i) No reference SNP ID number; and ii) they
had not been recorded in the public database including the Human
Gene Mutations Database (HGMD; hgmd.cf.ac.uk), ClinVar or a publicly funded database
for LDLR mutations (LOVD; databases.lovd.nl/shared/variants/). The prediction of
the pathogenicity of previously undescribed mutations was performed
using the SIFT (sift.jcvi.org/), PolyPhen-2
(genetics.bwh.harvard.edu/pph2) and
MutationTaster (mutationtaster.org) tools, as well as Human Splicing
Finder for intronic variants (umd.be/HSF3/HSF.shtml) (25). The nomenclature of molecular variants
follows the Human Genome Variation Society guidelines (varnomen.hgvs.org/). Assessment of the pathogenicity
of novel sequence variants was performed taking into account the
recommendations of the American College of Medical Genetics and
Genomics (26). These previously
developed protocols have allowed effective identification of
pathogenic variants for various other hereditary diseases (27).
Variant validation
Variant validation was performed by PCR-direct
sequencing. Specific primers were designed for verification in each
case (Table SI). DNA sequencing was
performed using the ABI BigDye Terminator 3.1 kit (Thermo Fisher
Scientific, Inc.; cat. no. 4337456) on an ABI 3130xl Genetic
analyzer (Saint-Petersburg) or ABI 3500 automatic sequencer
(Moscow; both supplied by Applied Biosystems; Thermo Fisher
Scientific, Inc.) according to the manufacturer's
specifications.
Results
NGS-based genetic findings
The NGS-based technique allowed for the
identification of 32 different pathogenic/likely pathogenic
variants, 7 of which had not been previously reported, in 43
patients (Table II). The overall
mutation detection rate was 73%. Phenotypic characteristics of
mutation-positive patients are reported in Tables SII and SIII. All novel variants in the LDLR
gene identified in this study and their pathogenicity analyses are
presented in Table II, with their
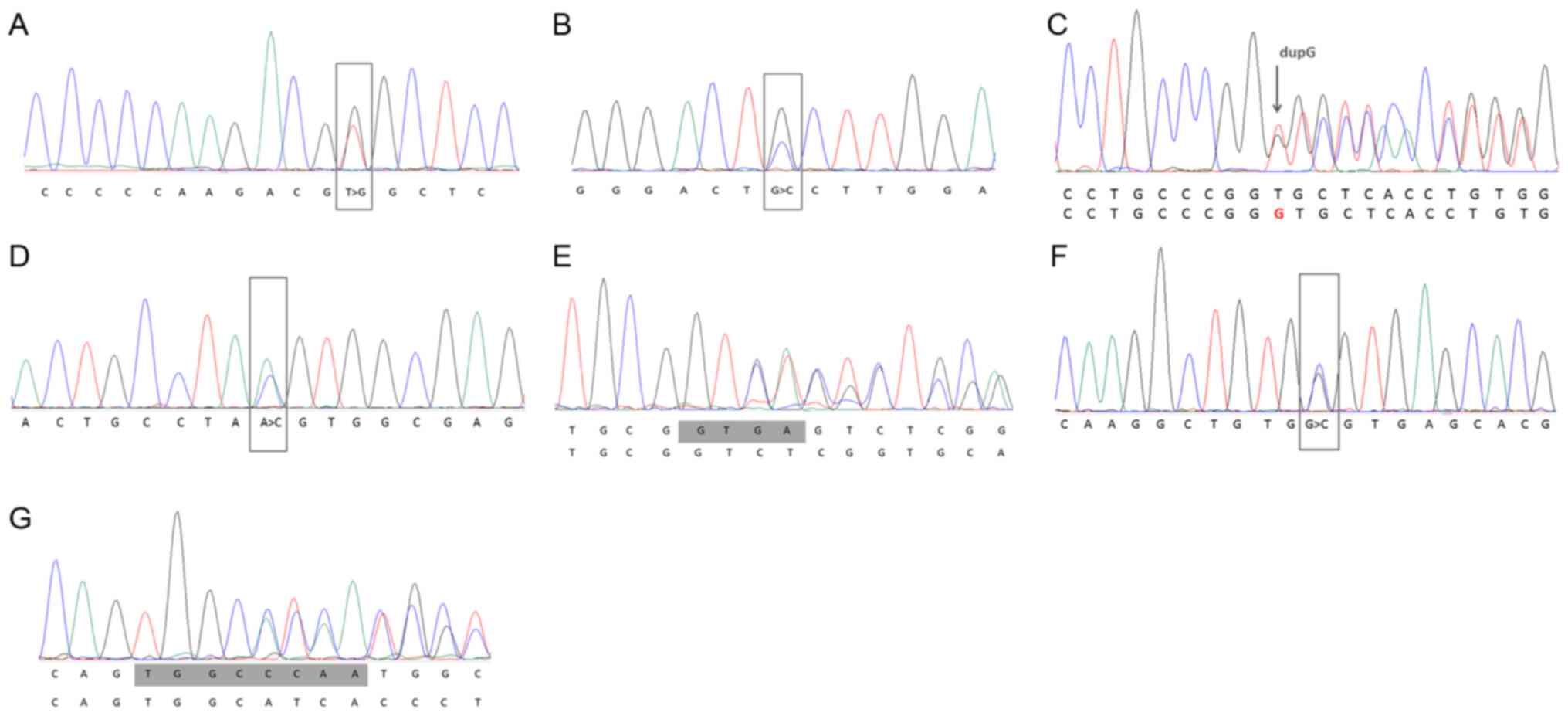
Sanger sequencing results shown in Fig.
