1. Introduction
Fanconi anemia (FA) was first described in 1927 by
Dr. Guido Fanconi, who observed a family of 3 siblings that
presented with several physical abnormalities and pernicious anemia
(Fig. 1) (1). FA is defined as a rare genetic disease
of chromosomal instability that affects the proteins involved in
DNA repair and the regulation of the cell cycle (2). In the majority of cases, patients with
FA present with an autosomal recessive inheritance pattern, where
the clinical effect is progressive depletion in bone marrow
function, congenital malformations and a high risk of developing
solid and hematological tumors at an earlier age than the general
population (2). FA is not the same
as Fanconi syndrome; the latter is a hereditary or acquired defect
of the proximal tubule that leads to the malabsorption of multiple
electrolytes and substances usually reabsorbed in this region
(3).
FA has an incidence of 1 in 300,000 live births and
a prevalence of 1-9 per million (4).
The carrier frequency varies according to the populations based on
the founding mutations; this is how the carrier prevalence reported
in the Afrikaans population in South Africa is 1 in 83(5), in Ashkenazi Jews is 1 in 100(6) and in the Spanish gypsies is 1 in 64 to
1 in 70(7), compared with the
general population, where it is ~1 in 189(8). In general, the male:female ratio of the
presentation of the disease is 1.2:1(9). This disease is a consequence, in the
majority of the cases, of biallelic mutations in the 22 genes that
been determined to be involved in DNA repair and genome stability,
termed complementation groups FANCA-FANCW (10,11). The
primary inheritance pattern is autosomal recessive (genes FANCA,
FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI,
FANCJ/BRIP1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4,
FANCQ/ERCC4, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7
and FANCW/RFWD3) (11,12),
except for FANCB, which exhibits X-linked recessive
inheritance (13), and
FANCR/RAD51, which presents a de novo autosomal
dominant inheritance pattern (14,15).
All proteins encoded by the aforementioned genes
participate in the FA/BRCA repair pathway, which detects damage
that covalently binds the two DNA strands (interstrand
crosslinking; ICL) and coordinates their repair through homologous
monoubiquitination and recombination (16).
ICLs are formed in DNA by the presence of exogenous
agents, such as cancer chemotherapeutics, as well as by endogenous
agents, such as alcohol metabolites, cigarette smoke, acetaldehyde
and malondialdehyde (17). These
lesions cause the blockade of transcription and replication forks,
making it impossible to separate double-stranded DNA; at this
point, the response to DNA damage and the homologous recombination
repair processes that act in the S-phase are activated (11). Individuals without compromises in
this group of genes manage to eliminate ICLs; however, patients
with FA do not have the optimal machinery. Unrepaired ICLs lead to
DNA breakage and nonhomologous end-joining of free ends, which are
visible and countable on metaphase chromosomes, in a chromosome
breakage study (17). Consequently,
there is an accumulation of damage in the genome, chromosomal
instability, a high risk of cancer and congenital malformations in
the majority of those individuals affected (16). Recently, FA proteins were discovered
to fulfill other noncanonical functions in maintaining the
integrity of genetic information and cellular metabolism, which
explains the complexity of the FA/BRCA pathway, and its
dysregulation in the etiology of the phenotype (18).
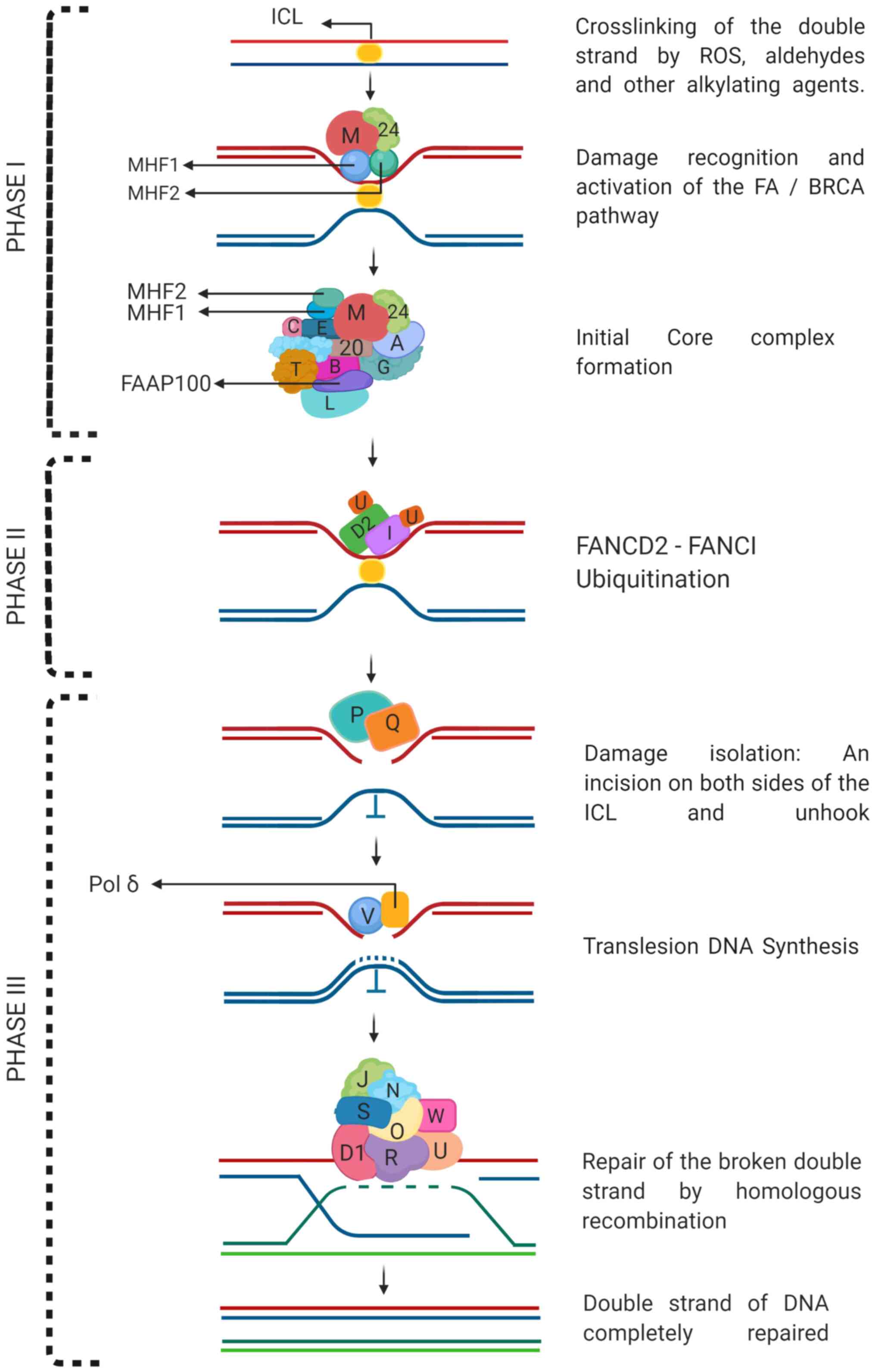
The canonical function of FANC proteins is to repair
ICLs, which can be divided into three phases: i) damage
recognition, AF core complex activation, and FAND2 and FANCI
monoubiquitination; ii) FANCD2-FANCI complex formation; and iii)
activation of the DNA repair complex and repair (18,19)
(Fig. 2).
