1. Introduction
Acute lung injury (ALI) is an injury to the
pulmonary capillary endothelial cells and pulmonary epithelial
cells following severe infection, shock, trauma or major surgery,
resulting in diffuse interstitial lung infiltration and
noncardiogenic pulmonary edema. This can ultimately result in acute
hypoxic respiratory insufficiency or failure (1), which can be a serious threat to a
patient's health. The global incidence of ALI ranges from
7.2/100,000 to 78.9/100,000, with a mortality rate of ~40% and
persistent pulmonary dysfunction occurs in ~50% of survivors, which
in-turn results in significant social implications and financial
burdens (2). Despite the notable
growth in the understanding of ALI over the past few decades,
several aspects remain unknown; there are no reliable diagnostic
measures to accurately identify individuals at risk of ALI, and
there are no effective interventional measures or treatments that
have been shown to prevent its occurrence. Researching the
pathogenesis of ALI could help to provide novel interventional and
treatment strategies for management of ALI, which may result in
reduced mortality rates and an improved quality of life for
patients, whilst also reducing the economic burden. In recent
years, in addition to the widely recognized role of platelets in
thrombosis and hemostasis, several researchers have focused on
their involvement in the inflammatory process as immune cells
(3-6).
The following is a review of the relevance of platelet involvement
in the development of ALI based on the possible factors involved in
the progression of inflammation after platelet activation, with the
aim of identifying therapeutic directions for targeting ALI through
the inhibition of platelet pathways.
2. Advances in the pathogenesis of acute
lung injury and interventional treatment
ALI has several causative factors, including those
that can directly affect the development of infection in the lungs,
such as accidental aspiration of gastric contents, pulmonary
contusion, inhalation of toxic gases, drowning and oxygen toxicity.
Moreover, other factors can indirectly lead to pulmonary infection,
such as severe non-thoracic trauma, acute severe pancreatitis,
massive blood transfusion, extracorporeal circulation and diffuse
intravascular coagulation (7). The
pathology of ALI can be divided into two components, impaired gas
exchange and inflammatory damage. In the acute phase of ALI, the
alveolar-capillary barrier, consisting of the vascular endothelium,
mesenchyme and alveolar epithelium, is damaged, resulting in
increased vascular and alveolar epithelial permeability and the
flow of protein-rich fluid into the interstitium and alveoli,
leading to impaired gas exchange (8). Following the occurrence of ALI,
numerous inflammatory cells are activated and induce the production
of several cytokines and inflammatory mediators, such as IL-6, IL-8
and TNF-α (9), which cause further
damage to the body. The development of ALI involves complex
regulatory mechanisms, and several experiments have demonstrated
the central role of inflammation in the progression of ALI
(10,11). The majority of studies have focused
on the recruitment and activation of neutrophil adhesion. For
example, Bhatia et al (12)
reported that inflammation initiates chemokine-driven neutrophil
activation, followed by neutrophil-derived production of reactive
oxygen species and proteases, among other molecules, which cause
damage to tissue cells; Saffarzadeh et al (13) demonstrated that neutrophils directly
induce epithelial and endothelial cell death through activation of
the p38 MAP kinase pathway and the Raf-MEK-ERK kinase pathway,
resulting in lung injury; Jeyaseelan et al (14) suggested that hyaluronic acid
produced by damaged tissues binds to Toll-like receptor (TLR)4 to
initiate the inflammatory response in acute respiratory distress
syndrome (ARDS) and that the TLR pathway activates lung
macrophages, which release pro-inflammatory mediators and trigger
an inflammatory cascade that activates and leads to the chemotaxis
of neutrophils and other inflammatory cells to the airways. In
addition, certain signaling pathways also act directly on
neutrophils to regulate their role in the development of ALI, as
suggested by the study by Huang et al (15), who demonstrated that
hypoxia-inducible factor 1α may regulate the activation of NOD-like
receptor protein 3 inflammatory vesicle activation and thus
regulate ALI.
3. Platelets and inflammatory diseases
Platelets are small pieces of cytoplasm shed from
mature megakaryocytes in the bone marrow, and contain glycogen,
mitochondria and at least three types of granules (dense granules,
lysosomes and α granules), which reside in the circulating blood,
and can themselves secrete granule contents when activated. The
most abundant particles are α granules, which contain a large
number of immunomodulatory cytokines and chemokines, including
platelet factor 4 (PF-4), β-thromboglobulin (β-TG), P-selectin,
macrophage inflammatory protein 1α and chemokine CCL5, which have
pro-inflammatory functions (16).
Dense granules store ADP, ATP and calcium ions, among other
components (17). Lysosomes contain
acid hydrolases that play a role in phagocytic cellular components
(18). It has been shown that
P-selectin expression is upregulated in inflammatory diseases, such
as hypertension and atherosclerosis (19), and platelet-derived P-selectin plays
a major role in the inflammatory and endothelial proliferative
response after arterial injury (20,21).
Platelets can also be involved in the progression of rheumatoid
arthritis by promoting platelet particle release through the
activation of type VI collagen binding to CD41 on the platelet
surface (22).
Immune system receptors, including TLRs 1-7 and 9,
are present on the platelet surface. TLRs are a family of pattern
recognition receptors expressed by phagocytes, such as neutrophils,
macrophages and dendritic cells (23), which recognize the Fc receptor of
immunoglobulins, and thus participate in allergic inflammation
(24), and promote atherosclerosis
and inflammation by inhibiting T-reg cell recruitment through the
activation of CD40(25). Platelets
also express thrombin (PAR1, 3 and 4), ADP (P2Y1 and
P2Y12), and the thromboxane A2 (TXA2) receptors
thromboxane receptor (TP)-α and TP-β, which when bound to their
respective ligands, can lead to platelet aggregation and the
secretion of bioactive mediators, which are involved in the
progression of inflammatory diseases (26).
Platelets can also be activated by direct contact,
such as platelets interacting with neutrophils via CD62P and with B
cells via pattern recognition receptors. They also interact with T
cells via the CD40/CD40L complex, and with endothelial cells and
erythrocytes via the integrin receptor (27). An in vitro study by Danese
et al (28) showed that
platelets rapidly adhere to human intestinal microvascular
endothelial cells when co-incubated with IL-1β, stimulating the
expression of vascular cell adhesion molecule 1 and intercellular
adhesion molecule-1 (ICAM-1; two major leukocyte receptors) on the
surface of endothelial cells, and the secretion of the neutrophil
chemokine IL-8, which plays an important role in inflammatory bowel
disease.