1.
| Table IICharacterization of genetic variants
identified in the present study and their pathogenicity
analysis. |
Table II
Characterization of genetic variants
identified in the present study and their pathogenicity
analysis.
| A,
Novela LDLR variants,
reported in this study |
|---|
| Patient ID | Genetic
variant | Number of
patients | Variant ID in dbSNP
or alternative database | Genomic position
(GCRh37/hg19) | Pathogenicity
analysisb MAF in
GnomAD | Functional
domain |
Pathogenicityc | SIFT | Mutation
taster | Poly Phen2 | (Refs.) |
|---|
| 1 | Missense Exon 4
c.325T>G p.(Cys109Gly) | 1 | 869387 in
ClinVar | Chr19:
11215907 | - | Ligand-binding | Likely pathogenic
(PS1 PM1 PM2 PM5 PP3) | D | D | D | - |
| 2 | Missense Exon 4
c.401G>C p.(Cys134Ser) | 1 | 869388 in
ClinVar | Chr19:
11215983 | - | Ligand-binding | Likely pathogenic
(PS1 PM1 PM2 PM5 PP3) | D | D | D | - |
| 3 | Frameshift Exon 4
c.433_434dupG p.(Val145Glyfs*35) | 1 | 870329 in
ClinVar | Chr19:
11216013 | - | Ligand-binding | Pathogenic (PVS1
PM2 PP3) | D | D | -- | - |
| 4 | Missense Exon 4
c.616A>C p.(Ser206Arg) | 1 | 869389 in
ClinVar | Chr19:
11216198 | - | Ligand-binding | Uncertain value
(PM2 PP3) | D | D | D | - |
| 5 | c.940+1_c.940+ 4
delGTGA (g.18154_18157 delGTGA) | 4 | 869390 in
ClinVar | Chr19:
11218191-11218194 | - | Splice donor site,
intron 6 | Pathogenic (PVS1
PM1 PM2 PP3) | -- | D | -- | - |
| 6 | Missense Exon 8
c.1186G>C p.(Gly396Arg) | 1 | 870321 in
ClinVar | Chr19:
11222315 | - | EGF precursor
homology B repeat | Pathogenic (PVS1
PM1 PM2 PM5 PP3) | D | D | D | - |
| 7 | Frameshift Exon 11
c.1684_1691del TGGCCCAA p.(Pro563Hisfs*14) | 1 | 869391 in
ClinVar | Chr19:
11226866-11226875 | - | EGF spacer | Pathogenic (PVS1
PM1 PM2 PP3) | D | D | -- | - |
| B, Genetic variants
in LDLR |
| 8 | Missense Exon 2
c.100T>G p.(Cys34Gly) | 1 | rs879254405 | Chr19:
11210931 | - | Ligand-binding | Pathogenic/ Likely
pathogenic | D | D | D | (12,59) |
| 9 | Frameshift Exon 4
c.316_328delCCC AAGACGTGCT p.(Lys107Argfs*95) | 1 | LDLR_001035 in LOVD
database | Chr19:
11215901-11215915 | - | Ligand-binding | Pathogenic | D | D | -- | (39) |
| 10 | Missense Exon 4
c.552T>G p.(Cys184Trp) | 1 | LDLR_000858 in LOVD
database | Chr19:
11216134 | - | Ligand-binding | Likely
pathogenic | D | D | D | (40) |
| 11 | Missense Exon 5
c.798T>A p.(Asp266Glu)d | 1 | rs139043155 | Chr19:
11217344 | 0.000032 | Ligand-binding | Pathogenic/ Likely
pathogenic | D | D | - | (50,55-58,65) |
| 12 | Missense Exon 6
c.887G>A p.(Cys296Tyr) | 1 | rs879254707 | Chr19:
11218137 | - | Ligand-binding | Likely
pathogenic | D | D | D | (51,62) |
| 13 | Nonsense Exon 6
c.888C>A p.(Cys296*) | 1 | rs879254708 | Chr19:
11218138 | - | Ligand-binding | Pathogenic | D | D | -- | (52) |
| 14 | Missense Exon 6
c.938 G>A p.(Cys313Tyr) | 1 | rs875989910 | Chr19:
11218188 | - | Ligand-binding | Pathogenic/ Likely
pathogenic | D | D | D | (63) |
| 15 | Missense Exon 7
c.986G>A p.(Cys329Tyr)d | 2 | rs761954844 | Chr19:
11221373 | 0.000016 | EGF precursor
homology repeat A | Likely
pathogenic | D | D | D | (12,39,50,65) |
| 16 | Nonsense Exon 7
c.1048C>T p.(Arg350*) | 1 | rs769737896 | Chr19:
11221435 | - | EGF precursor
homology repeat A | Pathogenic | D | D | -- | (32,51,53) |
| 17 | c.1186+1G>T | 1 | rs730880131 | Chr19:
11222316 | - | Splice donor site‡,
intron 8 | Pathogenic/ Likely
pathogenic | -- | D | -- | - |
| 18 | Missense Exon 9
c.1202T>A p.(Leu401His) | 3 | rs121908038 | Chr19:
11223969 | - | EGF spacer | Likely
pathogenic | D | D | D | (12) |
| 19 | Missense Exon 9
c.1277 T>C p.(Leu426Pro) | 1 | rs879254851 | Chr19:
11224044 | - | EGF spacer | Pathogenic/
conflicting- interpretations-of- pathogenicity | D | D | B | (66) |
| 20 | Frameshift Exon 10
c.1478_1479delCT p.(Ser493Cysfs*42) | 1 | rs869025453 | Chr19:
11113652-11113655 | 0.00003 | EGF spacer | Pathogenic/ Likely
pathogenic | -- | D | -- | (39,40) |
| 21 | Missense Exon 12
c.1730G>C p.(Trp577Ser) | 1 | rs138947766 | Chr19:
11227559 | 0.000008 | EGF spacer | Pathogenic/ Likely
pathogenic | D | D | D | (39) |
| 22 | Missense Exon 12
c.1775G>A p.(Gly592Glu) | 6 | rs137929307 | Chr19:
11227604 | 0.000044 | EGF spacer | Pathogenic/ Likely
pathogenic | D | D | D | (40,50,65) |
| 23 | Nonsense Exon 15
c.2230C>T p.(Arg744*) | 1 | rs200793488 | Chr19:
11233939 | 0.000004 | O-linked
sugars | Pathogenic | D | D | - | (62) |
| C, Genetic variants
in APOB |
| 24 | Missense Exon 26
c.9175C>T p.(Arg3059Cys)d | 1 | rs146377316 | Chr2: 21230565 | 0.000008 | LDLR binding | Unknown
significance | B | B | B | (35,42) |
| 25 | Missense Exon 26
c.10580G>A p.(Arg3527Gln)d | 1 | rs5742904 | Chr2: 21229160 | 0.000275 | LDLR binding | Pathogenic | B | D | D | (41) |
| 26 | Missense Exon 26
c.10580G>T p.(Arg3527Leu) | 1 | rs5742904 | Chr2: 21229160 | - | LDLR binding | Pathogenic | D | D | D | (39,40,41) |
| 27 | In-frame deletion
Exon 29 c.13480_ 13482delCAG p.(Gln4494del)d | 2 | rs562574661 | Chr2:
21001940-21001945 | 0.000384 | - | Likely pathogenic/
conflicting- interpretations-of- pathogenicity | - | B | - | (41) |
| D, Genetic variants
in ABCG5/8 |
| 28 | Nonsense
ABCG5 Exon 10 c.1336C>T p.(Arg446*) | 1 | rs199689137 | Chr2: 44050063 | 0.00018 | Cytoplasmic | Pathogenic | D | D | - | (43,45,46) |
| 29 | Missense
ABCG8 Exon 7 c.1083G>A p.(Trp361*) | 1 | rs137852987 | Chr2: 44099233 | 0.00102 | Cytoplasmic | Pathogenic | D | D | - | (47,67) |
| 30 | Missense
ABCG8 Exon 11 c.1629G>T p.(Arg543Ser)d | 1 | rs201690654 | Chr2, 44102425 | 0.000215 | Transmembrane | Unknown
significance | D | D | D | (48,49) |
| E, Genetic variant
in LIPA |
| 31 | c.894G>A p.
