1. Introduction
Uterine cervix cancer (UCC) is classed as the fourth
most common type of cancer that occurs uniquely in women, and
constitutes a serious global public health problem (1). With an estimated occurrence of 528,000
new cases in 2012 and an overall incidence rate of 14/100,000
women, UCC is the second most common type of cancer diagnosed and
the third leading cause of cancer-associated death in women in the
least developed countries (2,3). In
Brazil, UCC is third in prevalence, excluding non-melanoma skin
cancer (statistics for which may be incomplete), after breast and
colorectal cancer. The incidence of UCC has remained stable over
the last 5 years, with an estimated average risk of 15.57/100,000
women between 2014 and 2018(4).
Virtually all UCC cases are triggered by persistent
infection of the uterine cervix by human papillomavirus (HPV) high
risk genotypes, particularly HPV16 and HPV18 (5,6).
However, it is known that the virus alone is not sufficient to
cause this malignant disease (7).
Thus, additional factors are necessary for the progression of a
low-grade squamous intraepithelial lesion to a high-grade lesion,
and consequently to invasive cancer (8). Dysregulation of both viral and host
gene expression due to viral DNA integration into the cell's
genome, as well as epigenetic modifications are crucial events in
the carcinogenic process (7). In
addition, high-risk HPV infection can lead to aberrant expression
of oncogenic and tumor suppressor micro RNAs (miRNAs), most of
which have either c-Myc, p53 or E2F transcription factors as
downstream targets, and whose expression can be modulated by the E6
and E7 viral oncoproteins (9).
The unresolved long-term chronic inflammation caused
by HPV creates a microenvironment where complex interactions
involving cytokines, chemokines, free radicals, prostaglandins,
growth factors and enzymes, such as cyclooxygenase and matrix
metalloproteinases (MMPs) may also induce genetic and epigenetic
changes, affecting critical signaling pathways for maintenance of
cellular homeostasis (10).
Several epigenetic changes were identified during
HPV infection in both virus and host cell genomes, including
hypomethylation or hypermethylation of viral DNA and
hypermethylation of host cell tumor suppressor genes, as well as
histone modifications and changes in expression of non-coding RNAs
(ncRNAs) (11). The E6 and E7 viral
oncoproteins interact and/or alter the expression of several
cellular proteins involved in epigenetic regulation, altering the
transcriptional competence of the infected cells caused by the
changes in gene expression, increased activity of histone-modifying
enzymes and chromatin remodeling (11,12). It
has been observed that the loss of control of expression of E6 and
E7 genes during HPV infection is caused by the rupture of the E1
and/or E2 viral genes during integration of the viral DNA into the
host cell genome or due to the hypermethylation of the virus' early
promoter DNA, which is located in the long control region (LCR) of
the viral genome that regulates the expression of these genes
(13).
The LCR is a non-coding sequence of the HPV genome
responsible for regulating the expression of viral genes. The early
and late promoters of these genes, as well as the binding sites of
viral proteins E1 and E2, and several transcription factors of the
host cell, are located in this region of the viral genome. The E2
protein is the major intragenomic regulator of the virus and
modulates the expression of the viral E6 and E7 oncogenes by
binding to the E2BS site located in the LCR. The E2 binding site
has CpG islands with potential for methylation, which results in
the inhibition of its transcriptional regulatory function of the E6
and E7 genes, leading to overexpression of these viral oncogenes
(13,14).
In the present review, recent advances in our
understanding of the role of epigenetic changes in the process of
cervical carcinogenesis induced by high-risk HPV infections in the
initiation, progression and invasion of cervical cancer are
summarized.
2. Methods
The present literature review was performed using
PubMed (National Institutes of Health; ncbi.nlm.nih.gov/pubmed), Scopus (Elsevier; scopus.com/scopus/home.url) and Web of Knowledge
(Thomson Reuters; wok.mimas.ac.uk) electronic databases, and the
following key words were searched: ‘Epigenetics’, ‘Cervical
cancer’, ‘HPV-induced carcinogenesis’, ‘Regulation of genetic
expression in cervical cancer’, ‘Epigenetic changes in cervical
cancer’, ‘DNA Methylation in cervical cancer’, ‘Modifications of
histones in cervical cancer’, and ‘Non-coding RNA in cervical
cancer’. Several hundred articles were found in the surveyed
databases, and only the most relevant ones, published in
high-impact factor journals, and conducted by groups with
recognized knowledge in the area were selected.
3. Epigenetics
Epigenetic modifications are inherited
characteristics that are not caused by changes in the DNA sequence,
as they result from changes in gene expression due to changes in
DNA accessibility or chromatin structure (15). They are reversible modifications in
gene function, involving overexpression or silencing of genes by
mechanisms that do no result in DNA alterations, therefore being
responsible for changes in the phenotype without genotypic
alterations (16,17).
Such changes are caused by methylation or
acetylation of DNA, post-translational modification of histones, or
by the action of ncRNAs, which can be triggered by exogenous and
environmental factors that regulate the differentiation and
development of cells and organs (15). They are normal events of regular
occurrence that can be influenced by several factors including age,
environment, lifestyle, cellular stress and pathological factors
(18-20).
Although epigenetic modifications are a natural
adaptation mechanism to changes in the environmental conditions,
the complex regulation of gene expression promoted by these
modifications can lead to detrimental consequences in the organism
causing an inverse effect to what is expected, resulting in
accumulation of characteristics that diminish its adaptability,
which in-turn leads to pathological conditions, such as cancer
(21). The epigenetic modifications
may serve a critical role in cancer cells, promoting silencing of
tumor suppressor genes, activation of oncogenes and defects to DNA
repair mechanisms (22). Such
alterations may subvert the controlled division of healthy cells
through different molecular pathways, leading to unlimited capacity
for division, genomic instability, metabolic displacement, and
acquisition of characteristics of mesenchymal cells by increased
survival and displacement to distant sites from the original tissue
(23). At least three systems
including DNA methylation, histone modification, expression or
silencing of ncRNA encoding genes are currently considered the
primary systems responsible for initiating and supporting
epigenetic changes (24).
The tumorigenic process involves changes in the
transcriptional pathways of cells that lead to reprogramming with
remodeling of the 3D structures of the genome, which is used by
cancer cells to initiate tumors. This reprogramming is triggered by
hereditary chemical modifications of chromatin that result in the
formation of RNA-protein-DNA complexes that serve as the primary
factors responsible for the dysregulation of gene expression, and
this may be a cause and a consequence of cancer-related epigenetic
alterations (24).
Although UCC is directly related to a persistent
high-risk HPV infection, several epigenetic changes have been
identified in both the viral DNA and the genome of infected cells.
Hypermethylation of the E2 binding site located in the LCR of the
viral genome, hypomethylation of the overall DNA and
hypermethylation of host cell tumor suppressor genes have been
reported. In addition, histone modifications and acetylation, and
changes in ncRNA expression patterns are involved in the
carcinogenic process (11). Some
aspects of these epigenetic mechanisms in the initiation and
progression of UCC are discussed below.
Methylation of DNA
Methylation is the replacement of a hydrogen atom by
a methyl group by means of covalent attachment at the Carbon 5
position, predominantly of the nucleoside cytosine, preceding the
guanine nucleotide (CpG) catalyzed by DNA methyltransferase (DNMT)
enzymes (25). The methylation
levels in the CpG islands of regulatory gene promoters serve as an
epigenetic mechanism used by cells to regulate gene expression
(26). Methylation of the promoter
region of a gene generally results in its silencing, whereas
demethylation leads to an increase in its expression (27). This epigenetic mechanism evolved to
allow regulation of several biological processes aiming to maintain
homeostasis in situations of cellular stress. Thus, when these
mechanisms are deregulated for any reason, it may result in the
development and progression of several diseases, particularly
cancer (28).
DNA methylation in normal cells is involved in
regulating gene expression, including chromatin organization
(29). In contrast, global
hypomethylation of DNA in tumor cells is observed in repetitive
regions and hypermethylation in CpG islands of tumor suppressor
gene promoters, as well as increased maintenance of DNMT1 activity
(30,31).
The carcinogenic process of UCC is related to
several epigenetic mechanisms, including the hypermethylation of
the promoters of regulatory genes, which can result in the
activation of oncogenes and inactivation or loss of function of
tumor suppressor genes, additionally affecting the expression of
HPV genes, a primary risk factor of the disease (Fig. 1) (32).
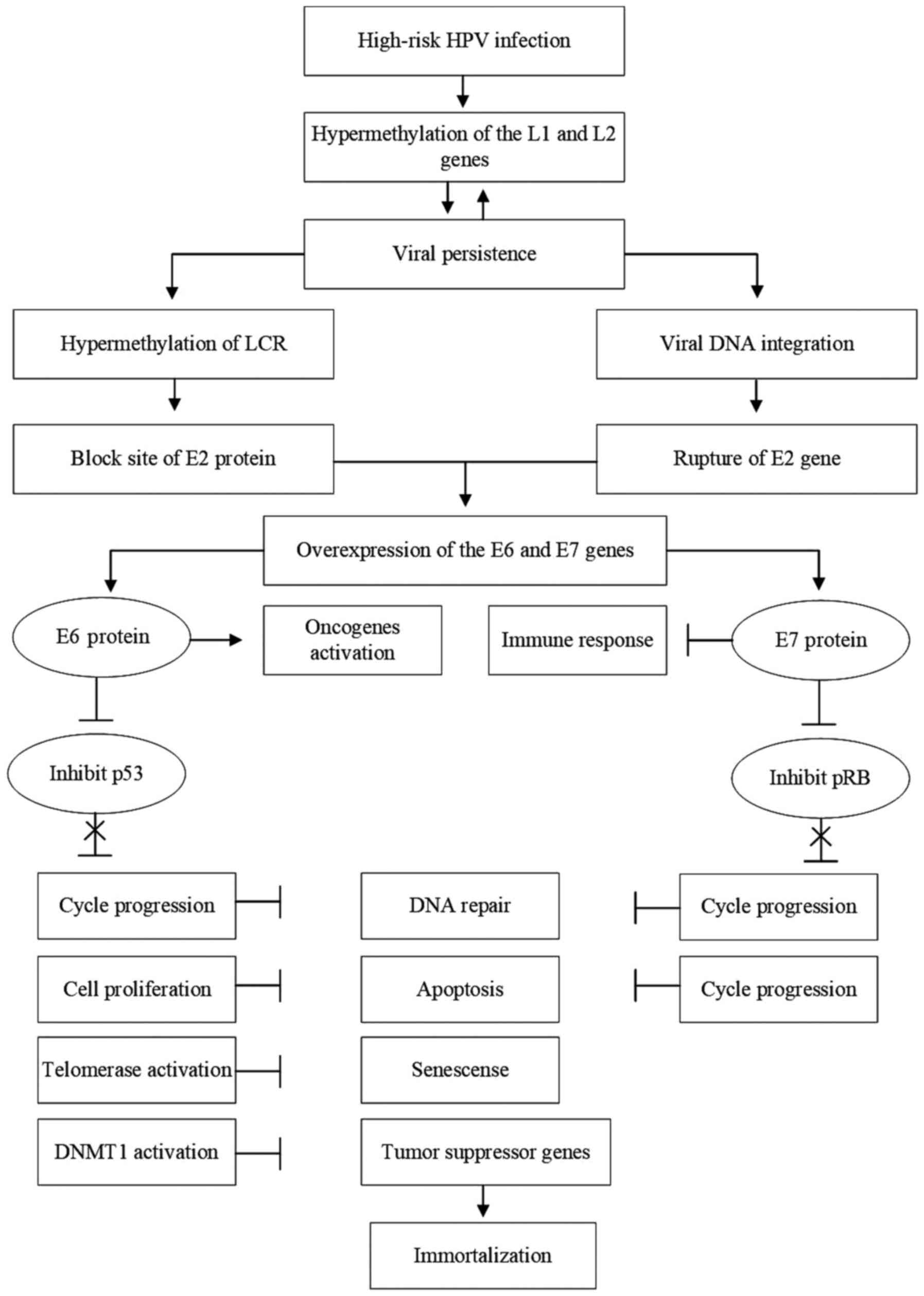 | Figure 1The role of epigenetic changes in the
development of UCC. The process begins with infection of the
epithelium that lines the cervix with high-risk HPVs. During
productive HPV infection, methylation of the L1 and L2 late viral
genes may occur, increasing the risk of viral persistence. Viral
persistence favors the methylation of both viral and cellular DNA,
also increasing the risk of malignant transformation of the
infected cell. Hypermethylation of the LCR of the viral genome
blocks the binding site of the viral E2 protein to this region,
preventing its regulatory function on the expression of the viral
oncogenes E6 and E7, whose products are directly associated with
carcinogenesis. Another crucial event in this process is the
integration of the viral DNA into the cell genome, which results in
the rupture of the viral E2 gene, interrupting the production of
the E2 protein and abolishing its regulatory function on the
expression of the viral genes E6 and E7, leading to overproduction
of viral oncoproteins. E6 protein can act directly by activating
the expression of oncogenes or inducing the degradation of the
tumor suppressor protein p53, resulting in progression of the cell
cycle, preventing DNA repair, increasing cell proliferation, and
inhibiting apoptosis. In addition, E6 activates telomerase,
prevents cell senescence and activates DNA methyltransferase, which
favors the methylation of cellular and viral genes, and promotes
the silencing of tumor suppressor genes. Conversely, E7 can act
directly by inhibiting the host's immune response, interfering with
the presentation of antigens, interferon signaling pathways and
maturation of T lymphocytes, as well as increasing the tolerance to
these T-cells. In addition, E7 induces degradation or inhibition of
the function of the cell's tumor suppressor pRB protein, which
results in cell cycle progression, inhibition of DNA repair
mechanisms and uncontrolled cell proliferation. Thus, the joint
action of viral proteins E6 and E7 leads to the immortalization of
cells infected by HPV, followed by the malignant transformation of
these cells. UCC, uterine cervix cancer; HPV, human papillomavirus;
LCR, long control region; DNMT1, DNA methyltransferase 1. |
However, the role of DNA methylation in silencing
tumor suppressor genes and its implications in the development of
diseases varies according to the ethnic characteristics of the
population. A meta-analysis of 15 studies covering a total of 950
UCC samples and 829 controls showed that methylation of the CDH1
gene promoter which encodes cadherin 1, a protein involved in
adhesion between cells, is associated with an increased risk of UCC
in Caucasian women, but not in Asian women (33). In addition to its role in the
initiation and progression of UCC, DNA methylation status can also
be used as a molecular marker for detection of cervical cancer,
from cervical scrapes and even in the urine. Although the cervical
scrapes showed better results, urine showed a significant increase
in levels of six of the methylation markers assessed, when compared
to the healthy controls, showing that this may be a promising
strategy for the detection of cervical cancer (34).
Cell DNA methylation
A wide range of cellular genes, particularly those
involved in cell cycle regulation, apoptosis, DNA damage repair and
cell adhesion, senescence and survival of cells, including
TP53 and RB1 tumor suppressor genes, are altered in
UCC due to aberrant methylation patterns of their promoters caused
by the action of the oncoproteins encoded by the E6 and E7 genes of
high-risk HPVs (6,35,36). In
HPV-infected cells expressing high levels of E6 and E7 viral
oncoproteins, there is a subversion of cell cycle control and DNA
repair mechanisms, as well as inhibition of senescence and cell
death by apoptosis (37). In
addition, these viral proteins also contribute to immune evasion by
inhibiting interferon-signaling pathways, reducing the ability of
antigen presentation and inducing tolerance of T cells (38). E6 and E7 viral oncoproteins act
together to promote the hypermethylation of cellular genes. E6
promotes degradation of p53 and release of Sp1 transcription
factors, which binds to the DNMT1 gene promoter, activating its
expression. On the other hand, E7 forms a stable complex with pRB,
releasing the transcription factor E2F, which binds to the DNMT1
gene promoter, activating its expression. Both mechanisms lead to
production of DNA methyl transferase enzymes, which promotes the
hypermethylation of CpG islands and the silencing of cellular genes
(6).
The Wnt-β-catenin signaling pathway is involved in
regulating the differentiation, proliferation, migration and
differentiation of cells. Therefore, dysregulation of this pathway
is associated with several types of cancer, including UCC.
High-risk HPVs, through their E6 and E7 oncoproteins, can activate
the Wnt-β-catenin pathway via several mechanisms. E6 and Dvl, both
cell proliferation regulatory proteins, bind to β-catenin and
promote its stabilization by increasing the transcriptional
activity of TCF, which prevents the phosphorylation of β-catenin
and its degradation. E6 can also protect β-catenin from degradation
by binding to the E6PA protein of the infected cell. In addition,
E6 and E7 bind to the catalytic subunit of protein phosphatase 2
(PP2A) to inhibit the phosphorylation of β-catenin by preventing
its degradation and promoting its stabilization in the cytoplasm.