In the first stage, FANCM, together with the non-AF
protein FAAP24 and the DNA-binding cofactors
histone-fold-containing FANCM-associated protein (MHF)1 and MHF2,
recognize the lesion site (ICL) and immediately recruit FA and
non-FA proteins to form the core complex (FANCA, FANCB, FANCC,
FANCE, FANCF, FANCG, FANCL, FANCT/UBE2T, FAAP100 and FAAP20), which
monoubiquitinates FANCD2 and FANCI (20-22).
In the second stage, the FANCD2-FANCI complex or ID complex is
formed, with ubiquitination that allows its displacement to the
site of damage (focus formation) and the coordination of the repair
of the ICL (23). Finally, in stage
three, the recruitment of FA and other non-FA proteins that make
DNA incisions on both sides of the ICL and unhook it (FANCP/SLX4
and FANCQ/XPF), translesion DNA synthesis (FANCV/REV7 and other
non-FA proteins), and repair by homologous recombination through
the formation of Holliday junction intermediaries occur for the
synthesis and final replacement of double-stranded sequences of the
DNA (FANCD1/BRCA2, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4,
FANCQ/ERCC4, FANCR/RAD51, FANCS/BRCA1, FANCU/XRCC2, FANCV/REV7 and
FANCW/RFWD3). During the S phase, the detection of ICLs leads to
their repair, and proteins such as ATR-CHK1 activate the cell cycle
control point to decrease the speed of DNA replication and allow
repair to be finished (16,19).
Early diagnosis of FA allows anticipation of
possible complications and thus affects the prognosis. Early
diagnosis may be based on clinical suspicions and the positive
findings of genetic analysis, and the chromosomal breakage test is
used to confirm the diagnosis; molecular tests such as western
blotting, multiplex ligation-dependent probe amplification (MLPA)
and gene sequencing studies using next-generation sequencing (NGS)
are also used as diagnostic methods (24,25).
This review presents the clinical, genetic and diagnostic aspects
of FA for healthcare professionals.
2. Clinical presentation
Patients with FA present with congenital
malformations, bone marrow failure that manifests as pancytopenia,
and a predisposition to cancer. Practically all systems are
affected by the disease; however, the clinical presentation has a
variable expressiveness (2). Not all
patients present malformations or pancytopenia at birth, and the
first manifestation of FA in these individuals may be solid tumors,
hematologic malignancies or other complications, such as
infertility (2,26). Physical abnormalities are found in
75% of all patients with FA and may be accompanied by a low birth
weight, short pre- and postnatal height and microcephaly (27,28). The
other 25% of patients represent a challenge for clinicians because
the absence of physical abnormalities can delay clinical suspicion,
and therefore, the timely diagnosis of the disease (29). Below are the most frequent
alterations found in the different organs.
Head and neck
The face of patients with FA has particular
identifiable characteristics, such as a triangular face, bilateral
epicanthic folds, micrognathia and middle facial hypoplasia. Ocular
findings such as microphthalmia, cataracts, astigmatism,
strabismus, hypotelorism, hypertelorism and ptosis have been
described (30,31). The neck may be short with low
implantation of the hairline, pterygium Colli, Sprengel deformity
and Klippel-Feil anomaly (2).
Cardiac and gastrointestinal
system
Cardiac malformations can be persistent arterial
ducts, atrial or ventricular septal defects, coarctation of the
aorta, common arterial trunk and situs inversus totalis (32). Only 5% of patients have
gastrointestinal abnormalities such as tracheoesophageal fistula,
esophageal, duodenal or jejunal atresia, imperforate anus, annular
pancreas and intestinal malrotation (33).
Bone and limb defects
Limb defects are the most suggestive for diagnosis.
However, limb defects are not observed in all patients, and may
involve both the upper and lower extremities unilaterally or
bilaterally (34). In the upper
extremities, the radius, thumbs and hands (hypoplasia of the thenar
and hypothenar eminence) are the most affected regions and less
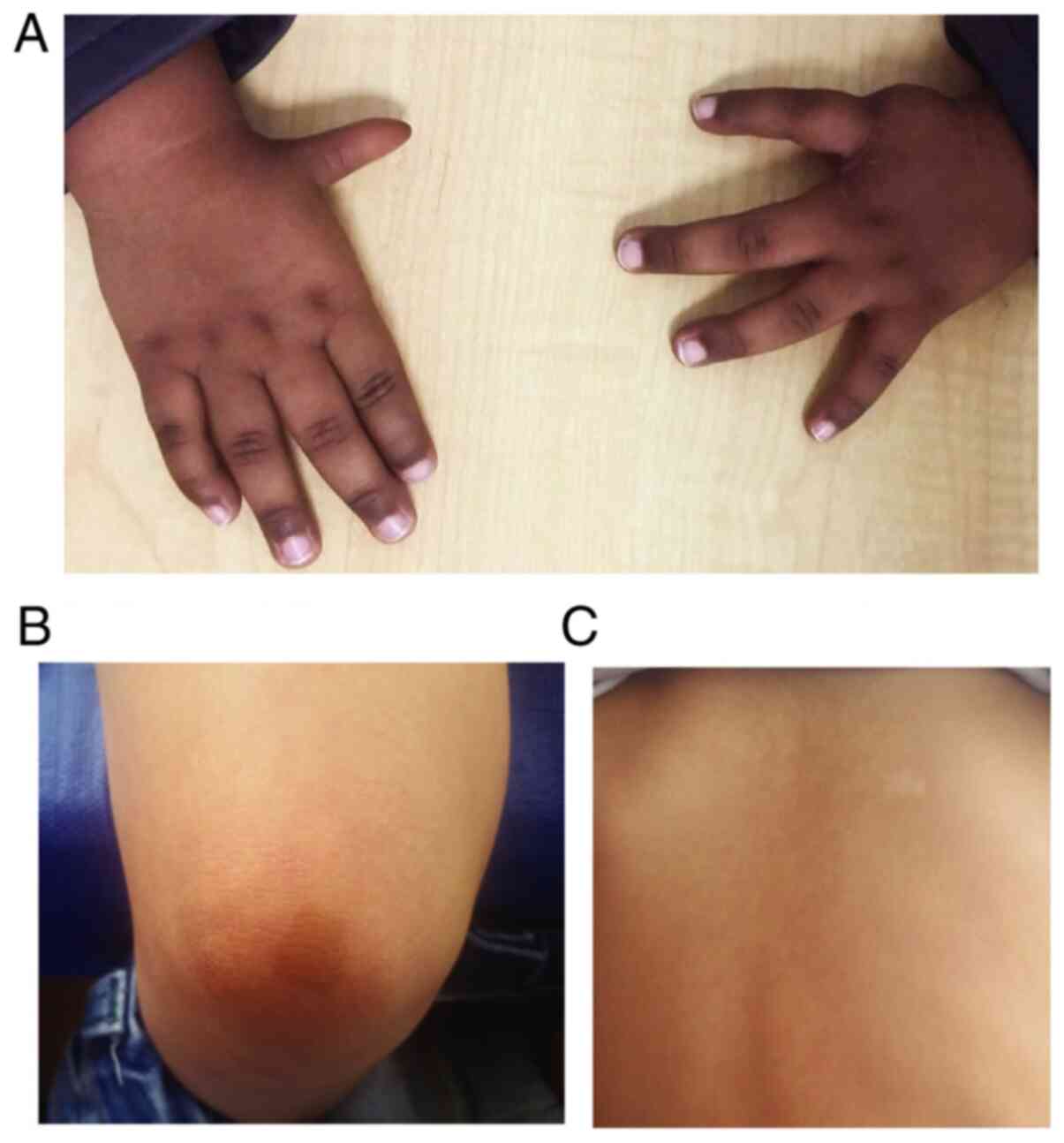
frequently, the ulna (Fig. 3A; most
common malformations of the thumbs and palms of patients with FA).