(Q298=) | 1 | rs116928232 | Chr10:
89222511 | 0.00083 | Exon skipping
mutation | Pathogenic | -- | D | -- | (38) |
| F, Genetic variant
in PCSK9 |
| 32 | Missense Exon 9
c.1486C>T p.(Arg496Trp) | 1 | rs374603772 | Chr1, 55524303 | 0.000044 | LDLR-binding | Unknown
significance/ conflicting- interpretations-of- pathogenicity | D | D | D | (68,69) |
In the adult group, pathogenic/likely pathogenic
variants were detected in 18 (58%) patients: 13 Patients had
FH-causing mutations in the LDLR gene, including 2 novel
variants; 3 patients had APOB mutations; and 2 patients had
ABCG5/G8 mutations. A total of 17 (95%) mutation positive
patients had tendon xanthomas and 13 (72%) had established
cardiovascular complications. In the children/adolescent group
pathogenic/likely pathogenic variants were detected in 25 (89%)
patients: 21 patients had FH-causing mutations in the LDLR
gene, including 5 novel variants, 2 patients had APOB
mutations and ABCG8 and LIPA mutations were found in
a single patient. Additionally, PSCK9 variants of unknown
significance were identified in patients from both groups.
In total, 23 mutations were found in the LDLR
gene in both cohorts: 7 Novel variants, 3 mutations that have
previously reported in Russia and 13 mutations that were identified
in other populations. A total of 2 LDLR mutations were
common between Saint-Petersburg and Moscow: p.(Leu401His) and
p.(Gly592Glu). These variants were detected in 3 and 6 patients,
representing 5 and 10% of all FH cases reported in the present
study, respectively.
Novel LDLR variants
Novel frame-shift variants in the LDLR gene
that lead to truncated proteins were considered certainly
pathogenic. Variant c.1684_1691delTGGCCCAA p.(Pro563Hisfs*14) was
found in a 16-year-old female with maximal TC levels of 9.6 mmol/l.
The c.433_434dupG p.(Val145Glyfs*35) variant was found in a
53-year-old woman with xanthomas and a history of myocardial
infarction (MI) at the age of 53. A maximal TC level of 10.3 mmol/l
was observed without statin therapy. The patient's father and uncle
both died from a MI at the age of 49 and 40, respectively, and were
expected to have suffered from FH.
A novel variant in exon 8, c.1186G>C
p.(Gly396Arg), was found in a young man (aged 29) who had no family
history of either hyperlipidemia or ASCVD. The patient had a
maximal TC level of 9.7 mmol/l. During the period of genetic
testing, the patient underwent a coronarography that showed
preclinical diffuse atherosclerosis (30%) of the anterior
interventricular artery. The p.(Gly396Arg) variant was predicted by
in silico tools as a pathogenic variant. The mutation was
encoded in the EGF-like domain of LDLR, which is known to be
important for receptor dissociation in endocytosis and receptor
recycling to the cell surface (28),
and thus could be regarded as pathogenic. It is hypothesized that
this variant was functional as another similar missense variant in
the codon, c.1186G>A p.(Gly396Ser), which had previously been
found in Finland, was predicted to be disease causing (29).
Another three new missense variants encoded in the
ligand-binding domain of LDLR, as well as one splicing variant,
were predicted by software tools as disease causing. c.325T>G
p.(Cys109Gly), c.401G>C p.(Cys134Ser) and c.616A>C,
corresponding to a known protein substitution c.618T>G
p.(Ser206Arg), were detected in children from families with a
history of hypercholesterolemia. These three patients, aged 11, 7
and 6 years old, respectively, demonstrated the highest LDL-C
levels in the children/adolescent group. A splicing variant in
intron 6 c.940+1_c.940+4delGTGA was detected in four unrelated
individuals. This variant alters a canonical splice donor site.
Previously, a similar splicing mutation, which affects 15
nucleotides (c.940_940+14del15), was described in a 21-year-old
Spanish man who suffered a premature MI at the age of 16 years
(30).
Discussion
Previously, NGS has been successfully used for
mutation screening of the LDLR gene and other FH-associated
genes in subjects with a clinical diagnosis of definite/possible
FH, as well as in patients with CAD with extremely high TC levels
(31,32). In the present pilot study using NGS
technology for the molecular diagnosis of FH in Russian
individuals, mutation screening of the LDLR, APOB,
PSCK9, LDLRAP1, ABCG5/8, APOE and
LIPA genes in a cohort of 59 probands was performed using
NGS technology, which has been widely reported to improve the
overall mutation detection rate (33-35).