Thus, transformation and carcinogenesis of human keratinocytes
induced by HPV requires activation of the Wnt-β-catenin,
contributing to the proliferation and invasion of tumor cells
(39).
The silencing by hypermethylation of the A-1 gene
promoter of the Adenomatous polyposis coli (APC), whose
product is a negative regulator that controls β-catenin via
interaction with E-cadherin, results in the abnormal accumulation
of β-catenin in cell lines infected with HPV16. Together, β-catenin
and E-cadherin participate in several prominent oncogenic
mechanisms in various types of cancer that are associated with
aberrant activation of Wnt-β. Demethylation of the A-1 gene
promoter results in increased APC expression levels and
reduced β-catenin expression, acting on two transcriptional targets
of the Wnt-β-catenin pathway: Matrix-7 metalloproteins and vascular
endothelial growth factor. This suggests that APC gene silencing by
hypermethylation of its promoter is involved in HPV-induced
carcinogenesis by promoting activation of Wnt-β-catenin (40).
The transcriptional silencing via CpG island
hypermethylation of the KIP1 and TP53 promoter genes
encoding the p27 and p53 proteins, respectively, both involved in
cell cycle regulation, is strongly associated with UCC when
compared with normal tissue (41). A
similar process occurs with the hypermethylation of the phosphatase
and tensin homolog gene promoter, which is also involved in the
regulation of cell cycle progression, resulting in the silencing of
this tumor suppressor gene. Reduced expression of these genes is
correlated with increased proliferation and cell motility via
inactivation of the PI3-kinase-dependent signaling pathway
(42).
The negative regulation by hypermethylation of the
RASSF1 promoter gene, which is involved in the activation of
the signaling pathway, which inhibits cell proliferation and
induces apoptosis, has been shown in numerous studies to increase
the risk of UCC (43). A similar
outcome is observed following silencing of the CDKN2A gene
by hypermethylation of its promoter. The CDKN2A gene encodes
the p16INK4a tumor suppressor protein, which interacts with cyclin
dependent kinases, CDK4 and CDK6. This interaction prevents pRB
phosphorylation with E2F release, leading to cell cycle arrest in
the G1 phase (44). Thus,
CDKN2A silencing by hypermethylation of its promoter results
in the phosphorylation of pRB with the release of E2F, which
in-turn activates gene transcription, in-turn promoting cell cycle
progression resulting in cell immortalization, and thus
contributing to the pathogenesis of UCC (6,45).
Hypermethylation of the CDH1 gene promoter,
is associated with HPV-induced carcinogenesis, as silencing of this
gene increases the risk of worsening cervical lesions. In addition,
the increased methylation density of this gene is associated with
UCC progression and metastasis, and has been suggested as a
possible epigenetic marker that can be used to predict the risk of
disease progression (46).
In a meta-analysis study, four genes were identified
as common targets for aberrant methylation in UCC including the
death-associated kinase protein-1 gene, which activates IFN-γ and
induces apoptosis; the retinoic acid receptor β (RARβ) gene, which
induces vitamin A production and is associated with cell growth and
differentiation; the Wnt-β inhibitory factor gene, whose product
inhibits the Wnt-β-catenin signaling pathway; and the
slit-orientation ligand gene 2, which is associated with cell
migration, all of which are silenced in UCC. The hypermethylation
of the promoters of these four genes occurs early in cervical
carcinogenesis and appears to be specific to UCC (47).
Hypermethylation of promoters EPB4L3 and FAM19A4
cell genes encoding a cell adhesion molecule and a cytokine that
attracts and enhances the phagocytic activity of macrophages,
respectively, is associated with an increased risk of UCC (48). Other cellular genes, such as
ADCYAP1, MAL (a T-cell differentiation protein) and
CADM1 are also silenced in cells derived from UCC, due to
hypermethylation of their promoters, and this condition is
associated with a greater risk of tumor progression (49). Moreover, PAX1 cell genes,
which encode the transcription factor paired box 1, SOX1 for
the sex determining region Y-box 1 and LMX1A for LIM
homeobox transcription factor 1α, all of which are involved in
controlling cell division and differentiation, showed significantly
higher methylation levels of their promoters in UCC cells when
compared to normal cervical tissues (50,51).
Finally, the RARβ gene, which is usually expressed in normal
epithelial tissue, acts as a tumor suppressor when interacting with
its ligand to inhibit cell migration (52).
In some cases, an inverse situation is observed in
which activation of oncogene expression by demethylation of its
promoters contributes to cervical carcinogenesis. For example,
serine/threonine kinase 31 (STK31) gene expression has been
shown to be regulated by demethylation of its promoter and serves a
crucial role in several types of cancer, increasing migration and
invasiveness without altering the proliferation of cancer cells
(53). The STK31 gene
promoter was found to be hypomethylated in the SiHa cell line
positive for high-risk HPV 16, and in the CaSki and HeLa cell lines
positive for high-risk HPV 18, and its expression was increased at
both the mRNA and protein level. In contrast, the STK31
promoter was hypermethylated, which resulted in silenced expression
in the C33A and HT-3 cell lines, both of which are derived from
UCC, but are HPV-negative. It is hypothesized that the E7 oncogene
of high-risk HPVs activates the expression of STK31 by promoting
the demethylation of its promoter, causing overexpression of this
gene, leading to an increase in the invasive capacity of cancer
cells (53). In addition, the
analysis of the methylation patterns of region 1 of the human
telomerase reverse transcriptase (hTERT) gene from 93
positive samples and 15 distinct HPV types revealed differences in
methylation patterns of this gene for different viral genotypes. A
positive association was identified between high-risk HPVs of the
α7 and α9 subtypes, and absence of methylation of the hTERT
gene promoter, indicating that it was being expressed (54).
HPV genome methylation
Studies have shown that epigenetic changes in the
HPV genome, particularly methylation, not only serves an important
role in the replicative cycle of the virus, but also in the
progression of low- and high-grade cervical intraepithelial lesions
in HPV-associated invasive cancer (55,56). It
has been found that methylation of CpG islands of the E2 viral
protein binding site, located in the LCR viral genome and promoters
of the late genes, may abolish the E2 regulatory function or
silence the expression of the L1 and L2 late genes,
thus contributing to HPV-induced carcinogenesis (55). It has also been observed that LCR
methylation is more frequent in UCC than in cervical
intraepithelial neoplasia (CIN). In addition, it was found that the
hypermethylation of CpG islands of LCR of the PV16 increases with
the severity of cervical lesions, being significantly higher in
invasive cancer (57).
High-risk HPV-induced carcinogenesis is primarily
caused by the overexpression of the E7 and E6 viral oncoproteins
after integration of the viral DNA into the host cell genome and
concomitant loss of the E2 viral protein regulatory function, due
to rupture and inactivation of that E2 gene that occurs
during integration. Therefore, the viral genome in most of the UCC
cells is in the integrated form (58). However, in part of the tumor cells,
the viral genome is found in the episomal form, but presents with
methylated promoters. This is due to the fact that the methylation
of the E2BS site of the LCR of the viral genome prevents binding of
the E2 protein to its target sequence, preventing its regulatory
action. This leads to overexpression of E6 and E7
viral oncogenes, creating a condition similar to what occurs after
integration of viral DNA into the cell genome, and also resulting
in carcinogenesis (14,58).
The analysis of LCR methylation levels of three
different high-risk HPV types (16, 18 and 45) in 137 samples of UCC
tissues positive for HPV revealed that HPV16 showed a higher
methylation density in all CpG islands of the LCR. The presence of
intact E1 and E2 was associated with higher levels of methylation
on all CpG islands of both HPV16 and HPV18. E1 and/or
E2 gene rupture was observed more frequently in the genomes
of HPV18 and HPV45, compared with HPV16. E1 gene rupture was
more frequent in HPV16, whereas E2 gene rupture was more
frequent in HPV18. A positive association was found between higher
methylation levels of the LCR and absence of disruption of
E1 or E2 genes for HPV 16 and 18. HPVs 18 and 45 are
highly phylogenetically related, and showed similar levels of
methylation, with the same being observed in relation to E2
gene rupture (13).
Methylation levels of the HPV genome, particularly
in the late L1 and L2 genes, vary during the viral replication
cycle, as well as during the different stages of HPV-associated
cervical lesions (55). Methylation
of the LCR seems to be correlated with persistent infection, as
when it is demethylated, the E1 and E2 proteins bind to the origin
of replication and initiate viral replication (59,60). The
E2 protein acts during the productive cycle of the infection,
activating the duplication of viral DNA and synchronizing the DNA
duplication of the virus with the cell DNA to guarantee the passage
of a copy of the viral genome to the daughter cells during cell
division. In persistent infection, the E2 protein is expressed in
the suppressor isoform (E8^E2), which functions as a negative
regulator of viral replication, since it represses the expression
of early genes, particularly E6 and E7 (58,60).
Both E2 activities are affected by hypermethylation of its binding
site, E2BSs located in the LCR of the viral genome (61).
Evidence indicates that hypermethylation of the L1
late gene of HPV16 is associated with a greater likelihood of
developing persistent infection. A recent study compared the
methylation status of the L1 gene from HPV 16 samples
isolated from women with transient and persistent infection. It was
revealed that the methylation status of the viral genome at
position 5,962, corresponding to the L1 gene was
significantly higher in samples of the virus isolated from women
with persistent infection compared with those isolated from women
with transient infection. This suggests that hypermethylation of
the L1 gene of HPV16 is associated with viral persistence
(62). A similar result has also
been reported in another study, in which a high level of
methylation was found in several CpG islands of the L1 gene
of HPV16 and this condition was shown to be associated with an
increased risk of persistent infection by HPV16(63).
It was found that the degree of L1 gene
methylation of HPV 16, 18 and 52 is associated with the severity of
cervical lesions associated with high-risk HPV genotypes. In
addition, it was found that the methylation of the L1 gene
promoter of HPV 16 and 18 was positively correlated with the degree
of methylation of the host genes, such as PAX1 and
SOX1 (64). PAX1 is a
tumor suppressor that regulates cell division and differentiation,
methylation and silencing of which is strongly associated with the
progression of premalignant lesions to UCC (65). SOX1 is a tumor suppressor that
is related to cell division and differentiation, and it is also
associated with cell growth and invasion in UCC by interfering with
the Wnt-β-catenin pathway. Therefore, the silencing of PAX1
and SOX1 by methylation of their promoters favors tumor
development (66).
A study found that CpG island methylation was
significantly more prevalent in the L1 gene promoter than in
the LCR of the HPV 16, 18 and 51 genomes. The intensity of DNA
methylation in the HPV 16 gene promoter L1 was correlated with the
severity of cervical injuries (56,67).
Methylation was detected in 13 CpG islands of the L1 gene of
HPV 16, with a gradual increase in methylation density proportional
to the severity of the lesions. This suggests an association
between L1 gene methylation and viral persistence,
contributing to the progression of pre-malignant lesions to
cervical cancer (63). It was found
that the methylation density of the CpG islands of the L1
gene promoters of HPV16 and HPV18 increased according to the
severity of lesions. Methylation of the CpG islands at position
5,608 of the L1 gene of HPV16 was associated with all
degrees of cervical intraepithelial lesions, whereas methylation of
CpG islands at position 5,617 was shown to be more strongly
associated with invasive cancer (68).
In a recent case-control study, the degree of
methylation of CpG islands within the late L1 and L2
gene promoters of 12 different types of high-risk HPVs was
evaluated in a total of 30 cases of precancerous lesions and 30
control HPV-infected cases without precancerous lesions. It was
found that the methylation density of L1 and L2 genes
was positively correlated with the presence of grade-3 CIN and
in situ adenocarcinoma for all 12 HPV types tested. The
authors concluded that methylation of HPV DNA is a general
phenomenon that marks the transition from HPV infection to
pre-malignant lesions and proposed the development of a combined
multiple-methylation assay that could be used as a screening test
for HPV-positive women to evaluate the risk of lesion progression
(69).
Histone modifications
In general, transcriptionally active genes are
characterized by promoters with dinucleotides and nucleosomes with
their unmethylated CpG islands. However, DNA methylation does not
only affect gene expression, as epigenetic regulation of gene
expression can also be influenced by histone modifications and
remodeling of nucleosomes (70).
Post-translational modifications of histone tails, such as
acetylation, methylation, phosphorylation, sumoylation and
ubiquitination affects the physical state and the transcriptional
competence of the chromatin. These changes in chromatin are crucial
in regulating cellular processes, including stem cell maintenance,
cell differentiation and cell fate, as well as cell cycle control
and epigenetic heritability of transcription programs (11,71).
Reduction of the levels of histone acetylation
serves an essential role in the neoplastic process through the
epigenetic silencing of tumor suppressor genes. Thus, inhibition of
histone deacetylase enzymes (HDACs) has become a promising approach
in cancer therapy (72).
Transcriptionally active genes generally have high histone
acetylation levels marked by low levels of trimethylation of lysine
residues of certain histones, as well as histone H2B
ubiquitylation. In contrast, transcriptionally inactive genes are
characterized by low acetylation levels and high lysine
trimethylation levels of certain histones and histone H2A
ubiquitylation (73). Histone
modifications and other modifications of chromatin components are
reversible and regulated by the action of enzymes termed ‘writers’,
which are responsible for modifications such as histone
acetyltransferase (HATs), histone methyltransferases, and histone
ubiquitinase, and the ‘erasers’, which can revert those changes,
including that of HDACs, histone demethylases, and histone
deubiquitinases (73,74).
The levels of HDAC10 expression were
significantly lower in patients who had UCC lymph node metastasis
compared with those without metastases. Overexpression of
HDAC10 in tumor-derived cells significantly inhibited cell
motility and metastasis. HDAC10 mechanistically reduces the
histone acetylation of the promoter regions of the MMP2 and
9 genes by suppressing the expression of these genes and preventing
enzyme production, thus maintaining adherence between cells.
Therefore, the reduction in HDAC10 expression enhances
histone acetylation and the transcriptional activation of the
MMP2 and 9 genes, in-turn promoting cell mobility, which
favors invasion and metastasis of cancer cells (75).
The regulation of HPV gene expression is strongly
influenced by histone modifications caused by both methylation and
acetylation, but differences are observed in relation to the
position of the lysine residues, which is acetylated according to
the presentation form of the viral genome in the host cells.
Important changes in histones, such as methylation and acetylation
at positions H3 lysine 27, H3 lysine 9 and H4 lysine 20 contribute
significantly to the regulation of HPV gene expression and an
increase in the neoplastic progression process as the cell
phenotypically progresses from a healthy state to cancerous during
carcinogenesis induced by both the episomal form and the integrated
form of the virus. However, trimethylation markers of H3 lysine 27
and trimethylation H3 lysine 9 decrease with neoplastic progression
in carcinogenesis mediated by the integrated form of the virus
(11,76,77). In
normal cells, the balance between HDACs and HATs means that cell
death and uncontrolled proliferation are kept under control.
However, in the case of UCC, which is mediated by HPV, the presence
of the E6 and E7 viral oncoproteins upsets this balance between
HDACs and HATS, resulting in uncontrolled cell growth and
proliferation of cancer cells (6,78).
In HPV-induced carcinogenesis, several regulatory
mechanisms for the transcription of cellular and viral genes are
controlled by histone modifications (11). Amongst the major cellular genes
involved in this process, the TP53 and RB tumor
suppressor products (p53 and pRB proteins) are the primary targets
of the E6 and E7 viral oncoproteins, respectively (79). Both tumor suppressors genes target a
wide range of genes and cellular mechanisms involved in multiple
biological processes including cell cycle arrest, DNA repair,
apoptosis, metabolism, autophagy and feedback mechanisms (80,81). It
has been shown that the high-risk HPV E6 protein association with
the Myc transcription factor of the cell triggers the
transactivation of the hTERT gene promoter by modulating
histone modifications through phosphorylation, thereby resulting in
increased production of the telomerase enzyme of the infected cell,
which contributes to immortalization, thus increasing the risk of
developing HPV-associated cancer (82).
Analysis of tumor suppressor genes, such as
RARβ2, E-cadherin and β-catenin in UCC tissues showed that
their promoters are deacetylated and that lack of acetylation
causes reduced or absent expression of these three genes, and this
favors the development of tumor metastasis. Treatment with HDAC
inhibitors, such as all-trans retinoic acid (ATRA) combined with
suberoylanilide-hydroxamic acid (SAHA) increased the enrichment of
acetylated histones in the promoter region of the genes. The
agonists of RARβ2 and valproic acid (VPA) significantly restored
expression of RARβ2 via epigenetic modulation. The VPA and ATRA
combination showed additional antitumor effects, reactivating
expression of RARβ2, E-cadherin, P21CIP1 and P53, and reducing the
expression levels of the STAT3 gene, which activates the
transcription of genes that promote cell proliferation and tumor
cell survival. These results suggest that treatment with HDAC
inhibitors and RARβ2 agonists may represent a novel approach for
treating UCC (72).