In the lower extremities, congenital dislocation of the hip,
syndactyly and talipes have been reported (30).
In 2% of patients, spina bifida, scoliosis and
abnormalities of the vertebrae (hemivertebrae, rib abnormalities,
coccygeal aplasia) may be observed (2). Bone abnormalities have also been
reported in the middle ear, associated with conductive hearing loss
or alterations in the conformation of the auricular pavilion, which
may be dysplastic or absent, low-set ears, narrow or absent
internal auditory canal, absent tympanic membrane, microtia and/or
fused ossicles (29).
Genitourinary system
Kidney malformations, such as horseshoe kidney,
ectopic, hypoplastic, dysplastic or absence of one kidney in
addition to hydronephrosis or hydroureter, have been reported in
patients with FA (30). Males can
present with hypospadias, micropenis, cryptorchidism, oligospermia
or azoospermia, and abnormal spermatogenesis is associated with
infertility (26). Females may
exhibit malposition of the uterus, bicornuate uterus and smaller
ovaries. Up to 50% of women are infertile, and when they achieve
pregnancy, they can have complications of rapid progression, such
as bone marrow failure, preeclampsia and premature delivery
(29).
Endocrinological system
In the endocrinological field, >60% of affected
individuals may present with a short stature due to growth hormone
deficiency, hypothyroidism, or glucose or insulin abnormalities.
Due to the endocrinological abnormalities, affected individuals
should be monitored with regard to their hormonal profile (35,36).
Cutaneous system
The alterations described in the skin are
generalized hyperpigmentation, hypopigmentation and cafe-au-lait
spots (Fig. 3B; Cafe-au-lait spots
and hypopigmentation in patients with FA evaluated by our FA
group). The areas of hyperpigmentation are primarily on the trunk,
neck, groin and armpits, with a mottled pattern of large patches
and diffuse boundaries (30).
Nervous system
Malformations such as small pituitary gland, an
absent corpus callosum, pituitary stalk interruption syndrome,
cerebellar hypoplasia, hydrocephalus and dilated ventricles in the
central nervous system have been described. Additionally, some
affected individuals may present with neurodevelopmental delays and
intellectual disability (33).
Hematological system
The cells of patients with FA exhibit chromosomal
instability generated by the presence of unrepaired damage during
the S-phase; stagnation in the G2 phase or passage to mitosis
without adequate DNA repair has been proposed as one of the
mechanisms that induces the depletion of hematopoietic cells by
cellular senescence and the presence of damage that eventually
leads to bone marrow failure, myelodysplastic syndrome or acute
leukemia (2,16).
The age of onset of bone marrow failure is very
variable, even in the same family, and it rarely manifests in the
lactation period. The average age of onset of hematological
symptoms is 7 years (9). Bone marrow
failure is one of the manifestations most commonly associated with
FA, so in patients with mild or imperceptible congenital
malformations, the diagnosis tends to be delayed until the onset of
cytopenia (37).
Generally, at the onset of the disease,
thrombocytopenia or leukopenia are present, followed by anemia in
fewer cases (38). In several cases,
macrocytosis and increased fetal hemoglobin are observed. As bone
marrow failure advances, progression to pancytopenia occurs;
therefore, patients with persistent and idiopathic FA cytopenia
should be suspected (39). According
to Kutler et al (40),
individuals with FA have a 90% risk of developing a hematologic
abnormality by the age of 40.
Patients with FA have a high risk of developing
myelodysplastic syndrome, which usually precedes acute myeloid
leukemia; this condition is associated with chromosomal findings in
the bone marrow, such as a gain of 3q, monosomy 7 or deletions in
7q (2,41).
As mentioned above, involvement of the hematopoietic
system is the most common clinical feature in FA. The failure of
the bone marrow occurs early in the life of those affected, and the
median survival rate is 21 years if it is not treated early
(9). Currently, the only curative
treatment to restore the function of the hematological system is
hematopoietic stem cell transplantation. However, this procedure
requires the intervention of a trained team due to the risk of
recurrence, graft-versus-host disease, and mortality (42). Recently, new treatment strategies,
such as gene therapy have been implemented, in which the
complications associated with hematopoietic stem cell
transplantation may be avoided (38). One of these therapies uses lentiviral
vectors to transfer a functional copy of a specific gene (the gene
that is mutated) to autologous hematopoietic stem cells. These
newly edited cells remove hematologic abnormalities in the patient
and restore bone marrow cell function. The mobilization of the stem
cells of the patient from the bone marrow into the peripheral blood
has been proposed to collect CD34+ cells, correct the
genetic alterations, and then infuse the cells into the patient
(38). To date, these therapies have
a good safety profile, but additional studies are required to
investigate the possible long-term effects.
Solid tumors
In patients with FA, solid tumors have an
accumulative incidence of 28% by the age of 40 years old (40,43).
Solid tumors commonly occurring in the anogenital area, and the
head and neck are 500 and 700 times more frequent in patients with
FA than in the healthy population, especially in cases with
transplanted hematopoietic stem cells (44). Tumors in the brain, in the liver
(secondary to androgen treatment) and in the kidney (as Wilms
tumor) can appear as other tumors (40,44).
With this condition in mind, the patients should be monitored
throughout a patient's life.
The most frequent carcinomas associated with FA are
squamous cell carcinoma of the head and neck, preferentially
located in the oral cavity, with the tongue being the most commonly
affected region (45). Carcinomas
appear at an earlier age than in the general population (20-40
years old) and the patients may exhibit exacerbated
radiosensitivity to therapy (46,47).
Patients with FA have a high risk of cancer associated with human
papillomavirus, which is why they should be vaccinated (48).
The defects in DNA repair in these patients makes
them extremely sensitive to chemotherapy and radiotherapy;
therefore, the suggested therapy is surgical resection of the
tumor. However, in cases where the diagnosis is belated and large
tumor sizes are encountered in surgical management, the use of
radiotherapy or chemotherapy is recommended as the only scheme or
in association with surgery (43,49).
Currently, the development of more secure and effective therapy
protocols for the treatment of squamous cell tumors of the head and
neck has been prioritized. Preclinical trials are being performed
with drugs already approved for the treatment of cancer, with
efficient cytotoxic and cytostatic activity that is nongenotoxic
for the cells of patients with FA (50).
Fig. 3 presents the
images of the most common alterations of 2 patients evaluated by
our FA group. Fig. 3A corresponds to
a 10-month-old male patient, with low height and weight at birth,
bilateral thumb hypoplasia, ectopic right kidney, renal tubular
acidosis, dysgenesis of the corpus callosum and colpocephaly,
hypochromic spots, very brown skin and mild pancytopenia. Fig. 3B and C correspond to a 3-year-old
male patient. Low height and weight, Left preaxial polydactyly,
bilateral hypoplasia of thenar eminence, café-au-lait spots,
hypochromic spots, mild VSD and aplastic anemia.