In the present study, pathogenic and likely pathogenic mutations
were revealed in 73% of patients with definite/possible FH.
Approximately the same percentage has been commonly reported in
studies where NGS technology has been applied for FH molecular
diagnosis (36,37). It is worth noting that previous
publications conducted in Russia applying the routine methods for
mutation detection with limited genes (LDLR and APOB)
have identified causative mutations in 20-50% of patients with FH
(12,14). Moreover, NGS technology has allowed
for the inclusion of additional genes that have been linked to FH,
which were previously missed by older mutation detection assays
(5). The present study was, to the
best of our knowledge, the first study to investigate the genetic
variation in ABCG5/8 and LIPA among Russian patients
with FH.
According to a previous publication, the mutation
detection rates in patients with suspected FH varies from 20-90%,
depending how rigorous the criteria used for selection of patients
were (7). It should be noted that in
the present study, the mutation detection rate was higher in the
children/adolescent group (89%). Only a few studies with a similar
cohorts have been performed. Van der Graaf et al (7) reported that 95% of children (aged 4-18
years) with plasma LDL-C >95th percentile for age and sex, and
an autosomal dominant pattern for inherited hypercholesterolemia,
have an FH causing mutation. This same study also reported that
only 4% of children with LDLR mutations exhibited physical
symptoms (tendon xanthomas and an arcus cornealis), which is
consistent with previous reports that have stated that only marked
differences in LDL-C levels can distinguish children with FH
(6,11). Early childhood (1-9 years) is the
optimal period for using TC or LDL-C levels to discriminate between
individuals with and without FH in the general population (11). Levels of both TC and LDL-C show
considerable overlap between adults with and without FH (17). A lower mutation detection rate in the
adult group may be due to polygenic inheritance, which may explain
phenotypic heterozygous FH being observed in several individuals
without monogenic mutations (5).
The present study identified several novel
LDLR variants as well as a number of previously reported FH
causing mutations in the LDLR and APOB genes. These
data expand upon the current knowledge of the spectrum of FH
mutations in Russian individuals. Of note, it was shown that the
p.(Gly592Glu) in the LDLR gene may be a major FH-associated
variant for individuals from the European portion of Russia. At the
same time, sequencing of noncanonical FH genes was established for
Russian patients with FH and rare ABCG5/8, PSCK9 and
LIPA mutations associated with a FH-like phenotype were
found.
Identified variants were located mostly in coding
regions which correspond to functional protein domains or produce
truncated proteins. These variants do not overlap with any genomic
regulatory regions. According to ENCODE in the UCSC Genome Browser,
Tracks H3K27Ac, DNase Clusters and Txn Factor ChIP (data not
shown), there was a highly probable presence of regulatory
sequences in exon 1, intron 1-2, 3'-untranslated region (UTR) of
the LDLR gene. Introns and 3'UTR were not studied, no
mutations were found in exon 1 in the present study. In the present
study, 2 identified variants were predicted to alter canonical
splice donor sites in introns 6 and 8 of the LDLR gene.
However, co-segregation analysis was not performed and there were
insufficient numbers of patients to analyze linkage disequilibrium.
All patients were carriers of the only one variant.
A proband with a family history of high TC levels
was homozygous for a c.894G>A splicing mutation in the
LIPA gene, one of the most frequent variants responsible for
cholesteryl ester storage disorder (CESD) (38). The variant has previously been found
to have an allele frequency of 0.11% (1 in 450 individuals) in a
large European population (38).
This mutation is known to predominantly result in a non-functional
transcript skipping of exon 8, causing the deletion of 24 amino
acids (p.Q298=). CESD is associated with reduced activity of
lysosomal acid lipase, an enzyme that is involved in intracellular
hydrolysis of cholesteryl esters and triglycerides. It has recently
been suggested that certain patients with a FH phenotype may have
CESD (38).
To the best of our knowledge, the present study is
the first to identify mutations in the APOB gene in citizens
in Saint-Petersburg. The most common mutations of the APOB
gene observed in European individuals, p.(Arg3527Gln), was also
found in one patient from Moscow, demonstrating an expected
frequency for this region (2-4.5%) (13). The mutation p.(Arg3527Gln) accounts
for 6-10% of all FH cases in Europe, but it was not found in the
patients from Saint-Petersburg with FH, neither in the present
study nor in a previous study where screening was established for
this sole mutation (12). The
present study also found a rare variant in the same codon,
p.(Arg3527Leu), in a patient from Saint-Petersburg. This variant
has been reported in single cases from the Netherlands and Poland
(39,40).
As APOB mutations can be located outside the
routinely analyzed APOB region, NGS allows for effective
mutation screening in the entire APOB coding sequence. A
rare mutation, p.(Arg3059Cys) reported in the Netherlands (35) was identified in one of the patients
from Moscow, who had a family history of FH. This mutation, similar
to p.(Arg3527Gln), maps to the region that binds with LDL, and the
uptake of LDL particles has been shown to be significantly reduced
when using LDL particles from carriers of both mutations compared
with controls (35,41).
A known APOB variant, p.(Gln4494del), has
been detected for the first time in a Russian individual, to the
best of our knowledge. This sequence results in a deletion of 3
nucleotides from exon 29 of APOB mRNA (c.13480_13482delCAG)
and leads to the deletion of 1 amino acid residue of the ApoB
protein, but otherwise preserves the integrity of the reading
frame. The p.(Gln4494del) variant, considered as pathogenic in the
UCL-FH mutation database, has been suggested as likely pathogenic
or of uncertain significance in certain studies (3,42).
Gln4494 in the ApoB tail is important for correct protein
conformation (41). Experimental
studies have shown that p.(Gln4494del) causes a 40-50% reduction in
the binding and uptake of LDL. A previous study investigating
secondary structure of the human ApoB using infrared spectroscopy,
as well as LDL particle size using dynamic light scattering and
electron microscopy, highlighted differences in the secondary
structure and in the particle size of the p.(Gln4494del) variant
when compared with the structure of the wild-type. These changes
may underlie reduced/defective LDL binding capacity of the
p.(Gln4494del) variant (41). These
findings also support the notion that this mutation is disease
causing.