Histone modifications are unevenly distributed
throughout the HPV16 genome in both UCC cells and keratinocytes
immortalized by HPV16. For example, H3K36me3 and H3K9Ac, which are
the most frequent modifications in cellular genes, are more common
in the early region of HPV16, whereas the H3K9me3, H4K20me3,
H2BK5me1 and H4K16Ac modifications are more frequently observed in
the late region. In addition, a region harboring the early
polyadenylation signal of the HPV16, pAE, exhibited high levels of
histone H3 acetylation. Treatment with HDAC inhibitors increased
the expression of early and late HPV16 mRNAs by 2-8x in cancer
cells and immortalized keratinocytes, with a simultaneous increase
of acetylated histone levels in both the host cell DNA and in the
HPV16 genome (83).
Analysis of HDAC3 expression in specimens of normal
cervical tissue, moderate (grade 2) and severe (grade 3) CIN and
UCC showed that the expression of HDAC3 was significantly higher in
the cancerous tissues compared with those of normal tissues, or
CIN2 and CIN3. This suggests HDAC3 may serve an important role in
the course of UCC carcinogenesis (84).
The E6 protein of high-risk HPVs can degrade p53,
activate telomerase and stimulate the expression of several cell
oncogenes (10). Evidence shows that
E6 of the HPV16 physically interacts with histone H3K4 demethylase
KDM5C, promoting its E6AP proteasomal degradation in an E3
ligase-dependent manner (85). CaSki
cells, cancer cells positive for HPV16, exhibit lower KDM5C levels
than cancer cells that are negative for HPV. It has been shown that
the CaSki cells contain enhancers in super-EGFR and the
c-MET oncogene, and that KTM5C overexpression reduces the
effects of these super-enhancers and expression of these oncogenes.
It is hypothesized that this phenomenon is due to modulation of
H3K4me3 and H3K4me1 dynamics, as well as decreased transcription of
the super-enhancers since deletion of KDM5C or E6 of the HPV16
activates these two elements. These results suggest that epigenetic
activation of the E6-mediated cell genome results in the expression
of important oncogenes, such as EGFR and c-MET (85).
4. The role of ncRNAs in HPV-induced
UCC
NcRNAs are single-stranded RNA transcripts that, in
general, do not encode proteins, although certain transcripts have
been shown to possess protein or peptide-coding potential. NcRNAs
are divided into three classes: miRNAs, long non-coding RNAs
(lncRNAs), and circular non-coding RNAs (circRNAs). These
transcripts are emerging as major players in tumorigenesis, due to
advances in biotechnology and high-throughput sequencing that have
enabled functional studies of these transcripts, providing a novel
perspective in the understanding and potential treatment of cancer
(86). The ncRNAs can be transported
via vesicles called exosomes released by almost all types of cells,
and can act as transport vehicles for molecules, including viral
proteins and genetic material, such as ncRNAs, which can affect
distant receptor cells, triggering inflammatory processes (87).
Regarding UCC, the process of carcinogenesis is
triggered by persistent infection with high-risk HPV and occurs
through a gradual progression from precursor lesions to invasive
cancer (88). Evidence obtained from
studies performed on tumor tissues and tumor-derived cell lines
show that the aberrant expression of ncRNAs serves critical roles
in the onset and progression of the disease (89). They can affect signaling pathways,
such as E6-p53, E7-pRb, PI3K-Akt, Notch and Wnt-β-catenin, amongst
others. Thus, ncRNAs can serve as biomarkers or therapeutic
targets, and may possess value for use in clinical practice
(90,91).
Tumor cells develop epigenetic mechanisms that
allows them to acquire novel capabilities, such as resistance to
apoptosis, increased proliferation, immune modulation, migration,
survival, vascularization and invasion through the deregulation of
cell signaling pathways, thereby creating advantageous conditions
for cancer development (92). One of
the bases of these mechanisms is the expression of miRNAs, which
regulates the expression of genes at both the mRNA and protein
levels, degrading target mRNA and/or silencing their translation.
Several deregulated miRNA encoding genes are involved in the
initiation, progression and metastasis of various types of tumors
(93,94).
miRNAs
The process of tumorigenesis, including cervical
carcinogenesis induced by high-risk HPVs, can be influenced by
positive or negative regulation of both cellular and viral genes
mediated by miRNAs (95). The
increased expression of certain miRNAs serves a critical role in
the initiation and progression of UCC, since they positively
regulate the proliferation, mobility and invasiveness of cancer
cells, whilst inhibiting apoptosis and cell adhesion. These
transcripts are involved in the inactivation of tumor suppressor
genes or in the activation of cellular or HPV oncogenes, with
particular activity on transcription factors involved in the
expression of target genes. These miRNAs are upregulated in UCC and
in cell lines derived from UCC, due to the action of viral
oncoproteins (96).
In cells infected with high-risk HPVs, miRNAs
regulated by tumor suppressor genes, particularly TP53 and RB, have
protective functions against UCC, and act to control the cell
cycle, repair to DNA damage, senescence and apoptosis of the
infected cells. Thus, certain miRNAs, including miR-23b, miR-34a,
miR-107, miR143 and miR-206, expression of which is increased by
the tumor suppressor cellular protein p53, are downregulated in
UCC, due to the degradation of this protein by the action of the
viral oncoprotein E6 (97-99).
On the other hand, miR-15 and miR-16, whose expression is activated
by cellular protein pRB, is also downregulated in UCC due to the
degradation of this tumor suppressor protein by the action of the
E7 viral oncoprotein (100,101). The negative regulation of these
miRNAs by both mechanisms contributes to tumor initiation and
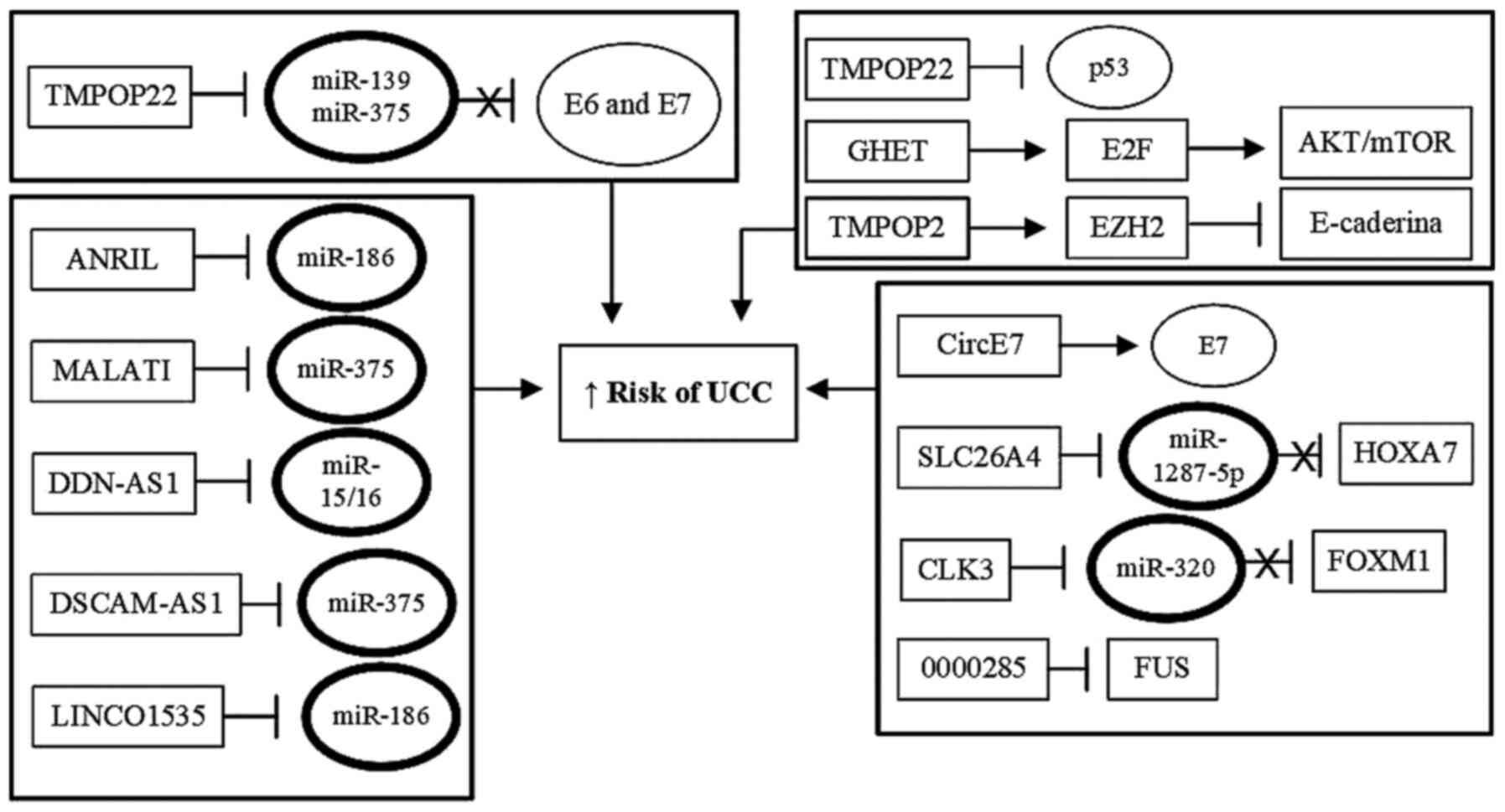
progression (Table I; Fig. 2).
 | Figure 2Role of HPV oncoproteins in
regulation of miRNAs. Viral oncoproteins E6 and E7 may also serve a
role in cervical carcinogenesis by regulating the expression of
miRNAs that function as tumor suppressors, by inhibiting the
functions of tumor suppressor cell proteins, such as p53 and pRB.
By abolishing the functions of the cellular proteins p53 and pRB,
viral proteins E6 and E7 prevent these two cellular proteins from
activating the expression of protective miRNAs against UCC, thus
preventing inhibition of the transcription of cellular oncogenes
associated with tumor initiation and progression. In addition, E6
and E7, acting either alone or together, can also directly activate
the expression of tumor-inducing miRNAs which increases expression
of oncogenes or inhibits the expression of tumor suppressor genes.
This results in increased proliferation, immortalization,
progression and invasion of tumor cells. UCC, uterine cervix
cancer; HPV, human papillomavirus; miRNA/miR, microRNA. |
 | Table IDownregulated tumor suppressor miRNAs
in UCC. |
Table I
Downregulated tumor suppressor miRNAs
in UCC.
| First author,
year | miRNA | Activator | Target gene | Physiolgocial
function | (Refs.) |
|---|
| Yeung et al,
2017 | 23b | p53 | SIX1 | Affects AKT/mTOR
signaling pathway as well as progress of epithelial-mesenchymal
transition. | (97) |
| Chen et al,
2017 | 34a | p53 | WNT-β | Inhibition of the
exchange of E-P cadherin, interfering in the WNT-β-catenin
pathway. | (98) |
| Dong et al,
2017 | 107 | p53 | MCL1 | Suppression of MCL1
expression, affecting ATR/Chk1 signaling path. | (99) |
| | | | MSI-2 | Inhibition of MSI-2
expression, which product promotes cell cycle progression. | |
| Dong et al,
2017 | 143 | p53 | GOLM1 | Inhibition of GOLM1
gene expression, responsible for encoding protein 1 in the Golgi
membrane. | (99) |
| | | | MSI-2 | Inhibition of MSI-2
expression. | |
| Chen et al,
2017 | 206 | p53 | G6PD | Suppression of
glucose-6-phosphate dehydrogenase gene expression. | (98) |
| Ofir et al,
2011 | 15a-5p | pRB | YAP1 | Inhibits
YAP1 oncogene expression, increases apoptosis, and reduces
tumor cell migration and invasion. | (100) |
| Ofir et al,
2011 | 16-1 | pRB | CCNE1 |
Post-transcriptionally suppression of the
CCNE1 gene expression, which product promotes cell cycle
progression. | (100) |
| Chen et al,
2017 | 34a | pRB | E2F3 | Repression of
BCL2 gene expression, inducing apoptosis. | (98) |
| Sannigrahi et
al, 2017 | 139-3p | - | E6 and E7
HPV | Inhibition of HPV16
E6 and E7 oncogenes expression. | (103) |
| Jiang et al,
2016 | 218 | - | SFMFBT1 | Inhibition of
SFMFBT1 gene expression, which induces
epithelial-mesenchymal transition and increases migration and
invasion of tumor cells. | (104) |
| | | | DCUN1D1 | Inhibition of
DCUN1D1 gene expression, the product of which increases
proliferation, migration and invasion, but does not induce
epithelial-mesenchymal transition. | |
The HPV-16 E6 oncoprotein, by degrading p53 in
infected cells, can cause aberrant methylation and contribute to
the development of UCC, as E6 positively regulates the DNMT1
gene. It was seen that miR-23b, which has tumor suppressor action,
is silenced in cells derived from UCC due to the methylation of its
promoter. miR-23b targets the c-MET oncogene, silencing the
gene that encodes this transcript increases c-MET expression, which
favors tumor initiation and progression (97). miR-34a and miR-206 acts as a tumor
suppressor in a p53-dependent manner, repressing the expression of
the BCL2 gene, leading to the induction of apoptosis and the
suppression of the c-MET oncogene and preventing its
transforming action. However, in the high-risk HPV-infected cells,
the E6 protein degrades p53 and reverses the protective effects of
these two miRNAs, which results in the inhibition of apoptosis of
the infected cell, in addition to activating the c-MET
oncogene. The positive regulation of Bcl2 and c-MET promotes the
progression of precancerous cervical lesions to UCC (98).
The Musashi 2 RNA-binding protein encoded by the
MSI-2 gene is highly expressed in UCC and presents an
inverse relationship with the expression of miR-107 and miR-143.
The Musashi 2 protein binds to the oncogene c-FOS mRNA,
increasing the expression of the c-FOS protein, which is associated
with increased proliferation, invasiveness of tumor cells, and
lower patient survival. miR-107 and miR-143 directly target the
MSI-2 gene, whose expression can be inhibited by the
presence of the functional p53 protein. However, the degradation of
p53 by the viral E6 protein neutralizes the tumor suppressing
action of these miRNAs (99). The
low expression of miR-143 is also associated with increased
expression of the GOLM1 gene encoding Golgi phosphoprotein 2, and
this condition results in increased proliferation, migration and
invasion of tumor cells. This is due to the absence of the p53
function, which prevents the tumor suppressor action of miR-143 on
GOLM1 expression (102).
Under normal conditions, miR-139-3p acts by
inhibiting the expression of HPV16 oncogenes, whose products, the
oncoproteins E6 and E7, target the cellular proteins p16, p21 and
p53. The silencing of these viral genes by miR-139-3p maintains
cell cycle arrest in the G2-M phase, inhibits proliferation and
migration, and induces apoptosis of cells infected by HPV16.
Increased DNA methylation of the promotor of miR-139-3p harboring
the gene PDE2A was observed in HPV-16-positive tissues and cancer
cell-lines (103).
miR-218 is downregulated in UCC, presenting an
inverse correlation between the expression of miR-218 and
expression of the DCUN1D1 gene that encodes the cancer-related
DCUN1D1 protein and the SFMFBT1 gene, both with a
tumorigenic role. The increased expression of SFMBT1 induced
epithelial-mesenchymal transition and increased the migration and
invasiveness of cancer cells, while the increased expression of
DCUN1D1 increased the proliferation, migration and invasiveness of
these cells, but did not induce epithelial-mesenchymal transition.
The HPV16 E6 protein inhibited the expression of miR-218 in UCC,
and restoration of miR-218 reversed the effects of E6 in activation
the expression of SFMBT1 and DCUN1D1 (104). In another study, it was found that
downregulation of miR-218 results in overexpression of the
IDO1 gene, which encodes the enzyme indoleamine
2,3-dioxigenase 1, which is associated with inhibition of Caspase-3
and apoptosis. Furthermore, this enzyme activates the JAK2/STAT3
signaling pathway, which leads to increased expression of immune
factors, such as TGF-β, VEGF, IL-6, PGE2 and COX-2, increasing the
viability of tumor cells, which favors tumor progression (105).
The miR-15/16 family members, including miR15a,
miR-15b, miR-16-1 and miR-16-2 act as tumor suppressors, promoting
the arrest of the cell cycle via a pRB-dependent mechanism, which
results in inhibition of the expression of cyclins A and E. These
cyclins are necessary for the activation of E2F, a key
transcription factor that activates the genes that promote cell
cycle progression (100). The low
expression of miR-15a-5p was observed in cervical cancer tumor
tissues with distant metastases and in cervical cancer cell lines.