In addition, the pathogenic mechanisms of platelets
in various lung diseases have been elucidated. Higher expression of
P-selectin, PF-4 and β-TG in the platelets of asthma patients, when
compared to that in controls, has been revealed by Kasperska et
al (29). High expression of
TXA2 and soluble (s)CD40L during platelet hyperfunction was found
in pulmonary cystic fibrosis disease, as summarized by O'Sullivan
and Michelson (30). In summary, it
can be concluded that platelets are not only widely recognized as a
major player in hemostasis and thrombosis, but also as immune cells
involved in the inflammatory response of the body, and can mediate
tissue damage.
4. Platelet activation is involved in the
pathogenic mechanism of ALI
Megakaryocytes are essential precursor cells for
platelet production, and the lungs are a reservoir of these cells
(31,32); therefore, platelets are present in
large numbers in pulmonary circulation. Despite the significant
protective role of platelets in hemostasis and inflammatory
defense, a large body of experimental and clinical data shows that
platelets play a dual role in ALI. On the one hand, platelets help
to maintain the integrity of the alveolar capillary base barrier,
which selectively limits the extravascular transfer of water,
proteins and red blood cells (33),
and contributes to pulmonary vascular repair (34). On the other hand, platelets can also
regulate ALI/ARDS pathogenic processes through complex mechanisms,
such as neutrophil recruitment, macrophage-dependent inflammation
and modulation of alveolar capillary permeability (35). The pathogenic role of platelets
depends on the balance of the inflammatory response in the body.
When the body is in a normal healthy state, platelets can maintain
the stability of the vascular endothelium and play an important
role in maintaining the healthy state of the body. When an organism
is in a diseased state, platelets are activated in various forms,
leading to a series of inflammatory reactions that cause damage to
the organism. Lê et al (36)
showed that the mouse ALI model relies heavily on platelet
activation, and that in a mouse model of ALI, several activated
platelets were detected in the lungs and plasma compared to the
levels in the normal control mice. Looney et al (37) found that depleted platelets protect
mice from severe lung injury, and when platelet activation was
blocked with aspirin, thromboxane production was reduced, as was
lung injury and mortality. There are several causes of ALI, and the
pathogenesis is not completely the same in different possible
stages; that is, the mechanism of platelet action in ALI induced by
different causes vaires itself. For example, Yasui et al
(38) showed that the occurrence of
lung injury may be due to platelet dysfunction in a rat model of
blunt traumatic brain injury, whereas Clark et al (39) found that increased
platelet–neutrophil binding leads to the occurrence of lung injury
in a model of sepsis due to bacterial infection. To conclude,
platelets are an important component involved in the progression of
ALI. In this section, the mechanisms through which platelet factors
are involved in ALI in terms of immune regulation of platelet
secretion, receptor-ligand signaling related to platelet
activation, and direct contact interactions between platelets and
related cells are discussed.
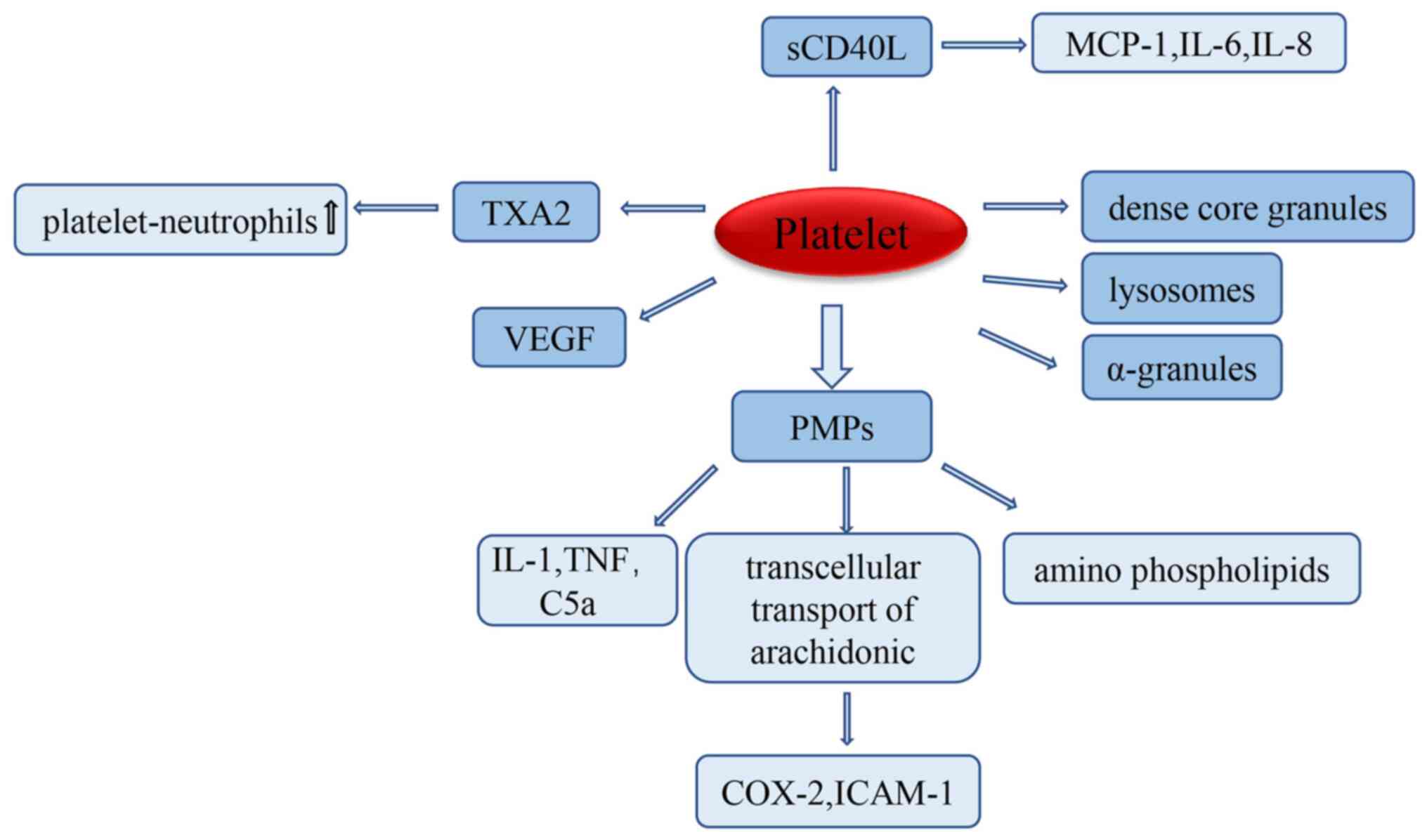
Platelets secrete active
substances
The binding of exogenous activators to platelet
membrane-bound proteins induces the secretion of platelet granules,
such as dense granules, lysosomes and α granules, which contain
adhesion molecules, factors related to coagulation and
fibrinolysis, as well as the secretion of calcium and pyrophosphate
(16). The expression of adhesion
molecules, such as P-selectin, CD31, GP IIb/IIIa, fibronectin and
thrombin-reactive proteins, can be upregulated to participate in
the development of ALI (40). When
ALI occurs, platelets are activated in the pulmonary
microvasculature, leading to platelet-microparticle (PMP)
secretion. Studies have shown that under inflammatory conditions,
the release of PMPs can activate immune cells, such as neutrophils
and non-immune cells, to synthesize and secrete several enzymes
that promote the progression of inflammation, such as proteases,
and active pro-inflammatory soluble mediators including IL-1, TNF
and complement C5a/C5a receptors, which further cause tissue damage
(41,42). Meanwhile, PMPs contain large amounts
of aminophospholipids, substrates of phospholipase A2, and are
associated with the production of lysophosphatidic acid, which in
turn affects the inflammatory process of platelets and is involved
in the pathogenic role of platelets in ALI (43). PMPs also support the transcellular
transport of arachidonic acid, upregulate the expression of
endothelial cyclooxygenase 2 and ICAM-1, and regulate the interface
between endothelial cells and platelets (44,45).