As mutations in the ABCG5 and ABCG8
genes have been shown to potentially cause noticeably elevated
cholesterol levels (43), these
genes were included in the present analysis. One proband had an
ABCG5 variant p.(Arg446*) causing a premature stop codon.
The patient presented with xanthomas and had a history of MI. This
rare mutation which causes sitosterolemia in homozygous individuals
was initially described in Italy in 2007 and later registered in
Japan and Germany (44-46).
High levels of blood TC and LDL-C as well as low levels of HDL-C
are typical for carriers (44,46).
From two identified ABCG8 variants, a terminating mutation
p.(Trp361*) is the most common mutation causing sitosterolemia
(47). The second detected variant
p.(Arg543Ser) appears to cause a destabilizing substitution of
conserved polar residues in the core of the transmembrane domain of
ABCG8(48). In vitro analysis
has shown that this mutation decreases the amount of mature ABCG8
protein (49). It should be noted
that a patient with this mutation suffered from severe CAD and had
a MI at the age of 30. Clinical manifestations of sitosterolemia
can be similar to those of FH as ABCG5/8 mutations are
paired with extreme hypercholesterolemia (45). Notably, LDL-C levels are
significantly increased in certain individuals with sitosterolemia,
although the mechanism underlying this is unknown (45), and thus, those cases are occasionally
misdiagnosed as FH.
In total, 16 LDLR mutations identified in the
present study have been previously reported and predicted to be
pathogenic/likely pathogenic. Of these, three variants have been
previously found in individuals from Saint-Petersburg and Moscow,
as well as in European countries: p.(Gly592Glu), p.(Cys329Tyr) and
p.(Leu401His). It is notable that p.(Gly592Glu) is the most
frequent mutation in Czech and Polish populations (50). The present study showed that this
mutation may be frequent or may even be considered major in Russian
individuals.
A further 13 LDLR mutations were described in
Russian patients for the first time. Of these, five resulted in
production of a truncated protein, and have been reported in
citizens of other European countries (36,39,40,51-54).
From the seven LDLR missense variants identified in the
present study, p.(Asp266Glu), known as FH Cincinnati-1, is the most
common in Germany and Austria (50,55),
second most common in the Czech Republic (56) and has also been found in Denmark,
Norway and USA (57,58). Several loss of cysteine residues,
p.(Cys34Gly), p.(Cys184Trp), p.(Cys296Tyr) and p.(Сys313Tyr),
encoded in the ligand binding domain of LDLR that are associated
with incorrect folding of the protein, have been previously
detected in European countries (39,40,51,58-63).
A novel mutation from the present study, c.616A>C, corresponded
to a known protein substitution c.618T>G p.(Ser206Arg). The
p.(Ser206Arg) variant is suggested to be a likely pathogenic
mutation as it is located in a strongly conserved ligand-binding
repeat (14). This mutation has been
found in an individual from Norway and another individual from the
Russian city of Petrozavodsk (14,58).
c.616A>C leading to the same protein substitution can be assumed
to be disease causing.
The present study has some limitations. For
example, analysis of introns/3'UTRs of the studied genes was not
performed, and co-segregation analysis could not be performed due
to an insufficient number of patients for linkage disequilibrium
studies.
In conclusion, the mutation spectrum for FH in
Russian individuals is similar to that of other European countries.
Evidence of this conclusion is that certain LDLR mutations
that had initially been found in Russian individuals (12,64) were
subsequently identified in European populations, such as
p.(Cys249*), p.(Trp422*), p.(Asp601Asn) and p.(Arg410Gly) (39,40,50,51).
Mutations in the LDLR gene are very diverse and can be
present in any part of the gene; therefore, it was not unexpected
that through using NGS, novel LDLR variants were discovered
in the present study. NGS allowed for the investigation of an
extended list of FH associated genes, and for the first time
revealed rare mutations in the ABCG5/8 and LIPA genes
in Russian patients with FH.
Supplementary Material
Primers for PCR and subsequent Sanger
sequencing.
Phenotypic characteristics of
mutation-positive adult patients.
Phenotypic characteristics of
mutation-positive patients from the children/adolescent group.
Acknowledgements
Not applicable.
Funding
This work was partly supported by the Russian
Science Foundation (grant no. 14-50-00069) and the state assignment
of Ministry of science and Higher Education of the Russian
Federation for RCMG.
Availability of data and materials
The data that support the findings of this study
are available from the corresponding author upon reasonable
request. The data are not publicly available due to privacy or
ethical restrictions. All novel variants have been submitted to the
ClinVar database (ncbi.nlm.nih.gov/clinvar/; IDs: 869387, 869388,
869389, 869390, 869391, 870321, 870329).
Author's contributions
EYZ, OSG, ASG, AMS, SNP designed and conceived the
methodology of the present study, organized experiments and
performed final interpretation of the data. MVM, SAU, VSG, SPU,
SGS, IVA and DMG collected the patient data and assisted with
clinical data interpretation. OVR, ONI, NAS, MAF and VVM performed
the experiments. YAB, AAP, VVM, MAF and ONI performed the
bioinformatics analysis and variant annotation. VVM wrote the first
draft of the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committees of
Center for Atherosclerosis and Lipid Disorders of North-Western
District Scientific and Clinical Center Named After L.G. Sokolov,
Medical Faculty of Saint-Petersburg State University, City Hospital
No. 40 (St. Petersburg, Russia) and Research Centre for Medical
Genetics (Moscow, Russia) where patients were treated and genetic
analysis was performed. Written informed consent was obtained from
all patients or their legal representatives before the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Defesche JC, Gidding SS, Harada-Shiba M,
Hegele RA, Santos RD and Wierzbicki AS: Familial
hypercholesterolaemia. Nat Rev Dis Primers. 3(17093)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Di Taranto MD, Giacobbe C and Fortunato G:
Familial hypercholesterolemia: A complex genetic disease with
variable phenotypes. Eur J Med Genet. 63(103831)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sharifi M, Futema M, Nair D and Humphries
SE: Genetic architecture of familial hypercholesterolaemia. Curr
Cardiol Rep. 19(44)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Berge KE, Tian H, Graf GA, Yu L, Grishin
NV, Schultz J, Kwiterovich P, Shan B, Barnes R and Hobbs HH:
Accumulation of dietary cholesterol in sitosterolemia caused by
mutations in adjacent ABC transporters. Science. 290:1771–1775.