Upregulation of miR-15a-5p suppressed the viability, migration and
invasion of tumoral cells. The oncogene yes-associated protein 1
was confirmed to be a target of this miRNA (101). However, the E7 protein of the
high-risk HPVs neutralizes the protective functions of these
transcripts, preventing their expression through the degradation of
cellular pRB. In addition, E7 is also capable of directly inducing
the expression of cyclins A and E, thus promoting the progression
of the cell cycle (106,107). miR-15 and miR-16 also possess tumor
suppressor functions in UCC, as they induce the silencing of the
TCF3 gene that encodes a transcription factor involved in
the activation of proliferation, migration and invasion of the
cancerous cells in this tumor (108).
Certain miRNAs are upregulated in UCC, acting by
silencing tumor suppressor genes or activating oncogenes (Table II). miR-20b is upregulated in cells
derived from UCC through the action of the HPV E6 oncoprotein,
whereas the tissue inhibitor of metalloproteinase 2 (TIMP-2), which
is a metastasis suppressor, has been identified as a novel target
of miR-20b and showed an inverse correlation with this miRNA.
Overexpression of miR-20b resulted in morphological changes in the
cells and induced epithelial-mesenchymal transition. The treatment
of cancer cells with miR-20b inhibitors decreased the migration and
invasion of these cells. TIMP-2 has been shown to be
regulated in an E6-dependent manner. This suggests that miR-20b is
activated by the viral protein E6, and inhibits TIMP-2
expression thus increasing the invasiveness of cancer cells
(109).
| Table IIUpregulated tumor-inducing miRNAs in
the UCC. |
Table II
Upregulated tumor-inducing miRNAs in
the UCC.
| First author,
year | miRNA | Activator | Target gene | Function in cells
derived from the UCC | (Refs.) |
|---|
| Cheng et al,
2017 | 20b | E6-HPV | TIMP2 | Induces production
of matrix metalloproteinases and increases migration and invasion
of tumor cells. | (109) |
| Kong et al,
2015; Cai et al, 2018 | 21-5p | E7-HPV | VHL | Inactivation of VHL
tumor suppressor gene, promoting proliferation and metastasis of
tumor cells. | (110,111) |
| Liu et al,
2016 | 27b | E7-HPV | PLK2 | Inhibition of
polo-like kinase-2 tumor suppressor gene, increasing proliferation
and inhibiting apoptosis. | (113) |
| Ding et al,
2014; Park et al, 2019; Coimbra et al, 2016 | 203 | E7-HPV | TP63 | Activation of TP63
expression, which encodes an isoform of this protein, ΔNp63 without
the transactivation domain, which exhibits tumor-inducing
function. | (114-116) |
| Ding et al,
2014 | 323 | E7-HPV | APPL1 | Activation of APPL1
gene expression, which encodes an adapter protein, both with
tumorigenic properties. | (114) |
| Cheng et al,
2016 | 106b | - | DAB2 | Inhibition of
DAB2 gene expression, increasing the potential of TGF-β1 to
induce cancer cell metastasis. | (120) |
| Natalia et
al, 2018 | 125a-5p | - | MARK1 | Inhibition of
MARK1 gene expression and phosphorylation of associated
proteins favoring cell migration. | (121) |
| Xu et al,
2015 | 135b | - | FOXO1 | Silencing of
FOXO1 gene, which encodes a transcription factor that
controls the progression of the cell cycle. | (122) |
| Li et al,
2018 | 141-3p | - | FOXA2 | Silencing of the
FOXA2 gene, which controls proliferation, epithelial-mesenchymal
transition, tumor growth, and metastatic invasion. | (123) |
| Zhu and Han
2019 | 150-5p | - | SRCIN1 | Promotes cell
proliferation and epithelial-mesenchymal transition by silencing
SRCIN1, an inhibitor of these processes. | (124) |
| Li et al,
2019 | 155-5p | - |
TP53INP1 | Inhibition of
TP53INP1, a tumor suppressor gene, which controls
proliferation, migration and invasion of tumor cells. | (125) |
| Yang et al,
2018 | 181a-5p | - | INPP5A | Inhibition of
apoptosis and increases proliferation and invasion of cancer cells
by silencing the INPP5A gene that controls these
processes. | (126) |
| Farzanehpour et
al, 2019 | 192 | - | CDH1 | Increases
expression of ZEB1 and ZEB2, which inhibits the expression of CDH1
gene, the gene encoding E-cadherin. | (127) |
| Hou et al,
2014 | 196a | - | FOXO1 and
p27Kip1 | Silencing of
FOXO1 and p27Kip1 genes, the products of which act as
inhibitors of the PI3K/Akt pathway, thus increasing cell
proliferation. | (128) |
| Chu et al,
2014 | 590-5p | - | CHL1 | Silencing of the
CHL1 gene reducing the production of a molecule, increasing
mobility, and invasion of tumor cells. | (131) |
The E7 protein of high-risk HPVs also serves an
oncogenic role by directly activating the expression of
tumor-inducing miRNAs, such as miR-21-5p, which induces
angiogenesis and tumor growth in UCC, as well as increasing
invasion and metastasis of tumor cells (110). The von Hippel-Lindau tumor
suppressor gene (VHL) has been identified as a direct target of
miR-21-5p, and VHL knockout results in abolishment of the
inhibitory function of miR-21-5p, and increases proliferation and
metastasis of UCC derived cells. This shows that miR-21-5p acts as
a tumor promoter in UCC through negative regulation of the
expression of VHL (111). In
addition, miR-21-5p positively regulates the expression of TNF-α,
which promotes tumorigenesis (112). E7 also upregulates miR-27b, which
inhibits the expression of the tumor suppressor gene polo-type
kinase-2, resulting in increased proliferation and inhibition of
apoptosis of cancer cells (113).
E7 is able to positively regulate the expression of
miR-203 and miR-323, which exhibit tumor-inducing functions.
miR-203 increases the expression of the TP63 gene, which encodes
p63, and miR-323 increases the expression of the APPL1 gene that
encodes an adapter protein, and both proteins posses tumorigenic
properties (114). p63 is a
transcription factor and a member of the same family as p53. Two
isoforms of this protein have been identified, one with a normal
transactivation domain and another, ΔNp63 with a truncated
transactivation domain, and thus has an opposite function to that
of p53, showing carcinogenic activity (115). miR-203 and ΔNp63 showed higher
levels of expression in cervical tissues obtained from UCC compared
with premalignant cervical lesions. This suggests that miR-203 acts
as an oncogene in UCC by activating ΔNp63 expression (116). miR-323 also acts as an oncogene in
UCC by activating the transcription of the APPL1 gene, which
encodes an adapter protein associated with tumor cell proliferation
and migration (114).
The expression levels of miR-203 and miR-323 are
upregulated in cells derived from UCC and are positively correlated
with the expression of the mRNA levels of oncoprotein E7 of HPV16.
The positive correlation between E7 and miR-323 expression is more
evident when the virus is in the episomal form, whereas for miR-203
this correlation is more evident when the virus is in its
integrated form. This indicates that the HPV16 E7 oncoprotein
increases the expression of miR-203 and miR-323, which contribute
to HPV 16-induced carcinogenesis. On the other hand, the expression
of miR-181c is downregulated in HPV-16 positive UCC only when the
virus is in its episomal form, and it is negatively correlated with
the expression of the CKS1B gene, which regulates G2/M
transition (117). The suppression
of CKS1B promotes dysregulation of the cell cycle and
prevents the repair of DNA damage, which results in the
accumulation of mutations generating genomic instability and
contributes to the transformation of the cell and acquisition of a
malignant phenotype (118). A
summary of the primary miRNAs regulated by the viral oncoproteins
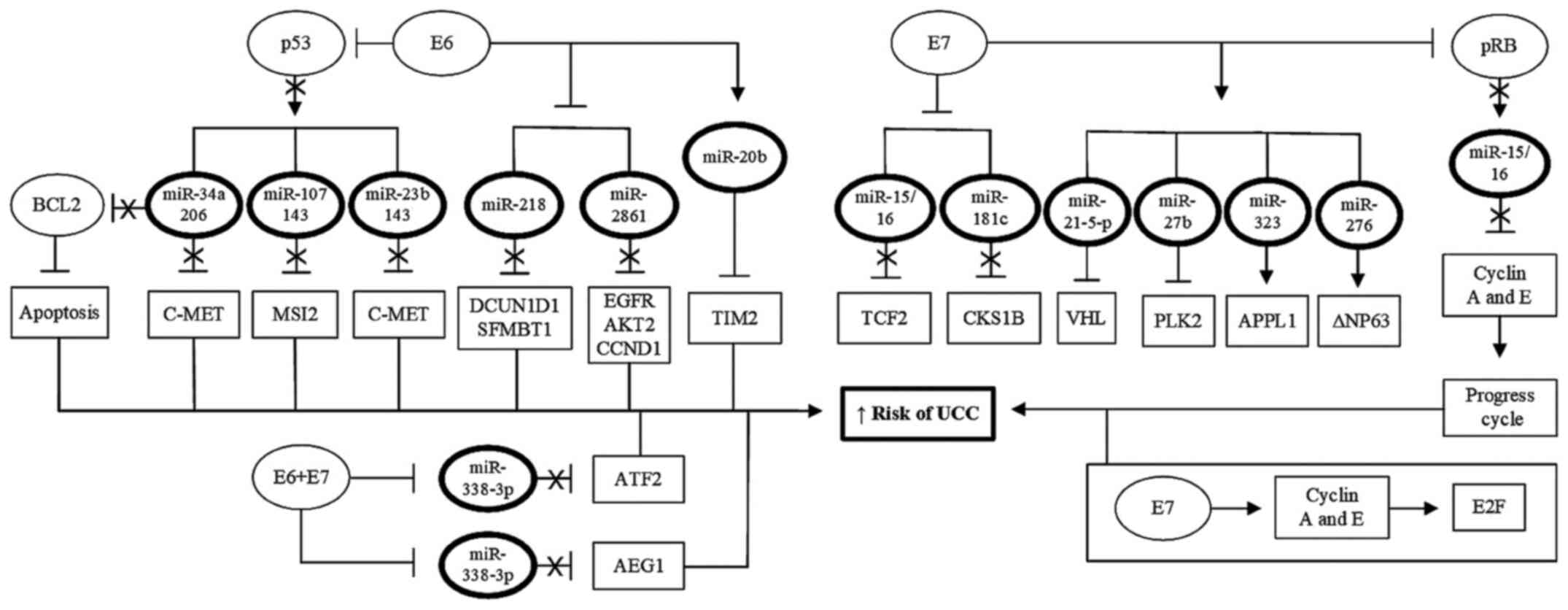
and their respective mechanisms of action is presented in Fig. 2.
The group of miRNAs with increased expression in
cells derived from UCC are associated with a greater risk of
disease initiation and progression, and are considered UCC-inducing
miRNAs. Bioinformatics analysis revealed that miR-106b-5p could
modulate the expression of GSK3B, VEGFA and PTK2
genes, all of which have an important role in the PI3K-Akt
signaling pathway (119). In
addition, miR-106b increases migration of cancer cells by
inhibiting the expression of the DAB2 gene, resulting in
TGF-β1-mediated induction of metastasis (120).
The expression of miR-125a-5p is upregulated in
cells derived from UCC, whereas the protein expression levels of
microtubule-1 affinity regulating protein kinase (MARK1) were
decreased. The UCC-derived HeLa and C-33A cell lines exhibited
increased migration after transfection with miR-125a-5p mimics and
the migration of these cells was also increased by inhibiting the
expression of the MARK1 gene. These results show that
miR-125a-5p acts as a tumor inducer in UCC, targeting the
MARK1 gene, expression of which is inhibited by miR-125a-5p,
thus favoring the migration of tumor cells (121).
miR-135b functions by silencing the tumor
suppressor gene FOXO1, which encodes a transcription factor that
controls the progression of the cell cycle. This condition favors
the proliferation of cancer cells (122). miR-141-3p is associated with
proliferation, epithelial-mesenchymal transition, tumor growth and
invasion with lymph node metastases. miR-135b functions by
targeting and silencing the tumor suppressor gene FOXA2,
whose product is a transcription factor that controls these
cellular processes (123).
miR-150-5p is upregulated in cells derived from UCC, and is
negatively correlated with the expression of the gene encoding SRC
kinase signaling inhibitor 1 (SRCIN1). Overexpression of
SRCIN1 inhibits proliferation and epithelial-mesenchymal
transition of cancer cells triggered by miR-150-5p mimics, and
increases apoptosis of cervical carcinoma cells. These results show
that miR-150-5p promotes cell proliferation and
epithelial-mesenchymal transition through silencing SRCIN1
(124).
miR-155-5p is upregulated UCC tissues compared with
normal tissues, and is inversely correlated with the expression of
the tumor suppressor TP53INP1. Transfection of miR-155-5p
inhibitors decreased proliferation, migration and invasion of
cancer cells in vitro, whereas miR-155-5p mimics had the
opposite effect. Knockdown of TP53INP1 mimicked the effects
of miR-155-5p on the activation of proliferation, migration and
invasion of tumor cells, whereas overexpression of TP53INP1
reversed these effects. These results show that miR-155-5p
functions as an oncogene in UCC by inhibiting the expression of the
tumor suppressor TP53INP1 (125).
miR-181a-5p inhibits apoptosis and increases
proliferation and invasion of cancer cells by negatively regulating
the INPP5A gene that encodes the enzyme inositol
polyphosphate-5-phosphatase A, which is involved in the activation
of several cellular processes (126). High levels of miR-192 in the serum
and tissues of patients with HPV positive UCC HPV has been
suggested as a possible diagnostic biomarker. miR-192 functions by
binding to the inhibitors of transcription factors of E-cadherin,
ZEB1 and ZEB2, promoting the activation of these transcription
factors, thus reducing the expression of the CDH1 gene,
which encodes E-cadherin. The reduction in E-cadherin expression
decreases the adhesion between cells, favoring cancer cell mobility
and the formation of metastases (127).
Expression levels of miR-196a are significantly
increased in tissues and cell lines derived from UCC compared with
the corresponding normal tissue. The positive regulation of
miR-196a is associated with advanced stage tumors and low overall
survival rates of patients. Positive regulation of miR-196a
increased G1/S phase transition and the proliferative capacity of
cancer cells, whereas forced suppression of miR-196a had the
opposite effect. This transcript has been shown to act as a tumor
inducer in UCC by inhibiting the expression of the FOXO1 and
p27Kip1 genes, whose products inhibit the PI3K/Akt signaling
pathway. Furthermore, the negative regulation of these
transcription factors by miR-196a leads to the activation of this
pathway, increasing cell proliferation and favoring the development
of the tumor (128). However, it
was shown that the gene encoding miR-196a-1 exhibits higher levels
of methylation in cells derived from UCC, compared with
premalignant lesions. Treatment with a demethylating agent
reactivated the expression of miR-196a in SiHa, HeLa and CaSki
cells, all of which are derived from UCC. In addition, expression
levels of miR-196a-1 were negatively correlated with methylation
levels in clinical samples. It has been shown that miR-196a-1
targets the AT-Hook 1 gene of the High Mobility Group, which
encodes a protein capable of modulating transcription by altering
the chromatin architecture. This suggests that the silencing of
miR-196a-1 gene by methylation from its promoter increases
HMGB1 expression, and may contribute to carcinogenesis in
UCC (129).
miR-466 acts as an oncogene, possibly serving a
role in increasing the expression of viral oncogenes.
Bioinformatics analysis showed that this transcript was homologous
to the LCR of the genomes of HPVs 16 and 18. High levels of miR-466
expression are strongly correlated with the progression and
invasive capacity of cancer cells, and is associated with lymph
node metastases and reduced patient survival (130). miR-590-5p also acts to promote
cervical carcinogenesis by targeting the CHL1 gene, which
encodes an adhesion molecule. Negative regulation of CHL1 by
miR-590-5p results in an increase in the proliferative and
migratory capacity of cancer cells, and inhibits differentiation
and apoptosis of these cells (131). A summary of the primary miRNAs with
tumor-inducing functions and their likely mechanisms of action are
presented in Fig. 3.
 | Figure 3Role of miRNAs in induction of
cervical carcinogenesis. Certain miRNAs possess tumor-promoting
functions in UCC by increasing the expression of both HPV and host
cell oncogenes, or inhibiting the expression of tumor suppressor
genes. In cells derived from UCC, expression of these miRNAs is
upregulated, together with the viral and cellular oncogenes
controlled by these transcripts, whereas the target tumor
suppressor genes of these miRNAs is downregulated in these cells.
This positive regulation of viral or cellular oncogenes, and the
inhibition of the expression of tumor suppressor genes, results in
increased proliferation, immortalization, progression and invasion
of tumor cells. UCC, uterine cervix cancer; HPV, human
papillomavirus; miRNA/miR, microRNA. |
The expression of certain miRNAs are reduced in UCC
cells, suggesting that these transcripts under normal conditions
exert protective functions against the tumor and act by negatively
regulating the expression of oncogenes or by increasing the
expression of tumor-suppressor genes. However, this function can be
abolished by mechanisms triggered by viral oncoproteins (132). MiRNAs with tumor-suppressive
function target cellular genes, particularly transcription factors
involved in regulation of the cell cycle, proliferation and
invasion of cancer cells, which are downregulated in UCC-derived
cells (133,134). A summary of the primary miRNAs that
possess tumor-suppressive functions in cervical carcinogenesis is
presented in Fig. 4.