3. Differential diagnoses
As mentioned above, FA has variable expressiveness
and can affect a range of systems. These characteristics overlap
with several clinical manifestations of other syndromes, which
often leads to a delay in an accurate diagnosis. Thus, according to
the phenotype of the patient, the attending physician must consider
different diagnoses.
At birth, malformations are the first signs that
allow health professionals to suspect exposure to teratogens or
congenital infection. Once acquired causes are ruled out, a genetic
etiology must be considered. A complete systematic physical
examination makes it possible to suspect a syndromic entity.
An example of differential diagnosis is esophageal
atresia with or without tracheoesophageal fistula, which can be
found at a low frequency in FA, and can also be related to the
VACTERL association, and syndromes such as trisomy 21 and
Klippel-Feil (51,52). However, in the case of a patient
without a history of malformations with idiopathic bone marrow
insufficiency, FA or other syndromes predisposing an individual to
bone marrow failure or cancer must be considered, or even syndromes
that affect DNA repair genes. FA is included in inherited bone
marrow failure syndrome (IBMFS), which present certain common
clinical signs that make diagnosis difficult. IBMFS typically
includes cytopenia of at least one hematopoietic cell lineage that
can progress to pancytopenia, in addition to an elevated risk of
hematologic and solid tissue cancer (53). IBMFS also includes Blackfan Diamond
anemia, congenital dyskeratosis, Shwachman-Diamond syndrome,
amongst other, less familiar conditions (9,53,54-58).
The clinical characteristics of each syndrome allow its diagnosis;
however, phenotypic overlap between them frequently occurs,
affecting the diagnosis and timely treatment (9,59). In
these cases, it is recommended to use differential diagnostic
methods to confirm a diagnosis of FA (9). Table I
describes the symptoms of certain conditions that are
differentially diagnosed for FA. Research on these types of
clinical conditions avoids incorrect or under diagnosis, and
adequately determines the specific follow-up and prognosis of the
affected individual.
 | Table ISyndromes with clinical
characteristics common to FA. |
Table I
Syndromes with clinical
characteristics common to FA.
| Diagnosis | Type of
inheritance | Genes | Clinical factors
common with Fanconi Anemia | Clinical factors
not common to FA | (Refs.) |
|---|
| Diamond-Blackfan
anemia | AD | RPS7, RPS17,
RPS19, RPS24, RPL5, RPL11, RPL35Aa | 1. Congenital
aregenerative anemia, generally macrocytic with erythroblastopenia.
2. Short stature, Pierre-Robin sequence, urogenital, and thumb
abnormalities. 3. Leukemia and increased risk of cancer. 4. Early
age of diagnosis. | Pure red cell
aplasia | (54) |
| Shwachman-diamond
syndrome | AR | SBDS,
EFL1 | 1. Hematological
disorder: Thrombocytopenia and anemia, increased fetal hemoglobin.
Some cases progress to bone marrow aplasia. 2. Presents as
ichthyosis, bone abnormalities, such as metaphyseal dysostosis, and
delayed motor neurodevelopment. | Pancreatic
lipomatosis, exocrine pancreatic insufficiency | (9) |
| Evans Syndrome
(immune pancytopenia) | - | - | 1. Chronic
hematological disorder, characterized by autoimmune hemolytic
anemia, immune thrombocytopenic purpura, occasionally autoimmune
neutropenia. 2. Manifests itself in childhood or adulthood. | Autoimmune
disorder. Hemolytic anemia and thrombocytopenia of immunological
origin | (9,55) |
|
Thrombocytopenia-absent radius
syndrome | AR | RBM8A | 1. Bilateral
absence of radius, thrombocytopenia, cardiac malformations. 2.
Patients may present abnormalities in the ulna, humerus,
phocomelia, and the lower extremities. | Thumbs are always
present | (56) |
| VACTERL
association | - | - | Association of
congenital malformations and at least three of the following:
vertebral defect, anal atresia, heart defects, tracheoesophageal
fistula, renal anomalies and anomalies in the extremities. | It does not present
with microcephaly, or hematological affection | (57) |
| Baller-Gerold
syndrome | AR | RECQL4 | 1. Association of
coronal craniosynostosis, facial and radial axis anomalies, such as
oligodactyly, aplasia, or hypoplasia of the thumb or radius. 2.
Risk of cancer, predominantly osteosarcoma. | Craniosynostosis,
with the coronal suture being the most commonly affected
region. | (58) |
4. Diagnostic methodologies
In the past, cases of FA were recognized based on
the association of aplastic anemia and birth defects. However,
overtime, the criteria have become more extensive, and a diagnosis
is now established based on a test using hypersensitivity to
clastogenic chemical agents such as diepoxybutane (DEB) or
mitomycin C (MMC), where damage is associated with the formation of
ICLs in DNA (8), or by the
identification of pathogenic variants in the genes associated with
FA through molecular studies. The most common tests for diagnosis
are described below.
Cytogenetics
The cells of patients with FA exhibit exacerbated
sensitivity to cytoreduction regimens used for bone marrow
transplantation and hypersensitivity to agents that cause DNA
interstrand crosslinks (60). This
characteristic is the basis for the chromosome breakage test, which
exposes lymphocytes or fibroblasts from individuals with suspected
FA to cisplatin, MMC or DEB in vitro (61,62).