This can lead to impairment of the alveolar-capillary barrier,
increased vascular and alveolar epithelial permeability, and the
influx of protein-rich fluid into the intercellular stroma and
alveoli, which is the primary pathological mechanism of ALI
(46). Furthermore, platelets are
stimulated to secrete the pro-inflammatory mediator TXA2, which is
an important mediator of platelet-neutrophil aggregation when ALI
occurs (47), and increased
platelet-neutrophil aggregation aggravates ALI damage (48). Platelets secrete vascular
endothelial growth factor (VEGF), a powerful angiogenic factor that
plays a key role in regulating angiogenesis, both by inducing
vascular endothelial cell proliferation, and by promoting cell
survival through the induction of the anti-apoptotic proteins Bcl-2
and A1(49). VEGF has the ability
to increase microvascular permeability by a factor of 20,000
compared to histamine, and therefore induces an increase in
endothelial permeability (50).
Endothelial permeability plays a key role in the pathogenesis of
ALI; therefore, it is hypothesized that platelet activation-induced
VEGF secretion may also play an important role in the progression
of ALI/ARDS.
CD40L is expressed at a low level in unstimulated
platelets, but can be rapidly upregulated on the platelet surface
after platelet stimulation. The platelet surface-expressed CD40L is
subsequently cleaved within minutes to hours, producing a soluble
fragment called sCD40L (51). The
expression of platelet-associated CD40L on the platelet surface and
exposure to CD40 receptor-containing vascular cells can initiate
various inflammatory responses, including the expression of
inflammatory adhesion receptors (such as E-selectin, vascular cell
adhesion molecule-1 and ICAM-1), the expression of tissue factors,
and the release of chemokines (monocyte chemotactic protein-1, IL-6
and IL-8) (52), all of which may
contribute to the development of ALI. sCD40L not only induces
monocytes to secrete the neutrophil chemotactic agent MIP-2 but
also interacts with membrane CD40 to directly activate neutrophils
(53); therefore, it is
hypothesized that sCD40L can lead to the substantial accumulation
and activation of neutrophils, accelerating the course of ALI.
However, the direct role of sCD40L in the pathogenesis of ALI
remains unclear. In summary, platelets as inflammatory cells can be
involved in the process of ALI through the synthesis and secretion
of a large number of active substances (Fig. 1).
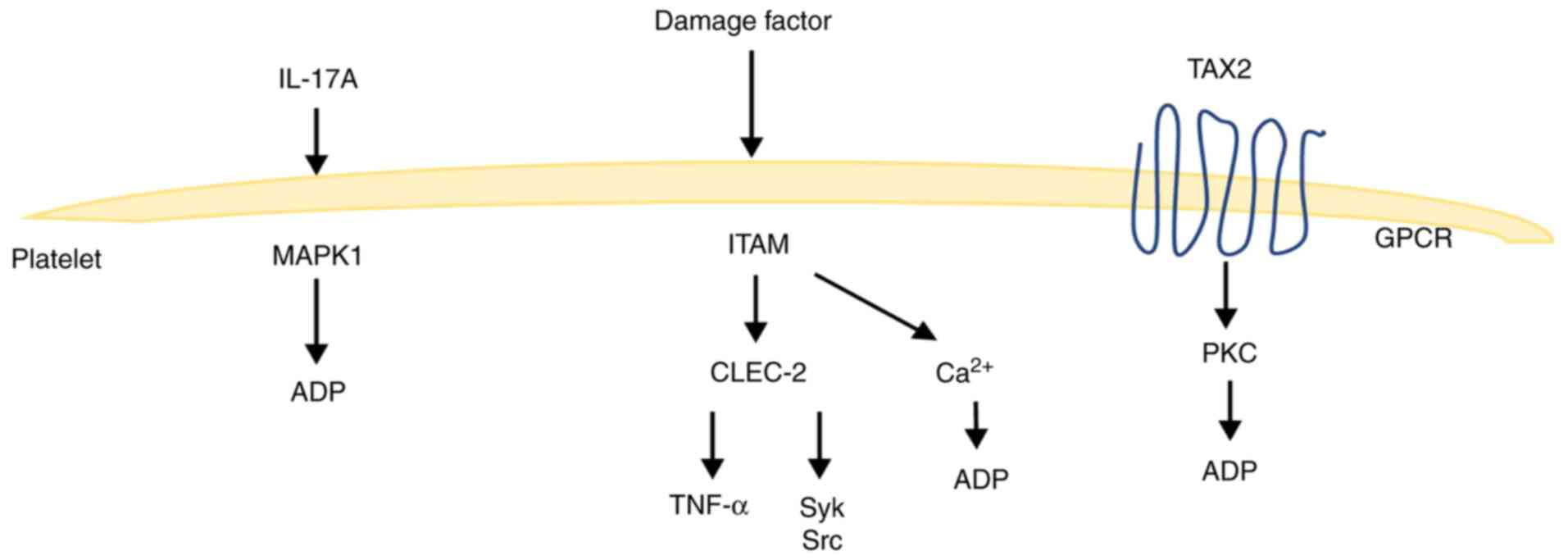
Platelet receptor ligand signaling
pathway
Although platelets are widely known for their
hemostatic and clotting effects, they are also critical for growth
and development, host defense, inflammation and tissue repair.