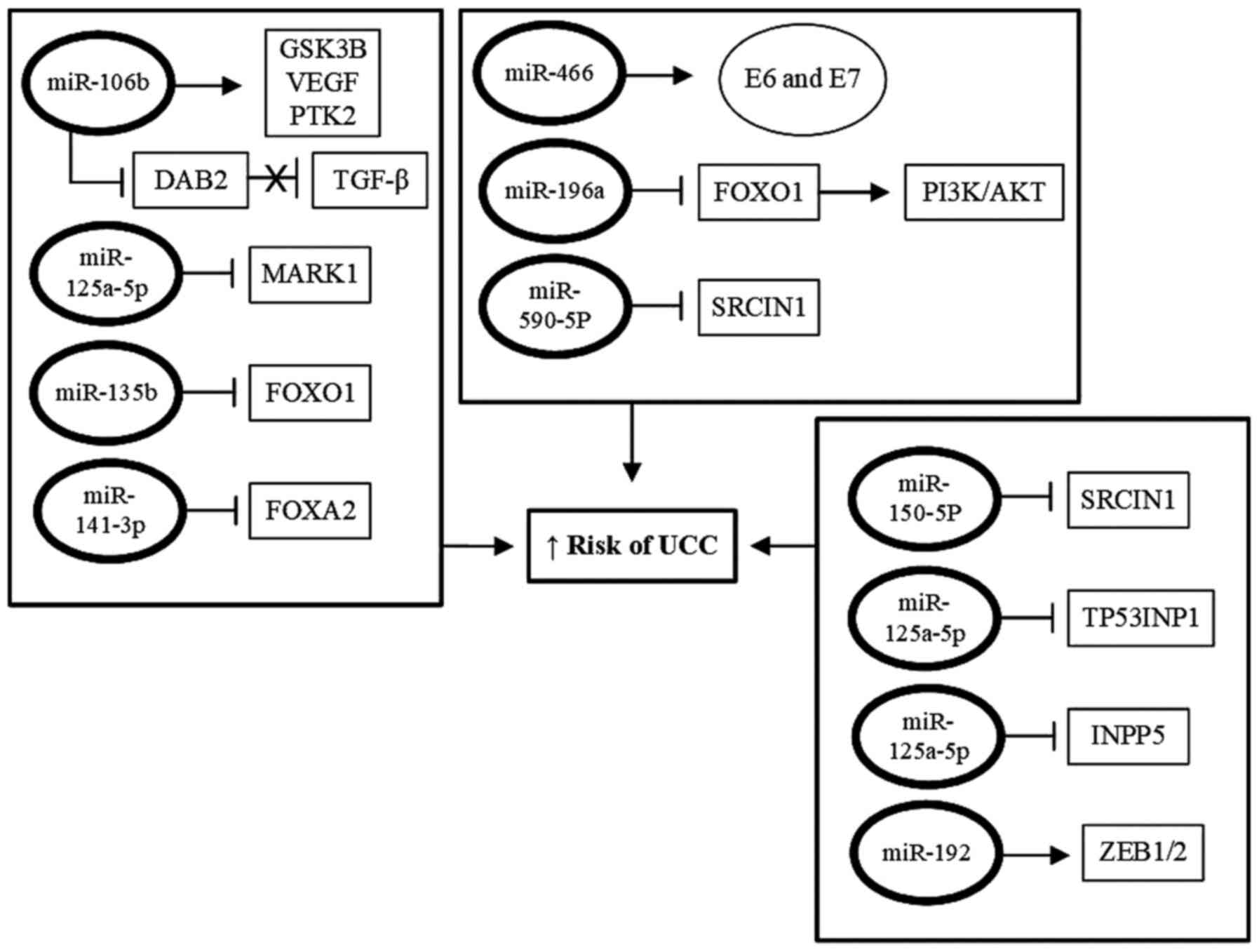
 | Figure 4Protective roles of miRNAs against
cervical carcinogenesis. Tumor suppressor miRNAs, under
physiological conditions, exhibit protective functions against the
development to UCC, acting as activators of the expression of tumor
suppressor genes, whose products act by suppressing the expression
of oncogenes. However, in UCC-derived cells, the expression tumor
suppressing miRNAs is downregulated, via different mechanisms, and
this effectively abrogates their suppressive functions on the
expression of oncogenes involved in proliferation, immortalization
and progression of UCC. UCC, uterine cervix cancer; miRNA/miR,
microRNA. |
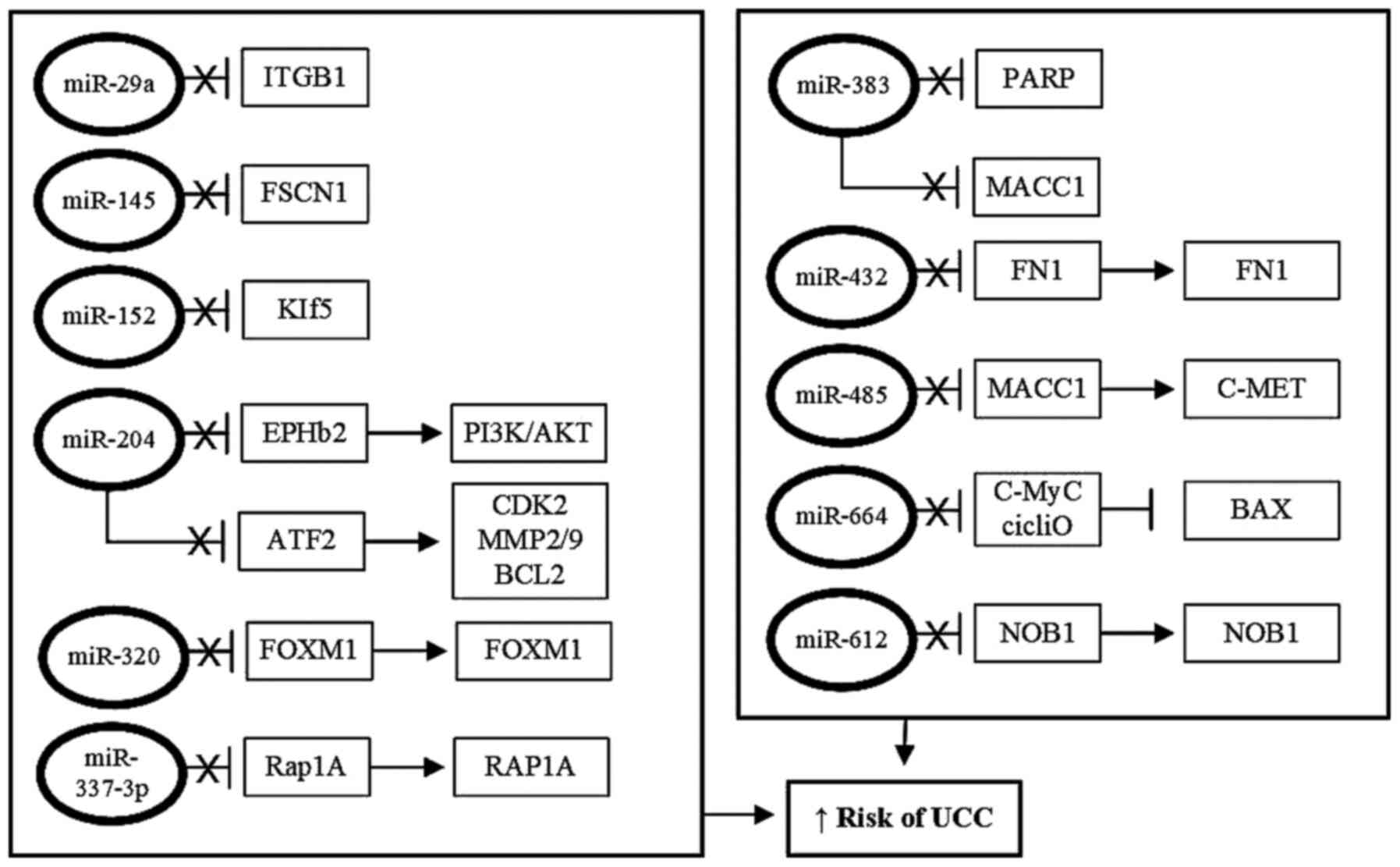
Studies have shown that miR-29a is downregulated in
UCC, resulting in upregulated expression of the oncogene
ITGB1, whose product, integrin β1, promotes proliferation
and migration of tumor cells, causing metastasis to lymph nodes
(135). This indicates that miR-29a
exerts a tumor suppressor function by directly inhibiting the
expression of ITGB1 and therefore, in its absence, tumor
progression occurs (94). miR-145 is
negatively regulated in UCC, and is inversely correlated with the
expression of the FSCN1 gene, which itself is upregulated in
tumor tissues when compared with normal tissues. Overexpression of
miR-145 significantly reduced the proliferation of HeLa cells and
reduced the expression of FSCN1. This shows that, under
physiological conditions, miR-145 functions as a tumor suppressor
by inhibiting the expression of the FSCN1 gene (136).
Reduced expression of miR-204 is also associated
with progression and metastasis to lymph nodes, as well as a low
survival rate in patients with UCC. Overexpression of miR-204
significantly suppressed the proliferation, migration and invasion
of cancer cells and promotes cell cycle arrest in the G0/G1. This
is due to the interaction of miR-204 with the Ephrin type B2
receptor (EphB2). Thus, under normal conditions, miR-204 inhibits
EphB2 to inhibit the PI3K/AKT signaling pathway (137). Low levels of miR-204 expression are
also correlated with overexpression of the transcription factor 12,
which results in increased expression of CDK2, cyclin E,
MMP2, MMP9 and BCL2, and reduction of expression of
BAX, increasing cell proliferation, migration and inhibiting
apoptosis, ultimately favoring tumor progression (138). Conversely, reducing the levels of
miR-204 in UCC results in increased expression of ATF2,
which, in-turn, increases the expression of BCL2 and
LC3I/II, inhibiting apoptosis and increasing proliferation
and autophagy of cancer cells (139). This indicates that under
physiological conditions, miR-204 negatively regulates ATF2
expression to inhibit proliferation, migration and invasion of
cervical cancer cells, and induces apoptosis of these cells
(139). miR-152 is downregulated in
UCC, and is negatively correlated with the expression of
Krüppel-like factor-5 (KLF5), expression of which is increased in
UCC. Thus, in the absence of the suppressor function of miR-152 on
the KLF5 gene, KLF5 protein expression is increased; KLF5
functions as a transcription factor involved in cell proliferation
(133).
miR-320 is downregulated in UCC cells, and
FOXM1 is upregulated. The low expression of miR-320 is
associated with increased viability, migration and invasion of
cancer cells. It has been shown that overexpression of miR-320
suppresses FOXM1 expression and reduces the viability,
migration and invasion of tumor cells. These results show that,
under physiological conditions, miR-320 functions as a tumor
suppressor by inhibiting FOXM1 expression (141). The inhibition of miR-320a results
in increased proliferation, migration and invasion of cells derived
from UCC. This is due to the absence of its suppressive function on
FOXM1, which encodes a transcription factor involved in
tumor promotion (141). miR-337-3p
also acts as a tumor suppressor in UCC, inhibiting the expression
of the Rap1A oncogene. Furthermore, reducing the expression of this
transcript results in the overexpression of Rap1A, leading to a
notable increase in Rap1A protein expression, which inhibits
apoptosis and increases proliferation, migration and invasion of
cancer cells (142).
The expression of miR-338-3p is substantially
reduced in cells derived from UCC, whereas expression of the
MACC1 gene is upregulated. Upregulated expression of
MACC1 is associated with advanced stage UCC and lymph node
metastasis, deep invasion and shorter overall patient survival. The
protein encoded by MACC1 is a growth factor that acts on the
MAPK signaling pathway, which is related to increased cell
proliferation and epithelial-mesenchymal transition. This shows
that miR-338-3p targets the MACC1 gene, downregulating its
expression to inhibit cell proliferation and epithelial-mesenchymal
transition. However, the reduction in miR-338-3p expression caused
by the action of viral oncoproteins, results in the opposite effect
(143). Another tumor suppressor
mechanism mediated by miR-338-3p involves the suppression of the
ATF2 gene, which is neutralized by the action of
oncoproteins produced by high-risk HPVs. This results is an
increase in the expression of ATF2 protein, which functions as an
activator of the mTOR signaling pathway, increasing the
proliferation and autophagy of infected cells. Restoration of
miR-338 expression inhibited the proliferation of UCC-derived SiHa
and HeLa cells, which are positive for HPV16 and HPV18,
respectively. In addition, the reduction of miR-338 expression
decreased the expression of phospho-(p-)mTOR and p-p70S6. This
suggests that under physiological conditions, miR-338 inhibits
proliferation and autophagy, by inhibiting the mTOR pathway,
through negative regulation of ATF2 gene expression.
However, this function is reversed by the action of viral
oncoproteins (144).
The expression levels of miR-375 are reduced in
UCC-derived cells, resulting in increased proliferation, migration
and invasion of the cancer cells, inducing angiogenesis, and
inhibiting apoptosis in vitro. Overexpression of miR-375
reversed these functions, resulting in the opposite effects.
Astrocyte gene-1 (AEG-1) has been identified as a target of
miR-375. The oncoproteins E6 and E7 of HPVs 16 and 18 downregulate
miR-375 expression, resulting in increased expression of the
AEG-1 oncogene, contributing to the initiation and
progression of UCC (145). The gene
encoding miR-375 showed higher levels of methylation in UCC
compared with premalignant lesions. In addition, treatment with a
demethylating agent increased the expression of miR-375 in SiHa and
HeLa cells. Interestingly, the expression levels of this transcript
were negatively correlated with the levels of methylation in
clinical samples. It has been observed that miR-375 targets the
Replication Factor C Subunit 3 gene, whose product is part of a
transcriptional complex. This indicates that silencing by
methylation of the miR-375 encoder gene promoter may facilitate the
process of carcinogenesis in UCC (129).
miR-381-3p is negatively regulated, whereas the
keratinocyte growth factor gene (FGF7) is upregulated in cells
derived from UCC, showing an inverse correlation between the
expression of miR-381-3p and FGF7. This shows that under
physiological conditions, miR-381-3p can upregulate FGF7
expression, inhibiting the proliferation and metastasis of cancer
cells (146). The levels of miR-383
expression are significantly lower in tissues derived from UCC
compared with tissues from precancerous lesions, and there was an
inverse correlation between miR-383 expression levels and that of
PI3K, AKT, mTOR, PARP2 and p70S6K. Cell viability, migration and
invasion were reduced in cells transfected with miR-383 mimics
following knockdown of the PARP2 gene, whereas treatment
with the miR-383 inhibitor increased these functions. These results
suggest that, under physiological conditions, miR-383 acts as a
tumor suppressor, negatively regulating the expression of
PARP2 and inhibiting the PI3K-AKT-mTOR signaling pathway.
Thus, in the absence of miR-383, expression of PARP2 is
increased, which activates the PI3K-AKT-mTOR signaling pathway,
leading to the initiation and progression of UCC (147).
miR-432 is downregulated in UCC cells, and is
inversely correlated with the expression of the gene encoding
fibronectin 1 (FN1). This suggests that miR-432 possesses
tumor suppressive activity via inhibition of FN1 gene expression.
Inhibition of miR-432 expression leads to overexpression of the
fibronectin-1 protein, which induces cancer cell proliferation and
invasion (134). A similar outcome
is observed with miR-485, expression of which is also reduced in
patients with UCC. Downregulation of this miRNA was associated with
advanced stage of the disease and lymph node metastasis. It was
also found that miR-485 may exert its tumor suppressive function in
cervical cancer by directly targeting MACC1 and inhibiting the
Met/AKT signaling pathway (148).
Conversely, downregulation of miR-664 in UCC resulted in increased
expression of the c-Myc oncogene and the cyclin D gene, in-turn
resulting in reduced expression of BAX and active Caspase-3.
This results in inhibition of apoptosis and increased cell
proliferation and tumor growth (149).
The expression of miR-2861 was significantly
reduced in 293T and HaCaT cells expressing the E6 protein of HPV16,
compared with the control cells. miR-2861 was also shown to be
downregulated in UCC cells, and its downregulation was negatively
correlated with tumor stage and lymph node metastases.