2000.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Iacocca MA and Hegele RA: Recent advances
in genetic testing for familial hypercholesterolemia. Expert Rev
Mol Diagn. 17:641–651. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wiegman A, Gidding SS, Watts GF, Chapman
MJ, Ginsberg HN, Cuchel M, Ose L, Averna M, Boileau C, Borén J, et
al: European atherosclerosis society consensus panel Familial
hypercholesterolaemia in children and adolescents: Gaining decades
of life by optimizing detection and treatment. Eur Heart J.
36:2425–2437. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Van der Graaf A, Avis HJ, Kusters DM,
Vissers MN, Hutten BA, Defesche JC, Huijgen R, Fouchier SW, Wijburg
FA, Kastelein JJ and Wiegman A: Molecular basis of autosomal
dominant hypercholesterolemia: Assessment in a large cohort of
hypercholesterolemic children. Circulation. 123:1167–1173.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Santos RD, Gidding SS, Hegele RA, Cuchel
MA, Barter PJ, Watts GF, Baum SJ, Catapano AL, Chapman MJ, Defesche
JC, et al: Defining severe familial hypercholesterolaemia and the
implications for clinical management: A consensus statement from
the international atherosclerosis society severe familial
hypercholesterolemia panel. Lancet Diabetes Endocrinol. 4:850–861.
2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Harada-Shiba M, Ohta T, Ohtake A, Ogura M,
Dobashi K, Nohara A, Yamashita S and Yokote K: Joint Working Group
by Japan Pediatric Society and Japan Atherosclerosis Society for
Making Guidance of Pediatric Familial Hypercholesterolemia.
Guidance for pediatric familial hypercholesterolemia 2017. J
Atheroscler Thromb. 25:539–553. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ramaswamia U, Humphries SE,
Priestley-Barnhamc L, Green P, Wald DS, Capps N, Andersong M, Dale
P and Morris AA: Current management of children and young people
with heterozygous familial hypercholesterolaemia-HEART UK statement
of care. Atherosclerosis. 290:1–8. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Martin AC, Gidding SS, Wiegman A and Watts
GF: Knowns and unknowns in the care of pediatric familial
hypercholesterolemia. J Lipid Res. 58:1765–1776. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zakharova FM, Damgaard D, Mandelshtam MY,
Golubkov VI, Nissen PH, Nilsen GG, Stenderup A, Lipovetsky BM,
Konstantinov VO, Denisenko AD, et al: Familial hypercholesterolemia
in St-Petersburg: The known and novel mutations found in the low
density lipoprotein receptor gene in Russia. BMC Med Genet.
6(6)2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Malyshev PP, Meshkov AN, Kotova LA and
Kuharchuk VV: Familial defect of apolipoprotein B-100: Molecular
disease basis and clinic-biochemical characteristics of the
patients. Cardiovascular Ther Prevention. 6:40–45. 2007.(In
Russian).
|
|
14
|
Korneva VA, Kuznetsova TY, Golovina AS,
Vasilyev VB and Mandelshtam MY: Familial hypercholesterolemia
mutations in Petrozavodsk: No similarity to St. Petersburg mutation
spectrum. BMC Med Genet. 14(128)2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hardin AP and Hackell JM: Committee on
Practice and Ambulatory Medicine. Age limits in pediatrics.
Pediatrics. 140(e20172151)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Watts GF, Sullivan DR, Poplawski N, van
Bockxmeer F, Hamilton-Craig I, Clifton PM, O'Brien R, Bishop W,
George P, Barter PJ, et al: Familial hypercholesterolaemia: A model
of care for Australasia. Atheroscler Suppl. 12:221–263.
2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Di Taranto MD, de Falco R, Guardamagna O,
Massini G, Giacobbe C, Auricchio R, Malamisura B, Proto M, Palma D,
Greco L and Fortunato G: Lipid profile and genetic status in a
familial hypercholesterolemia pediatric population: Exploring the
LDL/HDL ratio. Clin Chem Lab Med. 57:1102–1110. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From Fastq data to high-confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Auton A, Brooks LD, Durbin RM, Garrison
EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and
Abecasis GR: A global reference for human genetic variation.
Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lek M, Karczewski KJ, Minikel EV and
Samocha KE: Analysis of protein-coding genetic variation in 60,706
humans. Nature. 536:285–291. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu X, Wu C, Li C, Boerwinkle E, Jolla L
and Genome H: dbNSFP v3.0: A one-stop database of functional
predictions and annotations for human non-synonymous and splice
site SNVs. Hum Mutat. 37:235–241. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Barbitoff YA, Skitchenko RK, Poleshchuk
OI, Shikov AE, Serebryakova EA, Nasykhova YA, Polev DE, Shuvalova
AR, Shcherbakova IV, Fedyakov MA, et al: Whole-exome sequencing
provides insights into monogenic disease prevalence in Northwest
Russia. Mol Genet Genomic Med. 7(e964)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37(e67)2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Glotov OS, Serebryakova EA, Turkunova MS,
Efimova OA, Glotov AS, Barbitoff YA, Nasykhova YA, Predeus AV,
Polev DE, Fedyakov MA, et al: Whole-exome sequencing for monogenic
diabetes in Russian children reveals wide spectrum of genetic
variants in MODY-related and unrelated genes. Mol Med Rep.
20:4905–4914. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Arias-Moreno X, Velazquez-Campoy A,
Rodríguez JC, Pocoví M and Sancho J: Mechanism of low density
lipoprotein (LDL) release in the endosome: Implications of the
stability and Ca2+ affinity of the fifth binding module of the LDL
receptor. J Biol Chem. 283:22670–22679. 2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Koivisto UM, Viikari JS and Kontula K:
Molecular characterization of minor gene rearrangements in Finnish
patients with heterozygous familial hypercholesterolemia:
Identification of two common missense mutations (Gly823->Asp and
Leu380->His) and eight rare mutations of the LDL receptor gene.