Several of these processes are regulated by the immunoglobulin-like
receptor glycoprotein VI (GPVI) and C-type lectin receptor 2
(CLEC-2), which act based on signaling from the immunoreceptor
tyrosine-based activation motif (ITAM) (54). The ITAM receptor signaling pathway
is required for platelet activation by the extracellular matrix and
extravascular cells. Platelets express two types of ITAM receptors,
GPVI receptor for collagen and laminin and CLEC-2 receptor for
podoplanin (PDPN) (55) CLEC-2 is a
type II membrane protein that is highly expressed on megakaryocytes
and platelets, and expressed at low levels on peripheral blood
neutrophils, mediating phagocytosis and the release of
pro-inflammatory cytokines, including TNF-α (56). CLEC-2 interacts with PDPN to
activate platelets via the tyrosine kinase signaling pathway, and
in addition, GPVI interacts with ligands (collagen) to activate
platelets via the Syk pathway (57,58).
However, protein hydrolysis in the ITAM region causes the
activation of downstream protein phosphorylation, release of
calcium ions into the cytoplasm, activation of ATPase, catabolism
of ATP, and supply of energy for platelet activation and release
(59,60). These reactions may be involved in
the progression of ALI, but the exact mechanism underlying this
process needs to be elucidated by further studies.
Platelets express the thromboxane receptor TPα on
their surface, and both TPα and TPβ on endothelial cells activate
endothelial cells and G-protein-dependent downstream pathways, such
as increased expression of the PKC pathway-dependent adhesion
molecule ICAM-1(61). Meanwhile,
TXA2 binding to G protein-coupled TPs can lead to a wide range of
cellular responses, including integrin activation, platelet
aggregation, smooth muscle cell contraction and increased vascular
permeability, which can be involved in the development of ALI
(62). IL-17A can also promote
ADP-induced platelet activation through the extracellular
signal-regulated kinase 2 (ERK2; also known as MAPK1) signaling
pathway, which induces platelet responses involved in inflammatory
responses (63). Moreover, the link
between platelet-related signaling pathways and ALI needs to be
further elucidated (Fig. 2).
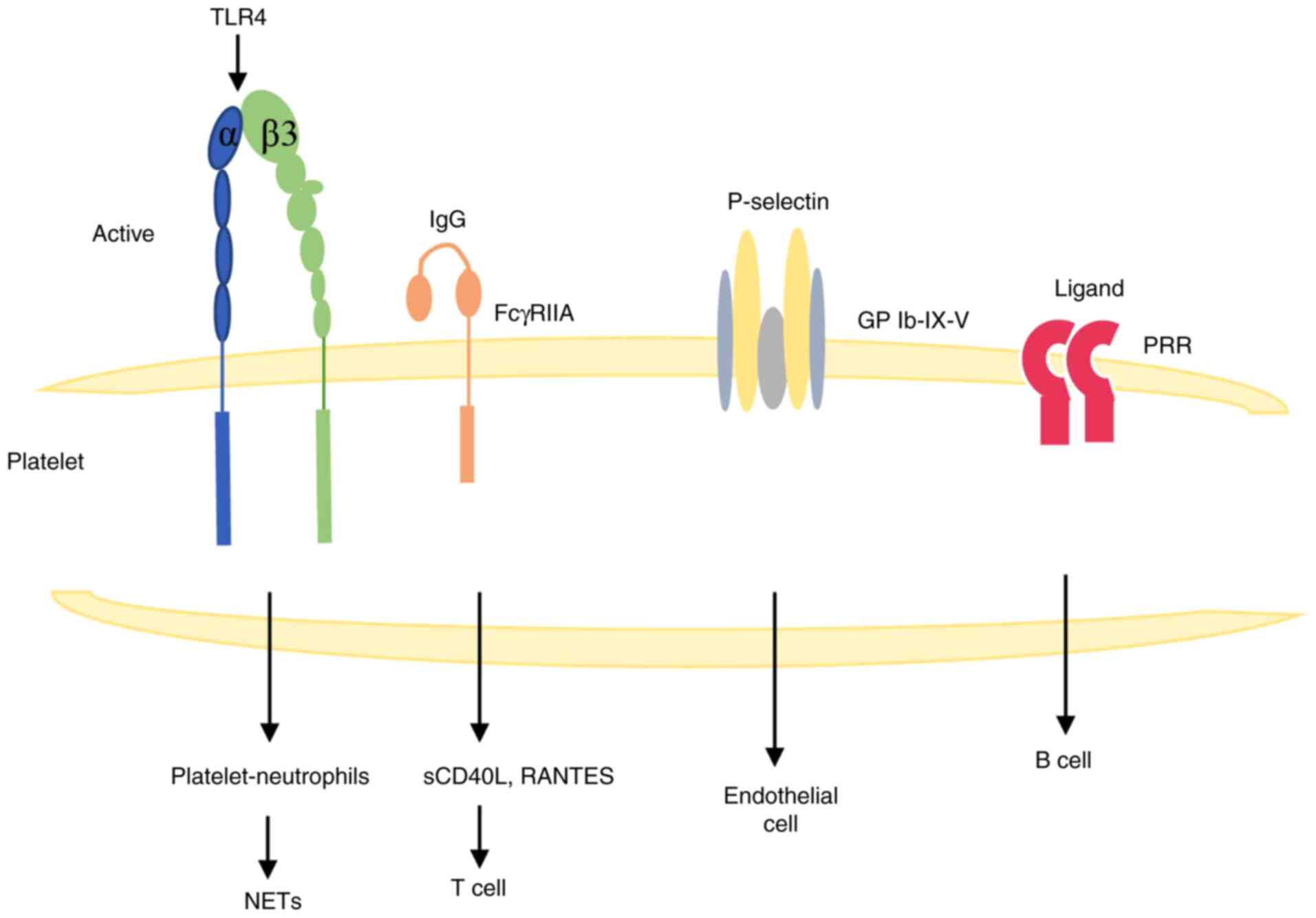
Direct contact between platelets and
other types of cells
Platelets are in a resting state under normal
conditions, but can be activated indirectly or directly during ALI
to exert their corresponding biological effects. In a model of
LPS-induced lung injury, platelet activation mediates an increase
in platelet surface CD62P and GPIIb/IIIa expression through the
activation of TLR4. Additionally, CD62P is a key ligand for
platelet binding to neutrophils and monocytes (39), which can enhance platelet-neutrophil
interactions during ALI, leading to the production of neutrophil
extracellular traps (NETs) (64).