The test consists of counting both spontaneous and
induced ruptures in the metaphase chromosomes of the patients after
exposure of the cells to the aforementioned agents, and comparing
those ruptures with those of a healthy control individual with
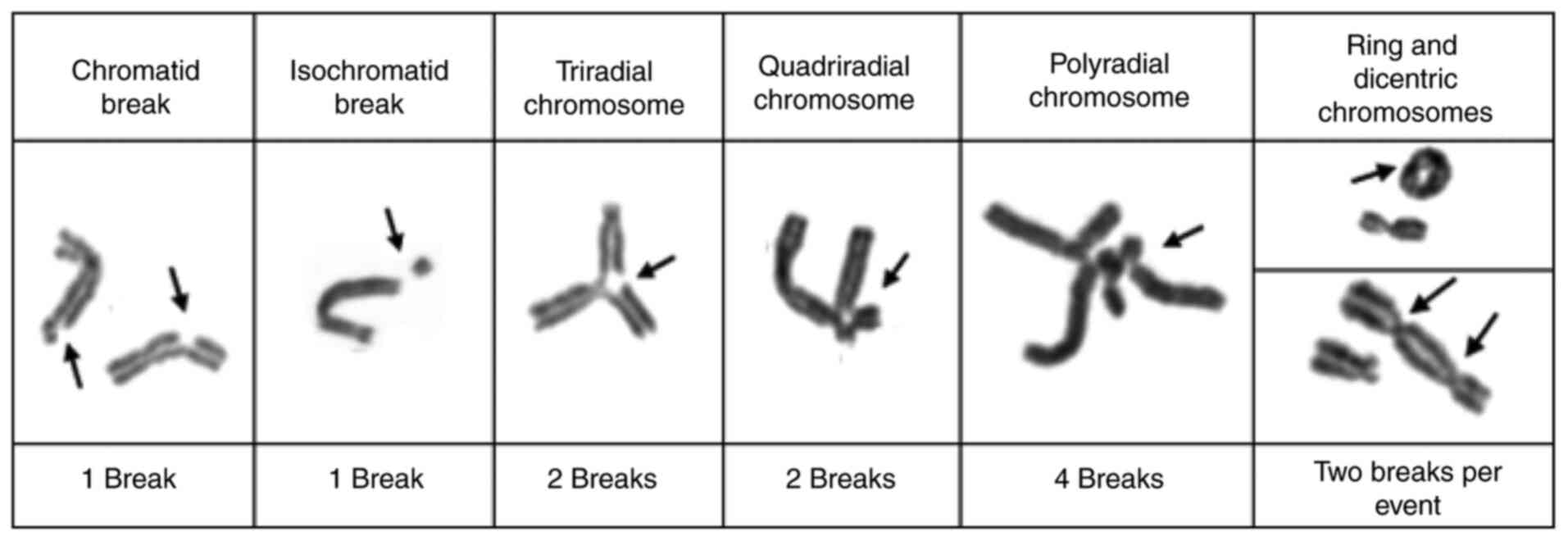
similar demographic characteristics. The number of chromosomal
breaks per cell, the presence of radial figures, and the proportion
of aberrant cells (one or more breaks/cell) are identified and
recorded. A patient with FA will exhibit a significant increase in
chromosomal breaks and radial figures compared with the control
individual, although there may be variations in this value in
patients with FA with somatic mosaicism (62,63). At
present, cytogenetic tests are considered the gold standard in the
diagnosis of FA (64). Fig. 4 shows the different fragility
expression chromosomal events that must be evaluated to establish
the diagnosis of FA.
In ~25% of cases of FA, patients may present with
somatic mosaicism, which reduces or eliminates lymphocyte
sensitivity to clastogenic agents. The presence of this condition
causes difficulties in the identification of affected individuals
utilizing the chromosomal breakage test (65). Somatic mosaicism in Mendelian
hematopoietic disorders is the process in which one pathogenic
mutation is reversed in a cell of a tissue, resulting in a group of
cells with the defect corrected (66). In FA, this process occurs primarily
in hematopoietic stem cells or lymphocyte progenitors. The
corrected cells proliferate and clonally expand, improving the
blood counts of the individual and thus reducing the incidence of
bone marrow failure and hematological malignancy (67).
The cytogenetic test analyzes peripheral blood T
cells; in this way, a high proportion of reversed T cells can lead
to a false-negative result. Some individuals without FA may present
with a proportion of T cells sensitive to DEB or MMC treatment,
which could be interpreted as mosaicism, generating false positives
(64). To resolve the overlap
between patients with mosaic FA and non-FA patients, and to
discriminate non-mosaics in patients with FA, it is proposed to use
the chromosome fragility index (CFI), which calculates the quotient
between the percentage of aberrant cells (cells with 1 or more
breaks) and the number of breaks in multi-aberrant cells (cells
with 2 or more breaks): [CFI = percentage of aberrant cells x
(number of aberrations/ number of multi-aberrant cells)]; Fig. 4 shows the establishment of the number
of breaks per chromosome or chromosomal event. For the Spanish
population, a patient with suspected FA and a CFI >55 is
considered to have FA, while within the group diagnosed with FA,
when a patient has <40% aberrant cells, it is considered mosaic
(64). In studies of patients
lymphocytes, where they have been reported as normal or
inconclusive and reversal mosaicism of their bone marrow mutation,
but FA is suspected, a test of sensitivity to ICL-inducing agents
in fibroblasts is recommended (62).
MLPA and array comparative genomic
hybridization (array-CGH)
In ~70% of cases of FA, the cause of the disease are
pathogenic variants in the FANCA gene. Although the majority
of these variants are produced by point mutations, up to 44% of
cases can be caused by small deletions/insertions and large
intragenic deletions. These types of variants have not only been
described in the FANCA gene but also in other genes related to the
disease (68).
MLPA allows the detection of intragenic deletions in
FANC genes and is recommended for the initial screening of patients
when the proportion of large intragenic deletions amongst mutated
alleles is high, as it happens for the FANCA gene (69). This test is useful to confirm or
dismiss compound heterozygosis, which explains the phenotype of the
patient. Additionally, the analysis for the search for large
intragenic deletions can be performed by array-CGH, which allows
establishing the extension of the deletions beyond the limits of
any FANC gene, and the exact points of breakage and loss in the
chromosome (68). The array-CGH test
is important as the additional loss of other genes involved in the
deletion may contribute to the phenotype of the patient (68).
Molecular test
With the advent of NGS, the identification of new
genes associated with FA has been achieved and has allowed the
analysis of several genes involved in different diseases, where
clinical diagnosis is not easy. Currently, 22 genes have been
confirmed to cause FA.
Within the molecular test, clinical exome sequencing
or the panel of genes specifically analyzes the exons of the genes
that are involved in the disease. However, despite having improved
coverage and specificity, the molecular test has the disadvantage
that not all panels include the same number of genes and have a low
cover of intronic regions (70). Due
to the above disadvantages, the specialist must ensure that the
requested study contains all the genes associated with FA. For
carriers or prenatal tests, the sequencing of a single gene or a
specific mutation is indicated.
Once the pathogenic variant associated with FA has
been identified, carriers in the family can be identified. The case
of an autosomal recessive inheritance pattern will require the
study of the parents to determine their carrier status and the risk
to offspring. In the case of identifying a female carrier under the
context of an X-linked recessive disease, the specialist should
advise not only on the risks but also on the options for having
healthy children, such as preimplantation diagnosis (2). In situations where the disease is
considered sporadic, the patient must attend a consultation with
his geneticist if they wish to have children.
5. Genetic counseling
Counseling is based around the inheritance pattern
and the genes involved in the disease. Parental consanguinity
should be questioned, as the autosomal inheritance pattern is most
commonly associated with this syndrome (2).
In autosomal recessive FA, each child of a couple
with a pathogenic variant has a 25% probability of inheriting both
pathogenic variants and being affected, a 25% probability of
inheriting both benign variants and being healthy, or 50%
probability of being a carrier by inheriting a single pathogenic
variant (heterozygous). When an affected individual is diagnosed
with FA, there is a possibility of another asymptomatic affected
individual amongst the siblings; here, cytogenetic tests should be
used on all siblings to rule out the disease, as a timely diagnosis
can improve the prognosis.
In autosomal dominant FA caused by a pathogenic
variant in the RAD51 gene, two cases have been reported,
each with a de novo variant, so the risk of having this same
disease for other family members is presumed to be very low
(14,15). Although only 2 cases are known, the
search should not be discarded, especially if clinical suspicion is
high.
Considering to whom the counseling is given in the
X-linked recessive inheritance is overriding because of the
probability of transmitting the pathogenic variant changes. If
genetic counseling is provided to an affected man with a healthy
partner, their daughters will be carriers of the pathogenic
variant, but not their sons, since they obtain the Y chromosome
from their father. Carrier women have a 50% chance of transmitting
the pathogenic variant in each pregnancy. According to this
scenario, male children who inherit the pathogenic variant of the
mother will be affected, whilst women will be healthy carriers.