Am J Hum Genet. 57:789–797. 1995.PubMed/NCBI
|
|
30
|
Maglio C, Mancina RM, Motta BM, Stef M,
Pirazzi C, Palacios L, Askaryar N, Boren J, Wiklund O and Romeo S:
Genetic diagnosis of familial hypercholesterolaemia by targeted
next-generation sequencing. J Intern Med. 276:396–403.
2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Norsworthy PJ, Vandrovcova J, Thomas ERA,
Campbell A, Kerr SM, Biggs J, Game L, Soutar AK, Smith BH,
Dominiczak AF, et al: Targeted genetic testing for familial
hypercholesterolaemia using next generation sequencing: A
population-based study. BMC Med Genet. 15(70)2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Radovica-Spalvina I, Latkovskis G,
Silamikelis I, Fridmanis D, Elbere I, Ventins KV, Ozola G, Erglis A
and Klovins J: Next-generation-sequencing-based identification of
familial hypercholesterolemiarelated mutations in subjects with
increased LDL-C levels in a latvian population. BMC Med Genet.
16(86)2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Johansen CT, Dubé JB, Loyzer MN, MacDonald
A, Carter DE, McIntyre AD, Cao H, Wang J, Robinson JF and Hegele
RA: LipidSeq: A next-generation clinical resequencing panel for
monogenic dyslipidemias. J Lipid Res. 55:765–772. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Vandrovcova J, Thomas ER, Atanur SS,
Norsworthy PJ, Neuwirth C, Tan Y, Kasperviciute D, Biggs J, Game L,
Mueller M, et al: The use of next-generation sequencing in clinical
diagnosis of familial hypercholesterolemia. Genet Med. 15:948–957.
2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Motazacker MM, Pirruccello J, Huijgen R,
Do R, Gabriel S, Peter J, Kuivenhoven JA, Defesche JC, Kastelein
JJ, Hovingh GK, et al: Advances in genetics show the need for
extending screening strategies for autosomal dominant
hypercholesterolaemia. Eur Heart J. 33:1360–1366. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Reiman A, Pandey S, Lloyd KL, Dyer N, Khan
M, Crockard M, Latten MJ, Watson TL, Cree IA and Grammatopoulos DK:
Molecular testing for familial hypercholesterolaemia-associated
mutations in a UK-based cohort: Development of an NGS-based method
and comparison with multiplex polymerase chain reaction and
oligonucleotide arrays. Ann Clin Biochem. 53:654–662.
2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang J, Dron JS, Ban MR, Robinson JF, Mc
Intyre AD, Alazzam M, Zhao PJ, Dilliott AA, Cao H, Huff MW, et al:
Polygenic versus monogenic causes of hypercholesterolemia
ascertained clinically. Arterioscler Thromb Vasc Biol.
36:2439–2445. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ashfield-Watt P, Haralambos K, Edwards R,
Townsend D, Gingell R, Wa Li K, Humphries SE and McDowell I:
Estimation of the prevalence of cholesteryl ester storage disorder
in a cohort of patients with clinical features of familial
hypercholesterolaemia. Ann Clin Biochem. 56:112–117.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Fouchier SW, Kastelein JJ and Defesche JC:
Update of the molecular basis of familial hypercholesterolemia in
The Netherlands. Hum Mutat. 26:550–556. 2005.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chmara M, Wasag B, Zuk M, Kubalska J,
Wegrzyn A, Bednarska-Makaruk M, Pronicka E, Wehr H, Defesche JC,
Rynkiewicz A and Limon J: Molecular characterization of Polish
patients with familial hypercholesterolemia: Novel and recurrent
LDLR mutations. J Appl Genet. 51:95–106. 2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fernández-Higuero JA, Etxebarria A,
Benito-Vicente A, Alves AC, Arrondo JL, Ostolaza H, Bourbon M and
Martin C: Structural analysis of APOB variants, p.(Arg3527Gln),
p.(Arg1164Thr) and p.(Gln4494del), causing Familial
Hypercholesterolaemia provides novel insights into variant
pathogenicity. Sci Rep. 5(18184)2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Alves AC, Etxebarria A, Soutar AK, Martin
C and Bourbon M: Novel functional APOB mutations outside
LDL-binding region causing familial hypercholesterolaemia. Hum Mol
Genet. 23:1817–1828. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Rios J, Stein E, Shendure J, Hobbs HH and
Cohen JC: Identification by whole-genome resequencing of gene
defect responsible for severe hypercholesterolemia. Hum Mol Genet.
19:4313–4318. 2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Mannucci L, Guardamagna O, Bertucci P,
Pisciotta L, Liberatoscioli L, Bertolini S, Irace C, Gnasso A,
Federici G and Cortese C: Beta-sitosterolaemia: A new nonsense
mutation in the ABCG5 gene. Eur J Clin Invest. 37:997–1000.
2007.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Tada H, Kawashiri MA, Takata M, Matsunami
K, Imamura A, Matsuyama M, Sawada H, Nunoi H, Konno T, Hayashi K,
Nohara A, et al: Infantile cases of Sitosterolaemia with novel
mutations in the ABCG5 gene: Extreme hypercholesterolaemia is
exacerbated by breastfeeding. JIMD Rep. 21:115–122. 2015.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Li Y, Salfelder A, Schwab KO, Grünert SC,
Velten T, Lütjohann D, Villavicencio-Lorini P, Matysiak-Scholze U,
Zabel B, Köttgen A and Lausch E: Against all odds: Blended
phenotypes of three single-gene defects. Eur J Hum Genet.
24:1274–1279. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lu K, Lee MH, Hazard S, Brooks-Wilson A,
Hidaka H, Kojima H, Ose L, Stalenhoef AF, Mietinnen T, Bjorkhem I,
et al: Two Genes that map to the STSL locus cause Sitosterolemia:
Genomic structure and spectrum of mutations involving Sterolin-1
and Sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am J Hum
Genet. 69:278–290. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
48
|
Lee JY, Kinch LN, Borek DM, Wang J, Wang
J, Urbatsch IL, Xie XS, Grishin NV, Cohen JC, Otwinowski Z, et al:
Crystal structure of the human sterol transporter ABCG5/ABCG8.