NETs are associated with a type of cell death distinct from
apoptosis and necrosis, a process known as NETosis (65), and the mechanism of their formation
is not fully understood. They may provide the conditions for
platelet capture and thrombosis in the pulmonary microcirculation
through exposure to their extracellular histones, neutrophil
granule proteins and extracellular DNA networks, ultimately leading
to pulmonary endothelial injury and participating in the
progression of ALI (66,67). Therefore, targeting NETs could be a
potential therapeutic modality for the treatment of lung
injury.
ALI leads to vascular injury, and various platelet
agonists and intrinsic factors lead to platelet activation. It has
been shown that platelet FcγRIIA can bind to endocrine IgG
complexes and release large amounts of sCD40L and RANTES (68), where sCD40L interacts with T cells
through direct contact with the CD40/CD40L complex, further
supporting the involvement of platelets in the inflammatory
response (69,70). Platelets can interact with B cells
via PRRs, and through PRR-mediated cell activation, inflammatory
signaling transduction and apoptosis induction occur, which are
involved in the progression of ALI (25,71).
Platelets can also interact with P-selectin-expressing endothelial
cells via the integrin receptor GPIb-IX-V to mediate the
endothelial cell inflammatory response (72). All of these factors can further
enhance the inflammatory response to activate platelets, and
platelets play an important role in ALI (Fig. 3).
5. Conclusion and future prospects
As a disease associated with high morbidity rates,
and poor diagnostic and prognostic measures, ALI has no definite
strategy for prevention and treatment; it can only be
improved/prevented by treating the primary disease, controlling the
systemic inflammatory response, and improving hypoxemia with
supportive therapy using non-invasive or invasive ventilators.
Owing to the poor prognosis of patients under the current treatment
strategies for ALI, and due to the persistent pulmonary dysfunction
observed in a significant proportion of survivors, patients have a
poor quality of life. Therefore, it is necessary to explore a
feasible means of intervention or treatment for ALI to save
patients' lives and improve their quality of life. Previous studies
have shown that the pathogenesis of ALI is centered on an
inflammatory response, and there is now increasing evidence that
platelets can act as inflammatory cells and participate in the
progression of various inflammatory diseases through various
pathways, such as the participation of platelets in the progression
of rheumatoid arthritis by amplifying inflammation through
collagen-dependent particles and mediating cerebral malaria through
PF-4 (22,73). Platelets are also involved in the
development of several types of lung diseases; for example, the
high expression of P-selectin, PF-4 and β-TG in platelets, as
discussed in this review, can increase the severity of disease in
asthma patients (29). Platelet
function is hyper-activated in pulmonary cystic fibrosis with high
expression of TXA2 and sCD40L (30). Furthermore, platelets also serve a
considerable role in the progression of ALI, and can influence the
course of ALI through a variety of pathways, such as the
immunoregulation of platelet secretion, receptor-ligand signaling
associated with platelet activation, and direct contact reactions
between platelets and associated cells. Several studies have now
found that anti-platelet reagents can reduce the severity of ALI;
for example Hagiwara et al (74) showed that platelet inhibition with
clopidogrel reduces LPS-induced ALI in rats by inhibiting the
P2Y12 receptor. In another experiment, Asaduzzaman et
al (75) also found that
platelet inhibition reduces lung injury by inhibiting
neutrophil-platelet aggregation. Cognasse et al (76) illustrated that the use of an
anti-GPIbα antibody, which was used as a specific platelet
inhibitor in a transfusion-associated ALI model, reduced the extent
of lung injury in a model of ALI. Moreover, with the recent
COVID-19 pandemic, it has been found that during the course of
pneumonia, anti-platelet use was associated with a lower risk of
severe disease (77). Furthermore,
at present, the primary genetic changes involved in lung diseases
are pulmonary fibrosis caused by paraquat and various lung cancers
(78-81);
however, the genetic alterations to platelets in ALI do not seem to
be involved, based on current studies (82-86),
and these need to be further analyzed by gene microarray analysis,
which is also the direction that our lab will take for future
research.
This review is only a summary of the possible
involvement of platelets in the progression of ALI. The immune
mechanisms by which platelets promote tissue damage and regulate
ALI are not yet fully understood, and further studies are needed to
elucidate the etiological roles of platelets as both mediators of
ALI and effector cells of ALI. Novel targets to intervene and
manage ALI may be discovered by exploring the mechanisms by which
platelets regulate or mediate lung injury, and intervening in the
relevant pathways in advance or inhibiting platelet activation,
thus blocking the vicious cycle. Moreover, future studies should
also focus on the mechanisms by which platelets act as inflammatory
cells during disease progression.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the National Natural
Science Foundation of China (grant no. 31801192).
Availability of data and materials
Not applicable.
Authors' contributions
YX and JP conceived the article. YX drafted the
manuscript and constructed the figures. JP revised the manuscript
and the figures. YYH, QCC, SP and SN analyzed the relevant
literature. LFZ edited the manuscript. All authors read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bernard GR, Artigas A, Brigham KL, Carlet
J, Falke K, Hudson L, Lamy M, Legall JR, Morris A and Spragg R: The
American-European consensus conference on ARDS. definitions,
mechanisms, relevant outcomes, and clinical trial coordination. Am
J Respir Crit Care Med. 149:818–824. 1994.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Phua J, Badia JR, Adhikari NK, Friedrich
JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, et
al: Has mortality from acute respiratory distress syndrome
decreased over time?: A systematic review. Am J Respir Crit Care
Med. 179:220–227. 2009.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jenne CN and Kubes P: Platelets in
inflammation and infection. Platelets. 26:286–292. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Maouia A, Rebetz J, Kapur R and Semple JW:
The immune nature of platelets revisited. Transfus Med Rev.