Overexpression of miR-2861 inhibited the proliferation and invasion
of cancer cells and increased apoptosis. It was shown that the
EGFR, AKT2 and CCND1 genes were all direct targets of
miR-2861, and restoring the functions of these genes neutralized
the suppressive effects of miR-2861. This suggests that, under
physiological conditions, miR-2861 is a tumor suppressor that acts
by inhibiting the transcription of EGFR, AKT2 and/or
CCND1 to regulate cell proliferation and migration. The
knockdown of the miR-2861 encoding gene mediated by the E6 protein
of HPV16 results in increased transcription of EGFR,
AKT2 and/or CCND1, thus contributing to initiation
and progression of UCC (150).
miR-612 has been shown to be associated with the
progression of other types of tumors; however, the expression of
miR-612 was downregulated in UCC tissues and cells, and this
reduced expression was associated with greater disease severity and
lymph node metastasis. Overexpression of miR-612 significantly
reduced the proliferation, migration and invasion of cancer cells
in vitro and delayed tumor growth in vivo. miR-612
was shown to target the NOB1 gene, which encodes an
RNA-binding protein; there was a negative correlation between
miR-612 expression and NOB1 protein expression in UCC samples.
Overexpression of NOB1 partially reversed the inhibitory
effects of miR-612 on tumor cells, suggesting that under
physiological conditions, miR-612 acts as a tumor suppressor in
UCC, by inhibiting the expression of the NOB1 gene (151).
LncRNAs
LncRNAs can modulate the progression of tumors
(Fig. 5). The role of lncRNA-ANRIL
was analyzed in cell lines derived from UCC, where it was shown to
be positively regulated with UCC progression. Knockdown of ANRIL
reduced proliferation and invasion, in addition to inducing
apoptosis of cervical cancer cells. Bioinformatics analysis showed
that miR-186 was a direct target of ANRIL, and ANRIL binds to
miR-186 to abrogate its tumor suppressor function. Expression of
miR-186 was reduced in cell lines derived from UCC and its
expression was negatively correlated with the expression of ANRIL.
This shows that lncRNA-ANRIL promotes UCC by sponging miR-186, to
prevent its tumor suppressor action (152). A similar mechanism was observed
with lncRNA-MALAT1 in relation to miR-375(153).
It has been shown that lncRNA DDN and PRKAG1 RNA
antisense 1 (DDN-AS1) are upregulated in UCC tissues and cell
lines, and it is positively associated with a poor prognosis in UCC
patients. Knockdown of DDN-AS1 suppressed UCC progression by
efficiently inhibiting the proliferation, migration and invasion of
cancer cells. It was shown that the increase in DDN-AS1 expression
was mediated by activation of the transcription factor TCF3. In
addition, DDN-AS1 increased TCF3 expression by competitively
binding to miR-15a and miR-16, generating a positive feedback loop
in which DDN-AS1 inhibits the suppressor function of miR-15a/16
over TCF3, which in-turn activates the expression of DDN-AS1,
increasing proliferation, migration and invasion of tumor cells in
UCC (109).
LncRNA-DSCAM-AS1 is associated with an increased
capacity for proliferation, migration and invasion of cancer cells
in UCC. It was found that DSCAM-AS1 physically binds to miR-877-5p,
preventing its tumor suppressing action in which it targets the
ATXN7L3 gene that encodes the histone acetylation protein
complex and activates transcription by chromatin remodeling.
Knockdown of DSCAM-AS1 or overexpression of miR-877-5p
inhibits UCC progression. This suggests that lncRNA-DSCAM-AS1 acts
by inhibiting the tumor suppressive function of miR-877-5p,
interfering with a miR-877-5p/ATXN7L3 axis to promote tumor
progression (1).
It has been shown that lncRNA-LINC01535 binds to
miR-214, neutralizing its tumor suppressor activity, preventing the
repression of the miR-214 target, the EZH2 gene, thus
resulting in increased expression of the EZH2 protein. This protein
was also found to directly repress miR-214 expression. Thus, by
positively regulating EZH2, LINC01535 further represses
miR-214 expression. The enhanced expression of LINC01535 promotes
the growth, migration and invasion of cancer cells in vitro
and in vivo. Knockdown of LINC01535 results in
overexpression of miR-214, thus regulating a miR-214/EZH2 axis.
Knockdown of EZH2 abrogates the effects LINC01535 in UCC.
Clinically, increased expression of LINC01535 is correlated with
advanced tumor stage and a poor prognosis (154).
LncRNA-TMPOP2 is overexpressed in cancer cells
derived from UCC and inhibits the expression of E-cadherin by
recruiting the transcription repressor EZH2, the promoter of
the E-cadherin gene (155).
High-risk HPVs have been shown to induce degradation of the tumor
suppressor p53, preventing this protein from binding to the
promoter of TMPOP2 to inhibit its transcription. In
addition, it was demonstrated that overexpression of TMPOP2
resulted in sequestration of tumor suppressor miRNAs, such as
miR-139 and miR-375, which target the mRNAs of E6 and E7,
increasing the expression of these viral proteins. Knockdown of
TMPOP2 reduced the expression of positive regulatory genes
of cell cycle progression, and induced arrest of the cycle, and
also inhibited the proliferation of HeLa cells. This suggests that
inhibition of the function of miR-139 and miR-375 by lncRNA-TMPOP2
increased the expression and transforming activity of E6 and E7
proteins (156).
The expression of lncRNA-CRNDE was increased in
tissues obtained from UCC and in several tumor-derived cell lines.
Knockdown of CRNDE significantly reduced the proliferation of
cancer cells, whereas overexpression of this transcript
significantly promoted the growth of cancer cells. CRNDE has been
found to bind to the p53 upregulated modulator of apoptosis
(PUMA), and PUMA is necessary for the CRNDE-mediated
increase in cancer cell proliferation. Thus, it was proposed that
lncRNA-CRNDE in combination with PUMA could be used for
clinical diagnosis, both as a prognostic factor for UCC and as a
potential therapeutic target in UCC (133).
A recent study showed that lncRNA-GHET1 is
upregulated in UCC cell lines. Loss of function analysis
demonstrated that the silencing of GHET1 inhibited
proliferation, migration and epithelial-mesenchymal transition of
these cells, as well as inhibiting the AKT/mTOR and Wnt/β-catenin
pathways. Conversely, the activation of these two pathways reversed
the inhibitory effect of knockdown of GHET1 on growth,
migration and epithelial-mesenchymal transition in the UCC cell
lines. It was also shown that GHET1 stabilizes E2F6 mRNA,
interacting with IGF2BP2, to activate the AKT/mTOR and
Wnt/β-catenin pathways. As knockdown of GHET1 resulted in
the inactivation of two signaling pathways, both of which are
important in cervical carcinogenesis, and inhibited the progression
of UCC, GHET1 may be a promising therapeutic target for management
of UCC (91).
CircRNAs
CircRNAs are a class of single-stranded RNA
molecules that possess a covalently closed loop structure. They are
produced by back-splicing of mRNA precursors or by jumping in the
mRNAs of the genes. They are widely expressed in eukaryotes and act
as critical regulators in several types of tumors. CircRNAs recruit
and reprogram the primary components of the tumor microenvironment,
regulating signaling pathways, modulating the immune response and
affecting tumorigenesis, angiogenesis, tumor progression and
metastasis (157). Their high
stability, abundance and evolutionary conservation suggest a
diverse set of properties with important implications for cellular
functions. These transcripts can act as efficient sponges of miRNAs
and proteins, and serve critical roles in the modulation of
transcription. In addition, numerous circRNAs are aberrantly
expressed in pathological conditions, and can be associated with
initiation or progression of cancer (158).
It has been shown that oncogenic HPVs are also
capable of generating circRNAs, as described in the CaSki cell
line, which is derived from cervical carcinoma and is transformed
by HPV 16. CircE7 is derived from the viral oncogene E7 and
is found primarily in the cytoplasm, where it is associated with
polysomes, and it is translated into the viral oncoprotein E7.
Silencing the viral gene E7 or the encoding circE7 resulted
in inhibition of cancer cell proliferation in vitro. CircE7
has only been detected in its episomal form in cancer cells
containing HPV DNA. These results suggest that the circRNAs
produced by HPV encode biologically functional proteins and they
are associated with viral-mediated transformation of cells
(159).
High levels of circCLK3 were found in UCC tissues
and were strongly associated with advanced stage UCC and stromal
invasion of the tumor. It was shown that circCLK3 functioned as a
sponge to absorb miR-320a, preventing its tumor suppressor
function, which under normal conditions would act by suppressing
the expression of the FOXM1 gene, which encodes the FOXM1
transcription factor and is associated with disease progression
(141).
CircSLC26A4 was found to be positively regulated in
tissue and cell lines derived from UCC, and this high expression
was associated with low patient survival. Loss of function
experiments showed that the knockdown of circSLC26A4 resulted in
inhibition of proliferation and invasion of cancer cells, as well
as of tumor growth both in vitro and in vivo. It was
found that circSLC26A4 acts as a sponge for miR-1287-5p, which,
in-turn, targets the HOXA7 mRNA that encodes the homeobox-7
transcription factor (Hox-A7). It was found that the RNA-binding
protein interacts with the QKI response elements in the introns of
the SLC26A4 gene, to promote the biogenesis of circSLC26A4.
This suggests that circSLC26A4 facilitates UCC progression,
preventing the suppressive action of miR-1287-5p on the
HOXA7 gene, increasing the expression of the transcription
factor Hox-A7(160).
The expression levels of circRNA_0000285 were shown
to be significantly higher in UCC samples compared with the
corresponding normal tissues. A positive correlation was identified
between the expression of this transcript and the expression of
FUS, a protein coding gene that binds to RNA, and possesses
high transcriptional activity. The proliferative and migratory
capacity of UCC-derived cells was notably reduced after silencing
of the gene encoding circRNA_0000285. In addition, the knockdown of
this gene resulted in strong inhibition of metastasis of UCC cells
in nude mice. This indicates that circRNA_0000285 increases the
proliferation and metastasis of cancer cells in UCC via the
positive regulation of FUS (161).
Finally, a study was performed using RNA sequencing
data and non-coding RNAs in cells derived from UCC, which included
102 lncRNAs, 15 miRNAs, 15 mRNAs and 522 pairs of interactions in a
competing endogenous (ce)RNA network. The analysis revealed that
the following genes were enriched in the network: Alcohol
dehydrogenase 7 (ADH7), a member of the vestigial family 3;
and cytochrome P450, family 26, subfamily B, polypeptide 1, which
is involved in the metabolic process of retinoic acid and in the
retinol metabolism pathway. The ADH7 gene was regulated by
miR-3065, which interacted with LINC01133 in the ceRNA network,
suggesting that these transcripts may serve an important role in
UCC progression (162). A summary
of the role of circRNAs in cervical carcinogenesis is presented in
Fig. 5.
5. Conclusions
Numerous clinical, epidemiological and molecular
studies have shown that persistent infection with high-risk HPV
genotypes is an indispensable, but not sufficient, prerequisite for
the development of UCC. This suggests that the cervical
carcinogenesis process induced by HPV depends on other associated
risk factors, possibly generated by stressful environmental
conditions, in addition to other factors the cells may experience,
creating conditions favorable for malignant transformation.
Therefore, for the development of cervical carcinogenesis,
additional genetic and epigenetic alterations are necessary as a
trigger for its initiation. These events act together through a
complex process, involving multiple factors, going through several
phases in the development of UCC. Such conditions disrupt cellular
homeostasis and contribute to generating genomic instability, which
favors the accumulation of mutations and increases the frequency of
epigenetic changes. Thus, these conditions affect the functions of
regulatory cell genes, which can result in activation of oncogenes
or inactivation of tumor suppressor genes by silencing the gene or
inactivating the function of their products.
These same changes can also occur in the viral
genome, particularly affecting the expression patterns of viral
oncogenes. It has been widely demonstrated that several types of
epigenetic changes can affect the expression of cellular and HPV
genes, participating in some manner in the process of
carcinogenesis, and contributing to the initiation and progression
of UCC. Thus, recent advances in cancer biology have shown that
other factors, in addition to genetic mutations, may be implicated
in the development of the disease. Epigenetic changes caused by
aberrant methylation and chromatin remodeling, due to histone
modifications, both in the cellular and viral DNA, serve an
increasingly relevant role in cervical carcinogenesis. In addition,
the participation of non-coding RNAs, such as miRNAs, lncRNAs and
circRNAs, has drastically increased the complexity of our
understanding of the molecular biology of cancer, whilst also
providing numerous novel potential druggable targets, particularly
in UCC. Therefore, these transcripts are the subject of numerous
studies, aiming to discover their value as biomarkers of diagnosis
and prognosis of the disease, as well as their therapeutic value
for the treatment of UCC.
Acknowledgements
Not applicable.
Funding
This study was funded in part by the Coordenação de
Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES) (grant
no. 001).
Availability of data and materials
Not applicable.
Authors' contributions
MLRDS and JVF were involved in the conception of
the study, literature review and drafting the manuscript. BHDRDA
and VDDA were involved in drafting the manuscript. TAADMF, RNDOC,
DCFL and JMGDA were involved in revising the manuscript critically
for important intellectual content. FLB, VSA and JCVDA were
involved in the literature review. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liang J, Zhang S, Wang W, Xu Y, Kawuli A,
Lu J and Xiu X: Long non-coding RNA DSCAM-AS1 contributes to the
tumorigenesis of cervical cancer by targeting miR-877-5p/ATXN7L3
axis. Biosci Rep. 40(BSR20192061)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
INCA: Estimativa 2018: Incidência de
câncer no Brasil/Instituto Nacional de Câncer Jose Alencar Gomes da
Silva. Coordenação de Prevenção e Vigilância, Rio de Janeiro,
2017.
|
|
5
|
Chen L, Qiu X, Zhang N, Wang Y, Wang M, Li
D, Wang L and Du Y: APOBEC-mediated genomic alterations link
immunity and viral infection during human papillomavirus-driven
cervical carcinogenesis. Biosci Trends. 11:383–388. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sen P, Ganguly P and Ganguly N: Modulation
of DNA methylation by human papillomavirus E6 and E7 oncoproteins
in cervical cancer. Oncol Lett. 15:11–22. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Senapati R, Senapati NN and Dwibedi B:
Molecular mechanisms of HPV mediated neoplastic progression. Infect
Agent Cancer. 11(59)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Steenbergen RD, Snijders PJ, Heideman DA
and Meijer CJ: Clinical implications of (epi)genetic changes in
HPV-induced cervical precancerous lesions. Nat Rev Cancer.
14:395–405. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Morel A, Baguet A, Perrard J, Demeret C,
Jacquin E, Guenat D, Mougin C and Prétet JL: 5azadC treatment
upregulates miR-375 level and represses HPV16 E6 expression.
Oncotarget. 8:46163–46176. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fernandes JV, de Medeiros Fernandes TA, de
Azevedo JC, Cobucci RN, de Carvalho MG, Andrade VS and de Araújo
JM: Link between chronic inflammation and human
papillomavirus-induced carcinogenesis (Review). Oncol Lett.
9:1015–1026. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Soto D, Song C and McLaughlin-Drubin ME:
Epigenetic alterations in human papillomavirus-associated cancers.
Viruses. 9(248)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Durzynska J, Lesniewicz K and Poreba E:
Human papillomaviruses in epigenetic regulations. Mutat Res Rev
Mutat Res. 772:36–50. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Amaro-Filho SM, Pereira Chaves CB, Felix
SP, Basto DL, de Almeida LM and Moreira MAM: HPV DNA methylation at
the early promoter and E1/E2 integrity: A comparison between HPV16,
HPV18 and HPV45 in cervical cancer. Papillomavirus Res. 5:172–179.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jacquin E, Baraquin A, Ramanah R,
Carcopino X, Morel A, Valmary-Degano S, Bravo IG, de Sanjosé S,
Riethmuller D, Mougin C and Prétet JL: Methylation of human
papillomavirus type 16 CpG sites at E2-binding site 1 (E2BS1),
E2BS2, and the Sp1-binding site in cervical cancer samples as
determined by high-resolution melting analysis-PCR. J Clin
Microbiol. 51:3207–3215. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Loscalzo J and Handy DE: Epigenetic
modifications: Basic mechanisms and role in cardiovascular disease
(2013 Grover Conference series). Pulm Circ. 4:169–174.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Shamsi MB, Firoz AS, Imam SN, Alzaman N
and Samman MA: Epigenetics of human diseases and scope in future
therapeutics. J Taibah Univ Med Sci. 212:205–211. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Vogt G: Facilitation of environmental
adaptation and evolution by epigenetic phenotype variation:
Insights from clonal, invasive, polyploid, and domesticated
animals. Environ Epigenet. 3(dvx002)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Marsit CJ: Influence of environmental
exposure on human epigenetic regulation. J Exp Biology. 218:71–79.
2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li Y and Tollefsbol TO: Age-related
epigenetic drift and phenotypic plasticity loss: Implications in
prevention of age-related human diseases. Epigenomics. 8:1637–1651.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zannas AS and Chrousos GP: Epigenetic
programming by stress and glucocorticoids along the human lifespan.