Nature. 533:561–564. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Graf GA, Cohen JC and Hobbs HH: Missense
mutations in ABCG5 and ABCG8 disrupt heterodimerization and
trafficking. J Biol Chem. 279:24881–24888. 2004.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Tichý L, Freiberger T, Zapletalová P,
Soska V, Ravcuková B and Fajkusová L: The molecular basis of
familial hypercholesterolemia in the Czech Republic: Spectrum of
LDLR mutations and genotype-phenotype correlations.
Atherosclerosis. 223:401–408. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Bertolini S, Pisciotta L, Rabacchi C,
Cefalù AB, Noto D, Fasano T, Signori A, Fresa R, Averna M and
Calandra S: Spectrum of mutations and phenotypic expression in
patients with autosomal dominant hypercholesterolemia identified in
Italy. Atherosclerosis. 227:342–348. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Lind S, Eriksson M, Rystedt E, Wiklund O,
Angelin B and Eggertsen G: Low frequency of the common Norwegian
and Finnish LDL-receptor mutations in Swedish patients with
familial hypercholesterolaemia. J Intern Med. 244:19–25.
1998.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Solberg K, Rødningen OK, Tonstad S, Ose L
and Leren TP: Familial hypercholesterolaemia caused by a non-sense
mutation in codon 329 of the LDL receptor gene. Scand J Clin Lab
Invest. 54:605–609. 1994.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Górski B, Kubalska J, Naruszewicz M and
Lubiński J: LDL-R and Apo-B-100 gene mutations in Polish familial
hypercholesterolemias. Hum Genet. 102:562–565. 1998.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Brænne I, Kleinecke M, Reiz B, Graf E,
Strom T, Wieland T, Fischer M, Kessler T, Hengstenberg C, Meitinger
T, et al: Systematic analysis of variants related to familial
hypercholesterolemia in families with premature myocardial
infarction. Eur J Hum Genet. 24:191–197. 2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Kuhrová V, Francová H, Zapletalová P,
Freiberger T, Fajkusová L, Hrabincová E, Slováková R and Kozák L:
Spectrum of low density lipoprotein receptor mutations in Czech
hypercholesterolemic patients. Hum Mutat. 18(253)2001.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Brusgaard K, Jordan P, Hansen H, Hansen AB
and Hørder M: Molecular genetic analysis of 1053 Danish individuals
with clinical signs of familial hypercholesterolemia. Clin Genet.
69:277–283. 2006.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Leren TP, Manshaus T, Skovholt U, Skodje
T, Nossen IE, Teie C, Sørensen S and Bakken KS: Application of
molecular genetics for diagnosing familial hypercholesterolemia in
Norway: Results from a family-based screening program. Semin Vasc
Med. 4:75–85. 2004.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Duskova L, Kopeckova L, Jansova E, Tichy
L, Freiberger T, Zapletalova P, Soskac V, Ravcukova B and Fajkusova
L: An APEX-based genotyping microarray for the screening of 168
mutations associated with familial hypercholesterolemia.
Atherosclerosis. 216:139–145. 2011.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Lind S, Rystedt E, Eriksson M, Wiklund O,
Angelin B and Eggertsen G: Genetic characterization of Swedish
patients with familial hypercholesterolemia: A heterogeneous
pattern of mutations in the LDL receptor gene. Atherosclerosis.
163:399–407. 2002.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Nauck MS, Köster W, Dörfer K, Eckes J,
Scharnagl H, Gierens H, Nissen H, Nauck MA, Wieland H and März W:
Identification of recurrent and novel mutations in the LDL receptor
gene in German patients with familial hypercholesterolemia. Hum
Mutat. 18:165–166. 2001.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Marduel M, Carrié A, Sassolas A, Devillers
M, Carreau V, Di Filippo M, Erlich D, Abifadel M, Marques-Pinheiro
A, Munnich A, et al: Molecular spectrum of autosomal dominant
hypercholesterolemia in France. Hum Mutat. 31:E1811–E1824.
2010.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Day IN, Whittall RA, O'Dell SD, Haddad L,
Bolla MK, Gudnason V and Humphries SE: Spectrum of LDL receptor
gene mutations in heterozygous familial hypercholesterolemia. Hum
Mutat. 10:116–127. 1997.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Voevoda MI, Kulikov IV, Shakhtshneider EV,
Maksimov VN, Pilipenko IV, Tereschenkov IP, Kobzev VF, Romaschenko
AG and Nikitin YP: The spectrum of mutations in the low-density
lipoprotein receptor Gene in the Russian Population. Genetics.
44:1191–1194. 2008.PubMed/NCBI
|
|
65
|
Benito-Vicente A, Uribe KB, Jebari S,
Galicia-Garcia U, Ostolaza H and Martin C: Validation of LDLr
Activity as a tool to improve genetic diagnosis of familial
hypercholesterolemia: A retrospective on functional
characterization of LDLr variants. Int J Mol Sci.
19(1676)2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Mak YT, Pang CP, Tomlinson B, Zhang J,
Chan YS, Mak TW and Masarei JR: Mutations in the low-density
lipoprotein receptor gene in Chinese familial hypercholesterolemia
patients. Arterioscler Thromb Vasc Biol. 18:1600–1605.
1998.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Hansel B, Carrié A, Brun-Druc N, Leclert
G, Chantepie S, Coiffard AS, Kahn JF, Chapman MJ and Bruckert E:
Premature atherosclerosis is not systematic in phytosterolemic
patients: Severe hypercholesterolemia as a confounding factor in
five subjects. Atherosclerosis. 234:162–168. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Du F, Hui Y, Zhang M, Linton MF, Fazio S
and Fan D: Novel Domain Interaction Regulates Secretion of
Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Protein. J
Biol Chem. 286:43054–43061. 2011.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Kaya E, Kayikçioğlu M, Vardarli AT, Eroğlu
Z, Payzın S and Can L: PCSK 9 gain-of-function mutations (R496W and
D374Y) and clinical cardiovascular characteristics in a cohort of
Turkish patients with familial hypercholesterolemia. Anatol J
Cardiol. 18:266–272. 2017.PubMed/NCBI View Article : Google Scholar
|