34:209–220. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Smyth SS, McEver RP, Weyrich AS, Morrell
CN, Hoffman MR, Arepally GM, French PA, Dauerman HL and Becker RC:
2009 Platelet Colloquium Participants. Platelet functions beyond
hemostasis. J Thromb Haemost. 7:1759–1766. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cox D, Kerrigan SW and Watson SP:
Platelets and the innate immune system: Mechanisms of
bacterial-induced platelet activation. J Thromb Haemost.
9:1097–1107. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Pugin J, Verghese G, Widmer MC and Matthay
MA: The alveolar space is the site of intense inflammatory and
profibrotic reactions in the early phase of acute respiratory
distress syndrome. Crit Care Med. 27:304–312. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349.
2000.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Grommes J and Soehnlein O: Contribution of
neutrophils to acute lung injury. Mol Med. 17:293–307.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nagase T, Ishii S, Kume K, Uozumi N, Izumi
T, Ouchi Y and Shimizu T: Platelet-activating factor mediates
acid-induced lung injury in genetically engineered mice. J Clin
Invest. 104:1071–1076. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Zemans RL, Colgan SP and Downey GP:
Transepithelial migration of neutrophils: Mechanisms and
implications for acute lung injury. Am J Respir Cell Mol Biol.
40:519–535. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bhatia M, Zemans RL and Jeyaseelan S: Role
of chemokines in the pathogenesis of acute lung injury. Am J Respir
Cell Mol Biol. 46:566–572. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Saffarzadeh M, Juenemann C, Queisser MA,
Lochnit G, Barreto G, Galuska SP, Lohmeyer J and Preissner KT:
Neutrophil extracellular traps directly induce epithelial and
endothelial cell death: A predominant role of histones. PLoS One.
7(e32366)2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jeyaseelan S, Chu HW, Young SK, Freeman MW
and Worthen GS: Distinct roles of pattern recognition receptors
CD14 and Toll-like receptor 4 in acute lung injury. Infect Immun.
73:1754–1763. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Huang JJ, Xia J, Huang LL and Li YC:
HIF-1α promotes NLRP3 inflammasome activation in bleomycin-induced
acute lung injury. Mol Med Rep. 20:3424–3432. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rendu F and Brohard-Bohn B: The platelet
release reaction: Granules' constituents, secretion and functions.
Platelets. 12:261–273. 2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Meng R, Wang Y, Yao Y, Zhang Z, Harper DC,
Heijnen HF, Sitaram A, Li W, Raposo G, Weiss MJ, et al: SLC35D3
delivery from megakaryocyte early endosomes is required for
platelet dense granule biogenesis and is differentially defective
in Hermansky-Pudlak syndrome models. Blood. 120:404–414.
2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Heijnen H and van der Sluijs P: Platelet
secretory behaviour: As diverse as the granules … or not? J Thromb
Haemost. 13:2141–2151. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Koyama H, Maeno T, Fukumoto S, Shoji T,
Yamane T, Yokoyama H, Emoto M, Shoji T, Tahara H, Inaba M, et al:
Platelet P-selectin expression is associated with atherosclerotic
wall thickness in carotid artery in humans. Circulation.
108:524–529. 2003.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wysokinski WE, Cohoon KP, Melduni RM,
Mazur M, Ammash N, Munger T, Konik E, McLeod T, Gosk-Bierska I and
McBane RD: Association between P-selectin levels and left atrial
blood stasis in patients with nonvalvular atrial fibrillation.
Thromb Res. 172:4–8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang K, Zhou X, Zhou Z, Mal N, Fan L,
Zhang M, Lincoff AM, Plow EF, Topol EJ and Penn MS: Platelet, not
endothelial, P-selectin is required for neointimal formation after
vascular injury. Arterioscler Thromb Vasc Biol. 25:1584–1589.
2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Boilard E, Nigrovic PA, Larabee K, Watts
GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O'Donnell E,
Farndale RW, Ware J and Lee DM: Platelets amplify inflammation in
arthritis via collagen-dependent microparticle production. Science.
327:580–583. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Semple JW, Italiano JE Jr and Freedman J:
Platelets and the immune continuum. Nat Rev Immunol. 11:264–274.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Hasegawa S, Tashiro N, Matsubara T,
Furukawa S and Ra C: A comparison of FcepsilonRI-mediated RANTES
release from human platelets between allergic patients and healthy
individuals. Int Arch Allergy Immunol. 125 (Suppl 1):S42–S47.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lapchak PH, Ioannou A, Kannan L, Rani P,
Dalle Lucca JJ and Tsokos GC: Platelet-associated CD40/CD154
mediates remote tissue damage after mesenteric ischemia/reperfusion
injury. PLoS One. 7(e32260)2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ghoshal K and Bhattacharyya M: Overview of
platelet physiology: Its hemostatic and nonhemostatic role in
disease pathogenesis. ScientificWorldJournal.
2014(781857)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yeaman MR: Platelets: At the nexus of
antimicrobial defence. Nat Rev Microbiol. 12:426–437.
2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Danese S, de la Motte C, Sturm A, Vogel
JD, West GA, Strong SA, Katz JA and Fiocchi C: Platelets trigger a
CD40-dependent inflammatory response in the microvasculature of
inflammatory bowel disease patients. Gastroenterology.
124:1249–1264. 2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kasperska-Zajac A, Brzoza Z and Rogala B:
Seasonal changes in platelet activity in pollen-induced seasonal
allergic rhinitis and asthma. J Asthma. 45:485–487. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
O'Sullivan BP and Michelson AD: The
inflammatory role of platelets in cystic fibrosis. Am J Respir Crit
Care Med. 173:483–490. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Weyrich AS and Zimmerman GA: Platelets in
lung biology. Annu Rev Physiol. 75:569–591. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lefrançais E and Looney MR: Platelet
biogenesis in the lung circulation. Physiology (Bethesda).
34:392–401. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tabuchi A and Kuebler WM:
Endothelium-platelet interactions in inflammatory lung disease.