Distinguishing heterozygous carriers from
noncarriers has genetic implications, as they may have an increased
risk of cancer. For example, carriers of heterozygous pathogenic
variants in FANCD1/BRCA2 have an increased risk of breast
and ovarian cancer (65). Knowing
the pathogenic variants of the family is a priority to identify
carriers or other affected members.
6. Other issues regarding diagnosis
FA is a rare disease, general knowledge of which is
still limited. At present, for several countries in the world,
including in our country of Columbia, the behavior of this disease
is not known in terms of its incidence, clinical characteristics,
genetics and treatment, reflecting the insufficient knowledge in
the medical community that exists in the clinic, despite the
existence of specialized guidelines produced by the Fanconi Anemia
Research Fund (initially created in 1999, now on the fifth edition,
which was published in 2020) (71),
and the abundance of pertinent literature (2,9,29,31,33,44,72).
In Colombia unlike in European countries, although
the healthcare system allows access to cytogenetics and molecular
studies, there is a significant lack of knowledge amongst
healthcare professionals regarding the clinical manifestations
associated with this condition. Therefore, referrals with
specialists trained to diagnose orphan diseases are not
prioritized. However, although the cytogenetic test is performed by
several diagnostic centers, personnel trained in these types of
tests are scarce, and there are no specialized reference centers
for these studies in fibroblasts. Therefore, the cytogenetic group
of Pontificia Universidad Javeriana in recent years has dedicated
itself to investigating this disease in Colombia. In the workgroup
experience with patients with FA, a delay has been observed in the
diagnosis of several cases and the timely treatment of
hematological complications, due primarily to the inadequate
application of confirmatory cytogenetic tests for FA, confusion of
the phenotype with other clinical entities, and the untimely
evaluation of the patient under the clinical geneticist criteria.
Thus, it is recommended that both the medical staff and the
cytogenetic diagnosis should be periodically trained and updated,
respectively.
7. Conclusions
FA is a hereditary disease in which there is a
compromise of genes involved in DNA repair. When the cell cannot
adequately repair its genetic information due to damage to this
machinery, various clinical manifestations, such as bone marrow
failure, congenital malformations and an increased predisposition
to cancer occur, which increases the morbidity and mortality of
these patients. Before the era of in-depth molecular studies,
multiple diseases with variable expressivity could not be
clinically confirmed, which limited the efforts of the
professionals to improve the prognosis of affected individuals.
Cytogenetic and molecular studies allowed for confirmation of
diagnoses of diseases with shared symptoms and has facilitated the
development of knowledge of the phenotype-genotype relationship, as
well as the pathophysiology of the diseases. Both clinical and
paraclinical criteria support early diagnosis, prognosis
evaluation, adequate monitoring and genetic counseling for
individuals and families of patients with FA.
Acknowledgements
We would like to thank Dr Jordi Surrallés of the
Autonomous University of Barcelona (Spain) and to Dr Javier Benítez
of the National Center for Oncological Research (CNIO; Madrid,
Spain) for their academic support in the development of the FA
research.
Funding
This review was partly funded by a Fanconi Anemia Grant financed
by Pontificia Universidad Javeriana (Columbia; grant no. ID
00006453. Ref: 2014/150).
Availability of data and materials
Not applicable.
Authors' contributions
OMM and ACP searched the literature, reviewed the
articles and collected the relevant data from selected papers. OMM,
ACP, AR wrote the manuscript. ACP and FSO reviewed the clinical
articles. All authors have read and approved the final manuscript.
Data authentication is not applicable.
Ethics approval and consent to
participate
Informed consents was signed by the parents of the
patients whose images are presented (approval no.
CIE-2014/150).
Patient consent for publication
Consent for publication was provided by the parents
of the patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wiedemann HR: Guido Fanconi (1892-1979) in
memoriam. Eur J Pediatr. 132:131–132. 1979.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mehta PA and Tolar J: Fanconi Anemia. In:
GeneReviews®. Adam MP, Ardinger HH, Pagon RA, et
al (eds). University of Washington, Seattle, WA, 2002.
|
|
3
|
Keefe P and Bokhari SRA: Fanconi Syndrome.
In: StatPearls. StatPearls Publishing, Treasure Island, FL,
2021.
|
|
4
|
Bagby GC: Multifunctional Fanconi
proteins, inflammation and the Fanconi phenotype. EBioMedicine.
8:10–11. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tipping AJ, Pearson T, Morgan NV, Gibson
RA, Kuyt LP, Havenga C, Gluckman E, Joenje H, de Ravel T, Jansen S,
et al: Molecular and genealogical evidence for a founder effect in
Fanconi anemia families of the Afrikaner population of South
Africa. Proc Natl Acad Sci USA. 98:5734–5739. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Strom CM, Crossley B, Redman JB, Quan F,
Buller A, McGinniss MJ and Sun W: Molecular screening for diseases
frequent in Ashkenazi Jews: Lessons learned from more than 100,000
tests performed in a commercial laboratory. Genet Med. 6:145–152.
2004.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Callén E, Casado JA, Tischkowitz MD,
Bueren JA, Creus A, Marcos R, Dasí A, Estella JM, Muñoz A, Ortega
JJ, et al: A common founder mutation in FANCA underlies the world's
highest prevalence of Fanconi anemia in Gypsy families from Spain.
Blood. 105:1946–1949. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rosenberg PS, Tamary H and Alter BP: How
high are carrier frequencies of rare recessive syndromes?
Contemporary estimates for Fanconi Anemia in the United States and
Israel. Am J Med Genet A. 155A:1877–1883. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Shimamura A and Alter BP: Pathophysiology
and management of inherited bone marrow failure syndromes. Blood
Rev. 24:101–122. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
D'Andrea AD: The Fanconi road to cancer.
Genes Dev. 17:1933–1936. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Niraj J, Färkkilä A and D'Andrea AD: The
Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol. 3:457–478.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ceccaldi R, Sarangi P and D'Andrea AD: The
Fanconi anaemia pathway: New players and new functions. Nat Rev Mol
Cell Biol. 17:337–349. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Meetei AR, Levitus M, Xue Y, Medhurst AL,
Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, et al:
X-linked inheritance of Fanconi anemia complementation group B. Nat
Genet. 36:1219–1224. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Ameziane N, May P, Haitjema A, van de
Vrugt HJ, van Rossum-Fikkert SE, Ristic D, Williams GJ, Balk J,
Rockx D, Li H, et al: A novel Fanconi anaemia subtype associated
with a dominant-negative mutation in RAD51. Nat Commun.
6(8829)2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang AT, Kim T, Wagner JE, Conti BA, Lach
FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al: A
Dominant Mutation in Human RAD51 Reveals Its Function in DNA
Interstrand Crosslink Repair Independent of Homologous
Recombination. Mol Cell. 59:478–490. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Nalepa G and Clapp DW: Fanconi anaemia and
cancer: An intricate relationship. Nat Rev Cancer. 18:168–185.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Stone MP, Cho YJ, Huang H, Kim HY, Kozekov
ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, et al:
Interstrand DNA cross-links induced by alpha,beta-unsaturated
aldehydes derived from lipid peroxidation and environmental
sources. Acc Chem Res. 41:793–804. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Milletti G, Strocchio L, Pagliara D,
Girardi K, Carta R, Mastronuzzi A, Locatelli F and Nazio F:
Canonical and Noncanonical Roles of Fanconi Anemia Proteins:
Implications in Cancer Predisposition. Cancers (Basel).