Mol Psychiatry. 22:640–646. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Osborne A: The role of epigenetics in
human evolution. Biosc Horizons. 10(hzx007)2017.
|
|
22
|
Stefansson OA and Esteller M: Epigenetic
modifications in breast cancer and their role in personalized
medicine. Am J Pathol. 183:1052–1063. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Llinàs-Arias P and Esteller M: Epigenetic
inactivation of tumor suppressor coding and non-coding genes in
human cancer: An update. Open Biol. 7(170152)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ramassone A, Pagotto S, Veronese A and
Visone R: Epigenetics and microRNAs in cancer. Int J Mol Sci.
19(459)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kurdyukov S and Bullock M: DNA methylation
analysis: Choosing the right method. Biology (Basel).
5(3)2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang X, Hu M, Lyu X, Li C, Thannickal VJ
and Sanders YY: DNA methylation regulated gene expression in organ
fibrosis. Biochim Biophys Acta Mol Basis Dis. 1863:2389–2397.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yang X, Han H, de Carvalho DD, Lay FD,
Jones PA and Liang G: Gene body methylation can alter gene
expression and is a therapeutic target in cancer. Cancer Cell.
26:577–590. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jin Z and Liu Y: DNA methylation in human
diseases. Genes Dis. 5:1–8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Moore LD, Le T and Fan G: DNA Methylation
and its basic function. Neuropsychopharmacology. 38:23–38.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ehrlich M: DNA hypomethylation in cancer
cells. Epigenomics. 1:239–259. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yang HJ: Aberrant DNA methylation in
cervical carcinogenesis. Chin J Cancer. 32:42–48. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lu Q, Ma D and Zhao S: DNA methylation
changes in cervical cancers. Methods Mol Biol. 863:155–176.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu G: CDH1 promoter methylation in
patients with cervical carcinoma: A systematic meta-analysis with
trial sequential analysis. Future Oncol. 14:51–63. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Snoek BC, Splunter AP, Bleeker MC, Ruiten
MC, Heideman DA, Rurup WF, Verlaat W, Schotman H, Gent MV, Trommel
NE and Steenbergen RD: Cervical cancer detection by DNA methylation
analysis in urine. Sci Rep. 9(3088)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
McCormick TM, Canedo NH, Furtado YL,
Silveira FA, de Lima RJ, Rosman AD, Almeida Filho GL and Carvalho
Mda G: Association between human papillomavirus and Epstein-Barr
virus DNA and gene promoter methylation of RB1 and CDH1 in the
cervical lesions: A transversal study. Diagn Pathol.
10(59)2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Cardoso MF, Castelletti CH, Lima-Filho JL,
Martins DB and Teixeira JA: Putative biomarkers for cervical
cancer: SNVs, methylation and expression profiles. Mutat Res.
773:161–173. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Fernandes JV, Araújo JM and Fernandes TA:
Biology and natural history of human papillomavirus infection. Open
Access J Clin Trials. 2013(5)2013.
|
|
38
|
Bashaw AA, Leggatt GR, Chandra J, Tuong ZK
and Frazer IH: Modulation of antigen presenting cell functions
during chronic HPV infection. Papillomavirus Res. 4:58–65.
2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang M, Wang M, Li X, Xie Y, Xia X, Tian
J, Zhang K and Tang A: Wnt signaling in cervical cancer? J Cancer.
9:1277–1286. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ayala-Calvillo A, Mojica-Vázquer LH,
García-Carrancá A and González-Maya L: Wnt/β-catenin pathway
activation and silencing of the APC gene in HPV-positive human
cervical cancer-derived cells. Mol Med Rep. 17:200–208.
2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lee J and Kim SS: The function of p27 KIP1
during tumor development. Exp Mol Med. 41:765–771. 2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Qi Q, Ling Y, Zhu M, Zhou L, Wan M, Bao Y
and Liu Y: Promoter region methylation and loss of protein
expression of PTEN and significance in cervical cancer. Biomed Rep.
2:653–658. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li JY, Huang T, Zhang C, Jiang DJ, Hong
QX, Ji HH, Ye M and Duan SW: Association between RASSF1A promoter
hypermethylation and oncogenic HPV infection status in invasive
cervical cancer: A meta-analysis. Asian Pac J Cancer Prev.
16:5749–754. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sherr CJ and Bartek J: Cell cycle-targeted
cancer therapies. Ann Rev Cancer Biol. 1:41–57. 2017.
|
|
45
|
Li X, Tao L, Tan Q, Dong Y, Pan X, Pang L,
Qi Y, Zou H, Liang W, Liu W, et al: CpG island methylation of the
CADM1 gene correlates with cervical carcinogenesis in the Uighur
and Han populations of Xinjiang, China. Int J Clin Exp Pathol.
9:6977–6987. 2016.
|
|
46
|
Holubeková V, Mendelová A, Grendár M,
Meršaková S, Kapustová I, Jašek K, Vaňochová A, Danko J and
Lasabová Z: Methylation pattern of CDH1 promoter and its
association with CDH1 gene expression in cytological cervical
specimens. Oncol Lett. 12:2613–2621. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Siegel EM, Riggs BM, Delmas AL, Koch A,
Hakam A and Brown KD: Quantitative DNA methylation analysis of
candidate genes in cervical cancer. PLoS One.
10(e0122495)2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lorincz AT: Virtues and weaknesses of DNA
methylation as a test for cervical cancer prevention. Acta Cytol.
60:501–512. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Kim MK, Lee IH, Lee KH, Lee YK, So KA,
Hong SR, Hwang CS, Kee MK, Rhee JE, Kang C, et al: DNA methylation
in human papillomavirus-infected cervical cells is elevated in
high-grade squamous intraepithelial lesions and cancer. J Gynecol
Oncol. 27(e14)2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Chang CC, Huang RL, Wang HC, Liao YP, Yu
MH and Lai HC: High methylation rate of LMX1A, NKX6-1, PAX1, PTPRR,
SOX1, and ZNF582 genes in cervical adenocarcinoma. Int J Gynecol
Cancer. 24:201–209. 2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Xu L, Xu J, Hu Z, Yang B, Wang L, Lin X,
Xia Z, Zhang Z and Zhu Y: Quantitative DNA methylation analysis of
paired box gene 1 and LIM homeobox transcription factor 1α genes in
cervical cancer. Oncol Lett. 15:4477–4484. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Flamini MI, Gauna GV, Sottile ML, Nadin
BS, Sanchez AM and Vargas-Roig LM: Retinoic acid reduces migration
of human breast cancer cells: Role of retinoic acid receptor beta.
J Cell Mol Med. 18:1113–1123. 2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yin FF, Wang N, Bi XN, Yu X, Xu XH, Wang
YL, Zhao CQ, Luo B and Wang YK: Serine/threonine kinases 31(STK31)
may be a novel cellular target gene for the HPV16 oncogene E7 with
potential as a DNA hypomethylation biomarker in cervical cancer.
Virol J. 13(60)2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Molano M, Moreno-Acosta P, Morales N,
Burgos M, Buitrago L, Gamboa O, Alvarez R, Garland SM, Tabrizi SN,
Steenbergen RD and Mejía JC: Association between type-specific HPV
infections and hTERT DNA methylation in patients with invasive
cervical cancer. Cancer Genomics Proteomics. 13:483–491.
2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Johannsen E and Lambert PF: Epigenetics of
human papillomaviruses. Virology. 445:205–212. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Simanaviciene V, Popendikyte V,
Gudleviciene Z and Zvirbliene A: Different DNA methylation pattern
of HPV16, HPV18 and HPV51 genomes in asymptomatic HPV infection as
compared to cervical neoplasia. Virology. 484:227–233.
2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wang W, Sun Z, Liu J, Wang G, Lu Z, Zhou
W, Qi T and Ruan Q: Increased methylation of human papillomavirus
type 16 DNA is associated with the severity of cervical lesions in
infected females from northeast China. Oncol Lett. 13:3809–3816.
2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
McBride AA and Warburton A: The role of
integration in oncogenic progression of HPV-associated cancers.
PLoS Pathog. 13(e1006211)2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Vinokurova S and von Knebel Doeberitz M:
Differential methylation of the HPV 16 upstream regulatory region
during epithelial differentiation and neoplastic transformation.
PLoS One. 6(e24451)2011.PubMed/NCBI View Article : Google Scholar
|
|
60
|
McBride AA: The papillomavirus E2
proteins. Virology. 445:57–79. 2013.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Filho SM, Bertoni N, Brant AC, Vidal JP,
Felix SP, Cavalcanti SM, Carestiato FN, Martins LF, Almeida LM and
Moreira MA: Methylation at 3'LCR of HPV16 can be affected by
patient age and disruption of E1 or E2 genes. Virus Res. 232:48–53.
2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Fertey J, Hagmann J, Ruscheweyh HJ, Munk
C, Kjaer S, Huson D, Haedicke-Jarboui J, Stubenrauch F and Iftner
T: Methylation of CpG 5962 in L1 of the human papillomavirus 16
genome as a potential predictive marker for viral persistence: A
prospective large cohort study using cervical swab samples. Cancer
Med. 9:1058–1068. 2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Niyazi M, Sui S, Zhu K, Wang L, Jiao Z and
Lu P: Correlation between methylation of human papillomavirus-16 L1
gene and cervical carcinoma in Uyghur women. Gynecol Obstet Invest.
82:22–29. 2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Huang J, Tan ZR, Yu J, Li H, Lv QL, Shao
YY and Zhou HH: DNA hypermethylated status and gene expression of
PAX1/SOX1 in patients with colorectal carcinoma. Onco Targets Ther.
10:4739–4751. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Luan T, Hua Q, Liu X, Xu P, Gu Y, Qian H,
Yan L, Xu X, Geng R, Zeng X and Li P: PAX1 Methylation as a
potential biomarker to predict the progression of cervical
intraepithelial neoplasia: A Meta-analysis of related studies. Int
J Gynecol Cancer. 27:1480–1488. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lin YW, Tsao CM, Yu PN, Shih YL, Lin CH
and Yan MD: SOX1 suppresses cell growth and invasion in cervical
cancer. Gynecol Oncol. 131:174–181. 2013.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Bowden SJ, Kalliala I, Veroniki AA, Arbyn
M, Mitra A, Lathouras K, Mirabello L, Chadeau-Hyam M, Paraskevaidis
E, Flanagan JM and Kyrgiou M: The use of human papillomavirus DNA
methylation in cervical intraepithelial neoplasia: A systematic
review and meta-analysis. EBioMedicine. 50:246–59. 2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Torres-Rojas FI, Alarcón-Romero LC,
Leyva-Vázquez MA, Ortiz-Ortiz J, Mendoza-Catalán MÁ,
Hernández-Sotelo D, Moral-Hernández OD, Rodríguez-Ruiz HA,
Leyva-Illades D, Flores-Alfaro E and Illades-Aguiar B: Methylation
of the L1 gene and integration of human papillomavirus 16 and 18 in
cervical carcinoma and premalignant lesions. Oncol Lett.
15:2278–2286. 2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Clarke MA, Gradissimo A, Schiffman M, Lam
J, Sollecito CC, Fetterman B, Lorey T, Poitras N, Raine-Bennett TR,
Castle PE, et al: Human papillomavirus DNA methylation as a
biomarker for cervical precancer: Consistency across 12 genotypes
and potential impact on management of HPV-positive women. Clin
Cancer Res. 24:2194–2202. 2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Shestakova EA and Nakatani Y:
Characterization of histone predeposition complexes from different
cellular compartments. J Investig Genomics. 2:25–28. 2015.
|
|
71
|
Bornelöv S, Reynolds N, Xenophontos M,
Gharbi S, Johnstone E, Floyd R, Ralser M, Signolet J, Loos R,
Dietmann S, et al: The nucleosome remodeling and deacetylation
complex modulates chromatin structure at sites of active
transcription to fine-tune gene expression. Mol Cell. 71:56–72.e4.
2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Feng T, Wang H, Su H, Lu H, Yu L, Zhang X,
Sun H and You Q: Novel N-hydroxyfurylacrylamide-based histone
deacetylase (HDAC) inhibitors with branched CAP group (Part 2).
Bioorg Med Chem. 21:5339–5354. 2013.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Li B, Carey M and Workman JL: The role of
chromatin during transcription. Cell. 128:707–719. 2007.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhang H, Dai X, Qi Y, He Y, Du W and Pang
JJ: Histone deacetylases inhibitors in the treatment of retinal
degenerative diseases: Overview and perspectives. J Ophthalmol.
2015(250812)2015.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Song C, Zhu S, Wu C and Kang J: Histone
deacetylase (HDAC) 10 suppresses cervical cancer metastasis through
inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J
Biol Chem. 288:28021–2833. 2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Kondo Y, Shen L, Cheng AS, Ahmed S,
Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B,
et al: Gene silencing in cancer by histone H3 lysine 27
trimethylation independent of promoter DNA methylation. Nat Genet.
40:741–750. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
77
|
Hiragami-Hamada K, Xie SQ, Saveliev A,
Uribe-Lewis S, Pombo A and Festenstein R: The molecular basis for
stability of heterochromatin-mediated silencing in mammals.
Epigenetics Chromatin. 2(14)2009.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Scarpini CG, Groves IJ, Pett MR, Ward D
and Coleman N: Virus transcript levels and cell growth rates after
naturally occurring HPV16 integration events in basal cervical
keratinocytes. J Pathol. 233:281–293. 2014.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Yeo-Teh NSL, Ito Y and Jha S: High-Risk
human papillomaviral oncogenes E6 and E7 target key cellular
pathways to achieve oncogenesis. Int J Mol Sci.
19(1706)2018.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Uchida C: Roles of pRB in the regulation
of nucleosome and chromatin structures. Biomed Res Int.
2016(5959721)2016.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Fischer M: Census and evaluation of p53
target genes. Oncogene. 36:3943–3956. 2017.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Zhang Y, Dakic A, Chen R, Dai Y, Schlegel
R and Liu X: Direct HPV E6/Myc interactions induce histone
modifications, Pol II phosphorylation, and hTERT promoter
activation. Oncotarget. 8:96323–96339. 2017.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Johansson C, Jamal Fattah T, Yu H, Nygren
J, Mossberg AK and Schwartz S: Acetylation of intragenic histones
on HPV16 correlates with enhanced HPV16 gene expression. Virology.
482:244–259. 2015.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhang L, Yuan C, Wang Y and Zhao S:
Histone deacetylases 3 (HDAC3) is highly expressed in cervical
cancer and inhibited by siRNA. Int J Clin Exp Pathol. 9:3600–3605.
2016.
|
|
85
|
Chen X, Loo JX, Shi X, Xiong W, Guo Y, Ke
H, Yang M, Jiang Y, Xia S, Zhao M, et al: E6 protein expressed by
high-risk HPV activates super-enhancers of the EGFR and c-MET
oncogenes by destabilizing the histone demethylase KDM5C. Cancer.
78:1418–1430. 2018.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Wang WT, Han C, Sun YM, Chen TQ and Chen
YQ: Noncoding RNAs in cancer therapy resistance and targeted drug
development. J Hematol Oncol. 12(55)2019.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Sadri Nahand J, Moghoofei M, Salmaninejad
A, Bahmanpour Z, Karimzadeh M, Nasiri M, Mirzaei HR, Pourhanifeh
MH, Bokharaei-Salim F, Mirzaei H and Hamblin MR: Pathogenic role of
exosomes and microRNAs in HPV-mediated inflammation and cervical
cancer: A review. Int J Cancer. 146:305–320. 2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Łaniewski P, Cui H, Roe DJ, Barnes D,
Goulder A, Monk BJ, Greenspan DL, Chase DM and Herbst-Kralovetz MM:
Features of the cervicovaginal microenvironment drive cancer
biomarker signatures in patients across cervical carcinogenesis.
Sci Rep. 9(7333)2019.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Gupta SM and Mania-Pramanik J: Retraction
note: Molecular mechanisms in progression of HPV-associated
cervical carcinogenesis. J Biomed Sci. 26(50)2019.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Zheng ZM and Wang X: Regulation of
cellular miRNA expression by human papillomaviruses. Biochim
Biophys Acta. 26:668–677. 2011.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Liu Z, Luo S, Wu M, Huang C, Shi H and
Song X: LncRNA GHET1 promotes cervical cancer progression through
regulating AKT/mTOR and Wnt/β-catenin signaling pathways. Biosci
Rep. 40(BSR20191265)2020.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Fouad YA and Aanei C: Revisiting the
hallmarks of cancer. Am J Cancer Res. 7:1016–1036. 2017.PubMed/NCBI
|
|
93
|
Oliveto S, Mancino M, Manfrini N and Biffo
S: Role of microRNAs in translation regulation and cancer. World J
Biol Chem. 8:45–56. 2017.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Zamani S, Sohrabi A, Hosseini SM,
Rahnamaye-Farzami M and Akbari A: Deregulation of miR-21 and
miR-29a in cervical cancer related to HPV infection. Microrna.