Vascul Pharmacol. 49:141–150. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ho-Tin-Noé B, Demers M and Wagner DD: How
platelets safeguard vascular integrity. J Thromb Haemost. 9 (Suppl
1):S56–S65. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Middleton EA, Weyrich AS and Zimmerman GA:
Platelets in pulmonary immune responses and inflammatory lung
diseases. Physiol Rev. 96:1211–1259. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lê VB, Schneider JG, Boergeling Y, Berri
F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S,
Antunes L, et al: Platelet activation and aggregation promote lung
inflammation and influenza virus pathogenesis. Am J Respir Crit
Care Med. 191:804–819. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Looney MR, Nguyen JX, Hu Y, Van Ziffle JA,
Lowell CA and Matthay MA: Platelet depletion and aspirin treatment
protect mice in a two-event model of transfusion-related acute lung
injury. J Clin Invest. 119:3450–3461. 2009.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yasui H, Donahue DL, Walsh M, Castellino
FJ and Ploplis VA: Early coagulation events induce acute lung
injury in a rat model of blunt traumatic brain injury. Am J Physiol
Lung Cell Mol Physiol. 311:L74–L86. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Clark SR, Ma AC, Tavener SA, McDonald B,
Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair
GD, et al: Platelet TLR4 activates neutrophil extracellular traps
to ensnare bacteria in septic blood. Nat Med. 13:463–469.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
40
|
Zarbock A, Polanowska-Grabowska RK and Ley
K: Platelet-neutrophil-interactions: Linking hemostasis and
inflammation. Blood Rev. 21:99–111. 2007.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Nurden AT: Platelets, inflammation and
tissue regeneration. Thromb Haemost. 105 (Suppl 1):S13–S33.
2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Xie RF, Hu P, Wang ZC, Yang J, Yang YM,
Gao L, Fan HH and Zhu YM: Platelet-derived microparticles induce
polymorphonuclear leukocyte-mediated damage of human pulmonary
microvascular endothelial cells. Transfusion. 55:1051–1057.
2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Barry OP, Pratico D, Lawson JA and
FitzGerald GA: Transcellular activation of platelets and
endothelial cells by bioactive lipids in platelet microparticles. J
Clin Invest. 99:2118–2127. 1997.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bulut D, Becker V and Mügge A:
Acetylsalicylate reduces endothelial and platelet-derived
microparticles in patients with coronary artery disease. Can J
Physiol Pharmacol. 89:239–244. 2011.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhou Q, Lian Y, Zhang Y, Li L, Li H, Shen
D, Zhou Y, Zhang M, Lu Y, Liu J, et al: Platelet-derived
microparticles from recurrent miscarriage associated with
antiphospholipid antibody syndrome influence behaviours of
trophoblast and endothelial cells. Mol Hum Reprod. 25:483–494.
2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bhattacharya J and Matthay MA: Regulation
and repair of the alveolar-capillary barrier in acute lung injury.
Annu Rev Physiol. 75:593–615. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zarbock A, Singbartl K and Ley K: Complete
reversal of acid-induced acute lung injury by blocking of
platelet-neutrophil aggregation. J Clin Invest. 116:3211–3219.
2006.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Bdeir K, Gollomp K, Stasiak M, Mei J,
Papiewska-Pajak I, Zhao G, Worthen GS, Cines DB, Poncz M and
Kowalska MA: Platelet-specific chemokines contribute to the
pathogenesis of acute lung injury. Am J Respir Cell Mol Biol.
56:261–270. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Medford AR and Millar AB: Vascular
endothelial growth factor (VEGF) in acute lung injury (ALI) and
acute respiratory distress syndrome (ARDS): Paradox or paradigm?
Thorax. 61:621–626. 2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Barratt S, Medford AR and Millar AB:
Vascular endothelial growth factor in acute lung injury and acute
respiratory distress syndrome. Respiration. 87:329–342.
2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
André P, Nannizzi-Alaimo L, Prasad SK and
Phillips DR: Platelet-derived CD40L: The switch-hitting player of
cardiovascular disease. Circulation. 106:896–899. 2002.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Henn V, Slupsky JR, Gräfe M,
Anagnostopoulos I, Förster R, Müller-Berghaus G and Kroczek RA:
CD40 ligand on activated platelets triggers an inflammatory
reaction of endothelial cells. Nature. 391:591–594. 1998.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Rahman M, Zhang S, Chew M, Ersson A,
Jeppsson B and Thorlacius H: Platelet-derived CD40L (CD154)
mediates neutrophil upregulation of Mac-1 and recruitment in septic
lung injury. Ann Surg. 250:783–790. 2009.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Coxon CH, Geer MJ and Senis YA: ITIM
receptors: More than just inhibitors of platelet activation. Blood.
129:3407–3418. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Watson SP, Herbert JM and Pollitt AY: GPVI
and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost.
8:1456–1467. 2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Kerrigan AM, Dennehy KM, Mourão-Sá D,
Faro-Trindade I, Willment JA, Taylor PR, Eble JA, Reis e Sousa C
and Brown GD: CLEC-2 is a phagocytic activation receptor expressed
on murine peripheral blood neutrophils. J Immunol. 182:4150–4157.
2009.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Badolia R, Kostyak JC, Dangelmaier C and
Kunapuli SP: Syk activity is dispensable for platelet GP1b-IX-V
signaling. Int J Mol Sci. 18(1238)2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Suzuki-Inoue K: Roles of the
CLEC-2-podoplanin interaction in tumor progression. Platelets: Jun
4, 2018 (Epub ahead of print). doi:
10.1080/09537104.2018.1478401.
|
|
59
|
Quintanilla M, Montero-Montero L, Renart J
and Martín-Villar E: Podoplanin in inflammation and cancer. Int J
Mol Sci. 20(707)2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Mammadova-Bach E, Gil-Pulido J,
Sarukhanyan E, Burkard P, Shityakov S, Schonhart C, Stegner D,
Remer K, Nurden P, Nurden AT, et al: Platelet glycoprotein VI
promotes metastasis through interaction with cancer cell-derived
galectin-3. Blood. 135:1146–1160. 2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Huang JS, Ramamurthy SK, Lin X and Le
Breton GC: Cell signalling through thromboxane A2 receptors. Cell
Signal. 16:521–533. 2004.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Goff CD, Corbin RS, Theiss SD, Frierson HF
Jr, Cephas GA, Tribble CG, Kron IL and Young JS: Postinjury
thromboxane receptor blockade ameliorates acute lung injury. Ann
Thorac Surg. 64:826–829. 1997.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zhang S, Yuan J, Yu M, Fan H, Guo ZQ, Yang
R, Guo HP, Liao YH and Wang M: IL-17A facilitates platelet function
through the ERK2 signaling pathway in patients with acute coronary
syndrome. PLoS One. 7(e40641)2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Caudrillier A, Kessenbrock K, Gilliss BM,
Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z and Looney MR:
Platelets induce neutrophil extracellular traps in
transfusion-related acute lung injury. J Clin Invest.