12(2684)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Che R, Zhang J, Nepal M, Han B and Fei P:
Multifaceted Fanconi Anemia Signaling. Trends Genet. 34:171–183.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Duxin JP and Walter JC: What is the DNA
repair defect underlying Fanconi anemia? Curr Opin Cell Biol.
37:49–60. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Singh TR, Saro D, Ali AM, Zheng XF, Du CH,
Killen MW, Sachpatzidis A, Wahengbam K, Pierce AJ, Xiong Y, et al:
MHF1-MHF2, a histone-fold-containing protein complex, participates
in the Fanconi anemia pathway via FANCM. Mol Cell. 37:879–886.
2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Shakeel S, Rajendra E, Alcón P, O'Reilly
F, Chorev DS, Maslen S, Degliesposti G, Russo CJ, He S, Hill CH, et
al: Structure of the Fanconi anaemia monoubiquitin ligase complex.
Nature. 575:234–237. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Smogorzewska A, Matsuoka S, Vinciguerra P,
McDonald ER III, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K,
D'Andrea AD, et al: Identification of the FANCI protein, a
monoubiquitinated FANCD2 paralog required for DNA repair. Cell.
129:289–301. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mehta PA, Davies SM, Leemhuis T, Myers K,
Kernan NA, Prockop SE, Scaradavou A, O'Reilly RJ, Williams DA,
Lehmann L, et al: Radiation-free, alternative-donor HCT for Fanconi
anemia patients: Results from a prospective multi-institutional
study. Blood. 129:2308–2315. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ebens CL, MacMillan ML and Wagner JE:
Hematopoietic cell transplantation in Fanconi anemia: Current
evidence, challenges and recommendations. Expert Rev Hematol.
10:81–97. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Krausz C, Riera-Escamilla A, Chianese C,
Moreno-Mendoza D, Ars E, Rajmil O, Pujol R, Bogliolo M, Blanco I,
Rodríguez I, et al: From exome analysis in idiopathic azoospermia
to the identification of a high-risk subgroup for occult Fanconi
anemia. Genet Med. 21:189–194. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Freire BL, Homma TK, Funari MFA, Lerario
AM, Leal AM, Velloso EDRP, Malaquias AC and Jorge AAL: Homozygous
loss of function BRCA1 variant causing a Fanconi-anemia-like
phenotype, a clinical report and review of previous patients. Eur J
Med Genet. 61:130–133. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bianchi FT, Berto GE and Di Cunto F:
Impact of DNA repair and stability defects on cortical development.
Cell Mol Life Sci. 75:3963–3976. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Auerbach AD: Fanconi anemia and its
diagnosis. Mutat Res. 668:4–10. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
De Kerviler E, Guermazi A, Zagdanski AM,
Gluckman E and Frija J: The clinical and radiological features of
Fanconi's anaemia. Clin Radiol. 55:340–345. 2000.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Giampietro PF, Adler-Brecher B, Verlander
PC, Pavlakis SG, Davis JG and Auerbach AD: The need for more
accurate and timely diagnosis in Fanconi anemia: A report from the
International Fanconi Anemia Registry. Pediatrics. 91:1116–1120.
1993.PubMed/NCBI
|
|
32
|
Tercanli S, Miny P, Siebert MS, Hösli I,
Surbek DV and Holzgreve W: Fanconi anemia associated with increased
nuchal translucency detected by first-trimester ultrasound.
Ultrasound Obstet Gynecol. 17:160–162. 2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Schneider M, Chandler K, Tischkowitz M and
Meyer S: Fanconi anaemia: Genetics, molecular biology, and cancer -
implications for clinical management in children and adults. Clin
Genet. 88:13–24. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Risitano AM, Marotta S, Calzone R,
Grimaldi F and Zatterale A: RIAF Contributors. Twenty years of the
Italian Fanconi Anemia Registry: Where we stand and what remains to
be learned. Haematologica. 101:319–327. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Petryk A, Kanakatti Shankar R, Giri N,
Hollenberg AN, Rutter MM, Nathan B, Lodish M, Alter BP, Stratakis
CA and Rose SR: Endocrine disorders in Fanconi anemia:
Recommendations for screening and treatment. J Clin Endocrinol
Metab. 100:803–811. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Rose SR, Myers KC, Rutter MM, Mueller R,
Khoury JC, Mehta PA, Harris RE and Davies SM: Endocrine phenotype
of children and adults with Fanconi anemia. Pediatr Blood Cancer.
59:690–696. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Tamary H, Nishri D, Yacobovich J, Zilber
R, Dgany O, Krasnov T, Aviner S, Stepensky P, Ravel-Vilk S, Bitan
M, et al: Frequency and natural history of inherited bone marrow
failure syndromes: The Israeli Inherited Bone Marrow Failure
Registry. Haematologica. 95:1300–1307. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Río P, Navarro S, Wang W,
Sánchez-Domínguez R, Pujol RM, Segovia JC, Bogliolo M, Merino E, Wu
N, Salgado R, et al: Successful engraftment of gene-corrected
hematopoietic stem cells in non-conditioned patients with Fanconi
anemia. Nat Med. 25:1396–1401. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Rosenberg PS, Alter BP and Ebell W: Cancer
risks in Fanconi anemia: Findings from the German Fanconi Anemia
Registry. Haematologica. 93:511–517. 2008.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kutler DI, Singh B, Satagopan J, Batish
SD, Berwick M, Giampietro PF, Hanenberg H and Auerbach AD: A
20-year perspective on the International Fanconi Anemia Registry
(IFAR). Blood. 101:1249–1256. 2003.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Savage SA and Walsh MF: Myelodysplastic
Syndrome, Acute Myeloid Leukemia, and Cancer Surveillance in
Fanconi Anemia. Hematol Oncol Clin North Am. 32:657–668.
2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Murillo-Sanjuán L, González-Vicent M,
Argilés-Aparicio B, Badell-Serra I, Rodríguez-Villa A,
Uria-Oficialdegui ML, López-Duarte M, Beléndez-Bieler C,
Sastre-Urgelles A, Sevilla-Navarro J, et al: Survival and toxicity
outcomes of hematopoietic stem cell transplantation for pediatric
patients with Fanconi anemia: a unified multicentric national study
from the Spanish Working Group for Bone Marrow Transplantation in
Children. Bone Marrow Transplant. 56:1213–1216. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kutler DI, Patel KR, Auerbach AD, Kennedy
J, Lach FP, Sanborn E, Cohen MA, Kuhel WI and Smogorzewska A:
Natural history and management of Fanconi anemia patients with head
and neck cancer: A 10-year follow-up. Laryngoscope. 126:870–879.
2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Alter BP and Giri N: Thinking of
VACTERL-H? Rule out Fanconi Anemia according to PHENOS. Am J Med
Genet A. 170:1520–1524. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Furquim CP, Pivovar A, Amenábar JM, Bonfim
C and Torres-Pereira CC: Oral cancer in Fanconi anemia: Review of
121 cases. Crit Rev Oncol Hematol. 125:35–40. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Rosenberg PS, Greene MH and Alter BP:
Cancer incidence in persons with Fanconi anemia. Blood.