8:110–115. 2019.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Vaschetto LM: miRNA activation is an
endogenous gene expression pathway. RNA Biol. 156:826–828.
2018.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Zhou K, Liu M and Cao Y: New insight into
microRNA fnctions in cancer: Oncogene-microRNA-tumor suppressor
gene network. Front Mol Biosci. 4(46)2017.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Yeung CLA, Tsang TY, Yau PL and Kwok TT:
Human papillomavirus type 16 E6 suppresses microRNA-23b expression
in human cervical cancer cells through DNA methylation of the host
gene C9orf3. Oncotarget. 8:12158–12173. 2017.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Chen AH, Qin YE, Tang WF, Tao J, Song HM
and Zuo M: miR-34a and miR-206 act as novel prognostic and therapy
biomarkers in cervical cancer. Cancer Cell Int.
17(63)2017.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Dong P, Xiong Y, Hanley SJB, Yue J and
Watari H: Musashi-2, a novel oncoprotein promoting cervical cancer
cell growth and invasion, is negatively regulated by p53-induced
miR-143 and miR-107 activation. J Exp Clin Cancer Res.
36(150)2017.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Ofir M, Hacohen D and Ginsberg D: miR-15
and miR-16 are direct transcriptional targets of E2F1 that limit
E2F-induced proliferation by targeting cyclin E. Mol Cancer Res.
9:440–447. 2011.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Chen X, Cao R, Liu H, Zhang T, Yuan X and
Xu S: MicroRNA-15a-5p-targeting oncogene YAP1 inhibits cell
viability and induces cell apoptosis in cervical cancer cells. Int
J Mol Med. 46:1301–1310. 2020.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Zhou M, Chen X, Wu J, He X and Ren R:
MicroRNA-143 regulates cell migration and invasion by targeting
GOLM1 in cervical cancer. Oncol Lett. 16:6393–6400. 2018.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Sannigrahi MK, Sharma R, Singh V, Panda
NK, Rattan V and Khullar M: Role of host miRNA Hsa-miR-139-3p in
HPV-16-induced carcinomas. Clin Cancer Res. 23:3884–3895.
2017.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Jiang Z, Song Q, Zeng R, Li J, Li J, Lin
X, Chen X, Zhang J and Zheng Y: MicroRNA-218 inhibits EMT,
migration and invasion by targeting SFMBT1 and DCUN1D1 in cervical
cancer. Oncotarget. 7:45622–45636. 2016.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Zhu L, Tu H, Liang Y and Tang D: miR-218
produces anti-tumor effects on cervical cancer cells in vitro.
World J Surg Oncol. 16(204)2018.PubMed/NCBI View Article : Google Scholar
|
|
106
|
McLaughlin-Drubin ME and Münger K: The
human papillomavirus E7 oncoprotein. Virology. 384:335–344.
2009.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Myklebust MP, Bruland O, Fluge Ø,
Skarstein A, Balteskard L and Dahl O: MicroRNA-15b is induced with
E2F-controlled genes in HPV-related cancer. Br J Cancer.
105:1719–1725. 2011.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Liu Z, Wu M, Shi H, Huang C, Luo S and
Song X: DDN-AS1-miR-15a/16-TCF3 feedback loop regulates tumor
progression in cervical cancer. J Cell Biochem. 120:10228–10238.
2019.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Cheng Y, Geng L, Zhao L, Zuo P and Wang J:
Human papillomavirus E6-regulated microRNA-20b promotes invasion in
cervical cancer by targeting tissue inhibitor of metalloproteinase
2. Mol Med Rep. 16:5464–5470. 2017.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Kong Q, Wang W and Li P: Regulator role of
HPV E7 protein on miR-21 expression in cervical carcinoma cells and
its functional implication. Int J Clin Exp Pathol. 8:15808–15813.
2015.PubMed/NCBI
|
|
111
|
Cai L, Wang W, Li X, Dong T, Zhang Q, Zhu
B, Zhao H and Wu S: MicroRNA-21-5p induces the metastatic phenotype
of human cervical carcinoma cells in vitro by targeting the
von Hippel-Lindau tumor suppressor. Oncol Lett. 15:5213–5219.
2018.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Xu L, Xu Q, Li X and Zhang X: MicroRNA-21
regulates the proliferation and apoptosis of cervical cancer cells
via tumor necrosis factor-α. Mol Med Rep. 16:4659–4663.
2017.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Liu F, Zhang S, Zhao Z, Mao X, Huang J, Wu
Z, Zheng L and Wang Q: MicroRNA-27b up-regulated by human
papillomavirus 16 E7 promotes proliferation and suppresses
apoptosis by targeting polo-like kinase2 in cervical cancer.
Oncotarget. 7:19666–19679. 2016.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Ding H, Wu YL, Wang YX and Zhu FF:
Characterization of the microRNA expression profile of cervical
squamous cell carcinoma metastases. Asian Pac J Cancer Prev.
15:1675–1679. 2014.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Park S, Lee S, Kim J, Kim G, Park KH, Kim
TU, Chung D and Lee H: ΔNp63 to TAp63 expression ratio as a
potential molecular marker for cervical cancer prognosis. PLoS One.
14(e0214867)2019.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Coimbra EC, Da Conceição Gomes Leitão M,
Júnior MR, De Oliveira TH, Da Costa Silva Neto J and De Freitas AC:
Expression profile of microRNA-203 and its ΔNp63 target in cervical
carcinogenesis: Prospects for cervical cancer screening. Anticancer
Res. 36:3939–3946. 2016.PubMed/NCBI
|
|
117
|
Mandal P, Saha SS, Sen S, Bhattacharya A,
Bhattacharya NP, Bucha S, Sinha M, Chowdhury RR, Mondal NR,
Chakravarty B, et al: Cervical cancer subtypes harbouring
integrated and/or episomal HPV16 portray distinct molecular
phenotypes based on transcriptome profiling of mRNAs and miRNAs.
Cell Death Discov. 5(81)2019.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Balasubramaniam SD, Balakrishnan V, Oon CE
and Kaur G: Key molecular events in cervical cancer development.
Medicina (Kaunas). 55(384)2019.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Yi Y, Liu Y, Wu W, Wu K and Zhang W: The
role of miR-106p-5p in cervical cancer: From expression to
molecular mechanism. Cell Death Discov. 4(36)2018.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Cheng Y, Guo Y, Zhang Y, You K, Li Z and
Geng L: MicroRNA-106b is involved in transforming growth factor
β1-induced cell migration by targeting disabled homolog 2 in
cervical carcinoma. J Exp Clin Cancer Re. 35(11)2016.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Natalia MA, Alejandro GT, Virginia TJ and
Alvarez-Salas LM: MARK1 is a novel target for miR-125a-5p:
Implications for cell migration in cervical tumor cells. Microrna.
7:54–61. 2018.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Xu Y, Zhao S, Cui M and Wang Q:
Down-regulation of microRNA-135b inhibited growth of cervical
cancer cells by targeting FOXO1. Int J Clin Exp Pathol.
8:10294–10304. 2015.PubMed/NCBI
|
|
123
|
Li JH, Zhang Z, Du MZ, Guan YC, Yao JN, Yu
HY, Wang BJ, Wang XL, Wu SL and Li Z: MicroRNA-141-3p fosters the
growth, invasion, and tumorigenesis of cervical cancer cells by
targeting FOXA2. Arch Biochem Biophys. 657:23–30. 2018.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Zhu J and Han S: miR-150-5p promotes the
proliferation and epithelial-mesenchymal transition of cervical
carcinoma cells via targeting SRCIN1. Pathol Res Pract.
215:738–747. 2019.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Li N, Cui T, Guo W, Wang D and Mao L:
miR-155-5p accelerates the metastasis of cervical cancer cell via
targeting TP53INP1. Onco Targets Ther. 12:3181–3196.
2019.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Yang M, Zhai X, Ge T, Yang C and Lou G:
miR-181a-5p promotes proliferation and invasion and inhibits
apoptosis of cervical cancer cells via regulating inositol
polyphosphate-5-phosphatase A (INPP5A). Oncol Res. 26:703–712.
2018.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Farzanehpour M, Mozhgani SH, Jalilvand S,
Faghihloo E, Akhavan S, Salimi V and Azad TM: Serum and tissue
miRNAs: Potential biomarkers for the diagnosis of cervical cancer.
Virol J. 16(116)2019.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Hou T, Ou J, Zhao X, Huang X, Huang Y and
Zhang Y: MicroRNA-196a promotes cervical cancer proliferation
through the regulation of FOXO1 and p27Kip1. Br J Cancer.
110:1260–1268. 2014.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Varghese VK, Shukla V, Jishnu PV,
Kabekkodu SP, Pandey D, Sharan K and Satyamoorthy K: Characterizing
methylation regulated miRNA in carcinoma of the human uterine
cervix. Life Sci. 232(116668)2019.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Zhou LL, Shen Y, Gong JM, Sun P and Sheng
JH: MicroRNA-466 with tumor markers for cervical cancer screening.
Oncotarget. 8:70821–70827. 2017.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Chu Y, Ouyang Y, Wang F, Zheng A, Bai L,
Han L, Chen Y and Wang H: MicroRNA-590 promotes cervical cancer
cell growth and invasion by targeting CHL1. J Cell Biochem.
115:847–853. 2014.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Guerrero-Gómez AO and Guerrero-Florez M:
MicroRNAs asociados al Cáncer de Cuello Uterino y sus lesiones
precursoras: Una revisión sistemática. Univ Salud. 18:345–363.
2016.
|
|
133
|
Zhang H, Lu Y, Wang S, Sheng X and Zhang
S: MicroRNA-152 acts as tumor suppressor microRNA by inhibiting
Krüppel-like factor 5 in human cervical cancer. Oncol Res.
27:335–340. 2019.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Wang S, Gao B, Yang H, Liu X, Wu X and
Wang W: MicroRNA-432 is downregulated in cervical cancer and
directly targets FN1 to inhibit cell proliferation and invasion.
Oncol Lett. 18:1475–1482. 2019.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Lu Y, Hu J, Sun W, Li S, Deng S and Li M:
miR-29c inhibits cell growth, invasion, and migration of pancreatic
cancer by targeting ITGB1. Onco Targets Ther. 9:99–109.
2015.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Ma L and Li LL: miR-145 contributes to the
progression of cervical carcinoma by directly regulating FSCN1.
Cell Transplant. 28:1299–1305. 2019.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Duan S, Wu A, Chen Z, Yang Y, Liu L and
Shu Q: miR-204 regulates cell proliferation and invasion by
targeting EphB2 in human cervical cancer. Oncol Res. 26:713–723.
2018.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Shu L, Zhang Z and Cai Y: microRNA-204
inhibits cell migration and invasion in human cervical cancer by
regulating transcription factor 12. Oncol Lett. 15:161–166.
2018.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Li N, Guo X, Liu L, Wang L and Cheng R:
Molecular mechanism of miR-204 regulates proliferation, apoptosis
and autophagy of cervical cancer cells by targeting ATF2. Artif
Cells Nanomed Biotechnol. 47:2529–2535. 2019.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Shi C and Zhang Z: MicroRNA-320 suppresses
cervical cancer cell viability, migration and invasion via directly
targeting FOXM1. Oncol Lett. 14:3809–3816. 2017.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Hong H, Zhu H, Zhao S, Wang K, Zhang N,
Tian Y, Li Y, Wang Y, Lv X, Wei T, et al: The novel
circCLK3/miR-320a/FoxM1 axis promotes cervical cancer progression.
Cell Death Dis. 10(950)2019.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Cao XM: Role of miR-337-3p and its target
Rap1A in modulating proliferation, invasion, migration and
apoptosis of cervical cancer cells. Cancer Biomark. 24:257–267.
2019.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Hua FF, Liu SS, Zhu LH, Wang YH, Liang X,
Ma N and Shi HR: miRNA-338-3p regulates cervical cancer cells
proliferation by targeting MACC1 through MAPK signaling pathway.
Eur Rev Med Pharmacol Sci. 21:5342–5352. 2017.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Lu R, Yang Z, Xu G and Yu S: miR-338
modulates proliferation and autophagy by PI3K/AKT/mTOR signaling
pathway in cervical cancer. Biomed Pharmacother. 105:633–644.
2018.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Jayamohan S, Kannan M, Moorthy RK,
Rajasekaran N, Jung HS, Shin YK and Arockiam AJ: Dysregulation of
miR-375/AEG-1 axis by human papillomavirus 16/18-E6/E7 promotes
cellular proliferation, migration, and invasion in cervical cancer.
Front Oncol. 9(847)2019.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Shang A, Zhou C, Bian G, Chen W, Lu W,
Wang W and Li D: miR-381-3p restrains cervical cancer progression
by downregulating FGF7. J Cell Biochem. 120:778–789.
2019.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Teng P, Jiao Y, Hao M and Tang X:
microRNA-383 suppresses the PI3K-AKT-MTOR signaling pathway to
inhibit development of cervical cancer via down-regulating PARP2. J
Cell Biochem. 119:5243–5252. 2018.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Wang S, Zhang Y, Yuan S and Ji X:
MicroRNA-485 targets MACC1 and inhibits cervical cancer cell
proliferation and invasion. Mol Med Rep. 18:2407–2416.
2018.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Lv M, Ou R, Zhang Q, Lin F, Li X, Wang K
and Xu Y: MicroRNA-664 suppresses the growth of cervical cancer
cells via targeting c-Kit. Drug Des Devel Ther. 13:2371–2379.
2019.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Xu J, Wan X, Chen X, Fang Y, Cheng X, Xie
X and Lu W: miR-2861 acts as a tumor suppressor via targeting
EGFR/AKT2/CCND1 pathway in cervical cancer induced by human
papillomavirus virus 16 E6. Sci Rep. 6(28968)2016.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Jin Y, Zhou X, Yao X, Zhang Z, Cui M and
Lin Y: MicroRNA-612 inhibits cervical cancer progression by
targeting NOB1. J Cell Mol Med. 24:3149–3156. 2020.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Zhang JJ, Wang DD, Du CX and Wang Y: Long
noncoding RNA ANRIL promotes cervical cancer development by acting
as a sponge of miR-186. Oncol Res. 26:345–352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Liu S, Song L, Yao H, Zhang L, Xu D, Gao F
and Li Q: miR-375 is epigenetically downregulated by HPV-16 E6
mediated DNMT1 upregulation and modulates EMT of cervical cancer
cells by suppressing lncRNA MALAT1. PLoS One.
11(e0163460)2016.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Song H, Liu Y, Jin X, Liu Y, Yang Y, Li L,
Wang X and Li G: Long non-coding RNA LINC01535 promotes cervical
cancer progression via targeting the miR-214/EZH2 feedback loop. J
Cell Mol Med. 23:6098–6111. 2019.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Sun NX, Ye C, Zhao Q, Zhang Q, Xu C, Wang
SB, Jin ZJ, Sun SH, Wang F and Li W: Long noncoding RNA-EBIC
promotes tumor cell invasion by binding to EZH2 and repressing
E-cadherin in cervical cancer. PLoS One. 9(e100340)2014.PubMed/NCBI View Article : Google Scholar
|
|
156
|
He H, Liu X, Liu Y, Zhang M, Lai Y, Hao Y,
Wang Q, Shi D, Wang N, Luo XG, et al: Human papillomavirus E6/E7
and long noncoding RNA TMPOP2 mutually upregulated gene expression
in cervical cancer cells. J Virol. 93:e01808–e18. 2019.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Ma Z, Shuai Y, Gao X, Wen X and Ji J:
Circular RNAs in the tumour microenvironment. Mol Cancer.
19(8)2020.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Bach DH, Lee SK and Sood AK: Circular RNAs
in Cancer. Mol Ther Nucleic Acids. 16:118–129. 2019.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Zhao J, Lee EE, Kim J, Yang R, Chamseddin
B, Ni C, Gusho E, Xie Y, Chiang CM, Buszczak M, et al: Transforming
activity of an oncoprotein-encoding circular RNA from human
papillomavirus. Nat Commun. 10(2300)2019.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Ji F, Du R, Chen T, Zhang M, Zhu Y, Luo X
and Ding Y: Circular RNA circSLC26A4 accelerates cervical cancer
progression via miR-1287-5p/HOXA7 axis. Mol Ther Nucleic Acids.
19:413–420. 2020.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Chen RX, Liu HL, Yang LL, Kang FH, Xin LP,
Huang LR, Guo QF and Wang YL: Circular RNA circRNA_0000285 promotes
cervical cancer development by regulating FUS. Eur Rev Med
Pharmacol Sci. 23:8771–8778. 2019.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Ding S, Huang X, Zhu J, Xu B, Xu L, Gu D
and Zhang W: ADH7, miR-3065 and LINC01133 are associated with
cervical cancer progression in different age groups. Oncol Lett.
19:2326–2338. 2020.PubMed/NCBI View Article : Google Scholar
|