122:2661–2671. 2012.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Holm S, Kared H, Michelsen AE, Kong XY,
Dahl TB, Schultz NH, Nyman TA, Fladeby C, Seljeflot I, Ueland T, et
al: Immune complexes, innate immunity, and NETosis in ChAdOx1
vaccine-induced thrombocytopenia. Eur Heart J. 42:4064–4072.
2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Brinkmann V, Reichard U, Goosmann C,
Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y and Zychlinsky A:
Neutrophil extracellular traps kill bacteria. Science.
303:1532–1535. 2004.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Fuchs TA, Brill A, Duerschmied D,
Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW,
Hartwig JH and Wagner DD: Extracellular DNA traps promote
thrombosis. Proc Natl Acad Sci USA. 107:15880–15885.
2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Worth RG, Chien CD, Chien P, Reilly MP,
McKenzie SE and Schreiber AD: Platelet FcgammaRIIA binds and
internalizes IgG-containing complexes. Exp Hematol. 34:1490–1495.
2006.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Antczak AJ, Vieth JA, Singh N and Worth
RG: Internalization of IgG-coated targets results in activation and
secretion of soluble CD40 ligand and RANTES by human platelets.
Clin Vaccine Immunol. 18:210–216. 2011.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Semple JW, Rebetz J, Maouia A and Kapur R:
An update on the pathophysiology of immune thrombocytopenia. Curr
Opin Hematol. 27:423–429. 2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Sowa JM, Crist SA, Ratliff TL and Elzey
BD: Platelet influence on T- and B-cell responses. Arch Immunol
Ther Exp (Warsz). 57:235–241. 2009.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Romo GM, Dong JF, Schade AJ, Gardiner EE,
Kansas GS, Li CQ, McIntire LV, Berndt MC and López JA: The
glycoprotein Ib-IX-V complex is a platelet counterreceptor for
P-selectin. J Exp Med. 190:803–814. 1999.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Srivastava K, Cockburn IA, Swaim A,
Thompson LE, Tripathi A, Fletcher CA, Shirk EM, Sun H, Kowalska MA,
Fox-Talbot K, et al: Platelet factor 4 mediates inflammation in
experimental cerebral malaria. Cell Host Microbe. 4:179–187.
2008.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Hagiwara S, Iwasaka H, Hasegawa A, Oyama
M, Imatomi R, Uchida T and Noguchi T: Adenosine diphosphate
receptor antagonist clopidogrel sulfate attenuates LPS-induced
systemic inflammation in a rat model. Shock. 35:289–292.
2011.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Asaduzzaman M, Lavasani S, Rahman M, Zhang
S, Braun OO, Jeppsson B and Thorlacius H: Platelets support
pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care
Med. 37:1389–1396. 2009.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Cognasse F, Tariket S, Hamzeh-Cognasse H,
Arthaud CA, Eyraud MA, Bourlet T, Berthelot P, Laradi S,
Fauteux-Daniel S and Garraud O: Platelet depletion limits the
severity but does not prevent the occurrence of experimental
transfusion-related acute lung injury. Transfusion. 60:713–723.
2020.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Zhou J, Lee S, Guo CL, Chang C, Liu T,
Leung KSK, Wai AKC, Cheung BMY, Tse G and Zhang Q: Anticoagulant or
antiplatelet use and severe COVID-19 disease: A propensity
score-matched territory-wide study. Pharmacol Res.
165(105473)2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Zhu Y, Wang J, Meng X, Xie H, Tan J, Guo
X, Han P and Wang R: A positive feedback loop promotes HIF-1α
stability through miR-210-mediated suppression of RUNX3 in
paraquat-induced EMT. J Cell Mol Med. 21:3529–3539. 2017.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Zhou N, Liu Q, Qi X, Zhang X, Ru Z, Ma Y,
Yu T, Zhang M, Li Y, Zhang Y and Cao Z: Paraquat exposure impairs
porcine oocyte meiotic maturation. Theriogenology. 179:60–68.
2021.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Zu Y, Ban J, Xia Z, Wang J, Cai Y, Ping W
and Sun W: Genetic variation in a miR-335 binding site in BIRC5
alters susceptibility to lung cancer in Chinese Han populations.
Biochem Biophys Res Commun. 430:529–534. 2013.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Zuo X, Xu W, Xu M, Tian R, Moussalli MJ,
Mao F, Zheng X, Wang J, Morris JS, Gagea M, et al: Metastasis
regulation by PPARD expression in cancer cells. JCI Insight.
2(e91419)2017.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Zu L, Xue Y and Wang J, Fu Y, Wang X, Xiao
G, Hao M, Sun X, Wang Y, Fu G and Wang J: The feedback loop between
miR-124 and TGF-β pathway plays a significant role in non-small
cell lung cancer metastasis. Carcinogenesis. 37:333–343.
2016.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Zou W, Chen L, Mao W, Hu S, Liu Y and Hu
C: Identification of inflammatory response-related gene signature
associated with immune status and prognosis of lung adenocarcinoma.
Front Bioeng Biotechnol. 9(772206)2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zou M, Xia S, Zhuang L, Han N, Chu Q, Chao
T, Peng P, Chen Y, Gui Q and Yu S: Knockdown of the Bcl-2 gene
increases sensitivity to EGFR tyrosine kinase inhibitors in the
H1975 lung cancer cell line harboring T790M mutation. Int J Oncol.
42:2094–2102. 2013.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Zou K, Tong E, Xu Y, Deng X and Zou L:
Down regulation of mammalian target of rapamycin decreases HIF-1α
and survivin expression in anoxic lung adenocarcinoma A549 cell to
elemene and/or irradiation. Tumour Biol. 35:9735–9741.
2014.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Zong L, Sun Q, Zhang H, Chen Z, Deng Y, Li
D and Zhang L: Increased expression of circRNA_102231 in lung
cancer and its clinical significance. Biomed Pharmacother.
102:639–644. 2018.PubMed/NCBI View Article : Google Scholar
|