101:822–826. 2003.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Bremer M, Schindler D, Gross M, Dörk T,
Morlot S and Karstens JH: Fanconi's anemia and clinical
radiosensitivity report on two adult patients with locally advanced
solid tumors treated by radiotherapy. Strahlenther Onkol.
179:748–753. 2003.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Katzenellenbogen RA, Carter JJ, Stern JE,
Butsch Kovacic MS, Mehta PA, Sauter SL, Galloway DA and Winer RL:
Skin and mucosal human papillomavirus seroprevalence in persons
with Fanconi Anemia. Clin Vaccine Immunol. 22:413–420.
2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Masserot C, Peffault de Latour R, Rocha V,
Leblanc T, Rigolet A, Pascal F, Janin A, Soulier J, Gluckman E and
Socié G: Head and neck squamous cell carcinoma in 13 patients with
Fanconi anemia after hematopoietic stem cell transplantation.
Cancer. 113:3315–3322. 2008.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Montanuy H, Martínez-Barriocanal Á,
Antonio Casado J, Rovirosa L, Ramírez MJ, Nieto R, Carrascoso-Rubio
C, Riera P, González A, Lerma E, et al: Gefitinib and Afatinib Show
Potential Efficacy for Fanconi Anemia-Related Head and Neck Cancer.
Clin Cancer Res. 26:3044–3057. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
de Jong EM, Felix JF, de Klein A and
Tibboel D: Etiology of esophageal atresia and tracheoesophageal
fistula: ‘mind the gap’. Curr Gastroenterol Rep. 12:215–222.
2010.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Beauregard-Lacroix E, Tardif J, Lemyre E,
Kibar Z, Faure C and Campeau PM: Genetic Testing in a Cohort of
Complex Esophageal Atresia. Mol Syndromol. 8:236–243.
2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Wegman-Ostrosky T and Savage SA: The
genomics of inherited bone marrow failure: From mechanism to the
clinic. Br J Haematol. 177:526–542. 2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Clinton C and Gazda HT: Diamond-Blackfan
Anemia. In: GeneReviews®. Adam MP, Ardinger HH, Pagon
RA, et al (eds). University of Washington, Seattle, WA,
2009.
|
|
55
|
Shaikh H and Mewawalla P: Evans Syndrome.
In: StatPearls. StatPearls Publishing, Treasure Island, FL,
2021.
|
|
56
|
Jameson-Lee M, Chen K, Ritchie E, Shore T,
Al-Khattab O and Gergis U: Acute myeloid leukemia in a patient with
thrombocytopenia with absent radii: A case report and review of the
literature. Hematol Oncol Stem Cell Ther. 11:245–247.
2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Solomon BD: VACTERL/VATER Association.
Orphanet J Rare Dis. 6(56)2011.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Van Maldergem L, Piard J, Larizza L and
Wang LL: Baller-Gerold Syndrome. In: GeneReviews®. Adam
MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G and
Amemiya A (eds). University of Washington, Seattle, WA, 2007.
|
|
59
|
Kallen ME, Dulau-Florea A, Wang W and
Calvo KR: Acquired and germline predisposition to bone marrow
failure: Diagnostic features and clinical implications. Semin
Hematol. 56:69–82. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Gluckman E, Devergie A, Schaison G, Bussel
A, Berger R, Sohier J and Bernard J: Bone marrow transplantation in
Fanconi anaemia. Br J Haematol. 45:557–564. 1980.PubMed/NCBI
|
|
61
|
Oostra AB, Nieuwint AW, Joenje H and de
Winter JP: Diagnosis of fanconi anemia: Chromosomal breakage
analysis. Anemia. 2012(238731)2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Auerbach AD: Diagnosis of Fanconi anemia
by diepoxybutane analysis. Curr Protoc Hum Genet. 85:8.7.1–8.7.17.
2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Esmer C, Sánchez S, Ramos S, Molina B,
Frias S and Carnevale A: DEB test for Fanconi anemia detection in
patients with atypical phenotypes. Am J Med Genet A. 124A:35–39.
2004.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Castella M, Pujol R, Callén E, Ramírez MJ,
Casado JA, Talavera M, Ferro T, Muñoz A, Sevilla J, Madero L, et
al: Chromosome fragility in patients with Fanconi anaemia:
Diagnostic implications and clinical impact. J Med Genet.
48:242–250. 2011.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Fargo JH, Rochowski A, Giri N, Savage SA,
Olson SB and Alter BP: Comparison of chromosome breakage in
non-mosaic and mosaic patients with Fanconi anemia, relatives, and
patients with other inherited bone marrow failure syndromes.
Cytogenet Genome Res. 144:15–27. 2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Revy P, Kannengiesser C and Fischer A:
Somatic genetic rescue in Mendelian haematopoietic diseases. Nat
Rev Genet. 20:582–598. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Nicoletti E, Rao G, Bueren JA, Río P,
Navarro S, Surrallés J, Choi G and Schwartz JD: Mosaicism in
Fanconi anemia: Concise review and evaluation of published cases
with focus on clinical course of blood count normalization. Ann
Hematol. 99:913–924. 2020.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Flynn EK, Kamat A, Lach FP, Donovan FX,
Kimble DC, Narisu N, Sanborn E, Boulad F, Davies SM, Gillio AP III,
et al: Comprehensive analysis of pathogenic deletion variants in
Fanconi anemia genes. Hum Mutat. 35:1342–1353. 2014.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Castella M, Pujol R, Callén E, Trujillo
JP, Casado JA, Gille H, Lach FP, Auerbach AD, Schindler D, Benítez
J, et al: Origin, functional role, and clinical impact of Fanconi
anemia FANCA mutations. Blood. 117:3759–3769. 2011.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Bogliolo M, Pujol R, Aza-Carmona M,
Muñoz-Subirana N, Rodriguez-Santiago B, Casado JA, Rio P, Bauser C,
Reina-Castillón J, Lopez-Sanchez M, et al: Optimised molecular
genetic diagnostics of Fanconi anaemia by whole exome sequencing
and functional studies. J Med Genet. 57:258–268. 2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Frohnmayer L, Van Ravenhorst S and
Wirkkula L (eds): Fanconi anemia Clinical Care Guidelines. 5th
edition. Fanconi Anemia Research Fund, Eugene, OR, 2020.
|
|
72
|
Rosenberg PS, Huang Y and Alter BP:
Individualized risks of first adverse events in patients with
Fanconi anemia. Blood. 104:350–355. 2004.PubMed/NCBI View Article : Google Scholar
|
|
73
|
de Winter JP and Joenje H: The genetic and
molecular basis of Fanconi anemia. Mutat Res. 668:11–19.
2009.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wang X, Andreassen PR and D'Andrea AD:
Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1
in chromatin. Mol Cell Biol. 24:5850–5862. 2004.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Lobitz S and Velleuer E: Guido Fanconi
(1892-1979): A jack of all trades. Nat Rev Cancer. 6:893–898.
2006.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Sasaki MS and Tonomura A: A high
susceptibility of Fanconi's anemia to chromosome breakage by DNA
cross-linking agents. Cancer Res. 33:1829–1836. 1973.PubMed/NCBI
|