1. Introduction
The cytoskeleton of neurons is the primary structure
that regulates cell shape, protein localization and transport from
the soma to dendrites and axons or vice versa. This structure
consists of microfilaments (MFs) (diameter of ~8 nm), intermediate
filaments (IFs) (diameter ranging from 7-11 nm) and microtubules
(MTs) (diameter of ~25 nm) (1).
MFs are primarily composed of actin, enriched in
cortical regions near the cell membrane and are particularly
concentrated in presynaptic terminals, dendritic spines and growth
cones. MFs are also called actin filaments; they are formed during
proliferation, are considered to be versatile due to their ability
to create and destroy themselves, used as construction materials
and bind to accessory proteins, regulating the creation and
destruction of filaments (2,3).
IFs are components of the cytoskeleton that extend
from the nucleus to the cell periphery, and their main function is
to provide mechanical resistance. IFs show an unusual degree of
cell specificity and are often used as markers of cell
differentiation. These filaments are subdivided into multiple
types, among which neurofilaments (neurons) and glial filaments
(glial cells) are noteworthy structures in the current discussion
(4). Neurofilaments (NFs) support
axons and dendrites (5) and play an
important role in determining the axonal caliber of myelinated
fibers (1). These filaments have
been a focus of research, as an increase in their presence is
potentially related to axonal damage and the severity of
neurological diseases (6).
MTs consist of dimers of α and β-tubulin, and these
dimers subsequently bind from one end to the other to form a
‘protofilament’ of tubulin. Additionally, 13 protofilaments are
aligned in parallel in a circular manner to form an empty tube that
is nm-µm long. The tube is polarized: it has head (+) and tail (-)
ends. Polymerization occurs by adding new subunits at the (+) end.
In this process, the units are removed from the (-) end and added
to the (+) end. This process requires various accessory factors;
two of the most important accessory factors are microtubule
accessory proteins (MAPs) and tau protein. These accessory proteins
appear to be involved both in the polymerization process and in the
stabilization of tubulin once it is polymerized (7).
Due to the significant role of the cytoskeleton in
cell transport, in addition to being essential for the normal
functioning of neurons, cytoskeletal anomalies result in neuronal
damage and cell death, which are the common denominators of
neurodegenerative diseases (8,9).
Although the causes of transport abnormalities may vary among the
various pathological conditions, in many circumstances,
deficiencies in transport are caused by a decrease in the stability
of its components, such as alterations in neurofilaments,
microtubule stability, loss of actin dynamics (9), and an increase in the phosphorylation
rates of some proteins that comprise this network (10), as is the case for the tau protein
(11). Based on these observations,
cytoskeletal organization defects may be a common feature that
contributes to neurodegeneration in pathologies such as Alzheimer's
disease (AD), cerebral ischemia and multiple sclerosis (MS).
Therefore, the potential benefits of cytoskeletal stabilizing
agents in improving axonal transport and nerve function in patients
with these diseases has been highlighted.
2. Cytoskeletal damage and
neurodegeneration
The process of destabilization and aggregation of
cytoskeletal proteins causes neurons to become unstable, and
interferes with the antegrade and retrograde transport of
biomolecules along their axons and dendrites, and, in some cases,
generates protein aggregates that lead to neurodegeneration
(12). For example, in AD and
cerebral ischemia, the hyperphosphorylation of the tau protein
leads to the formation of neurofibrillary tangles, axonal damage,
dendritic damage and subsequent cell death (13,14).
Axonal damage associated with the tau protein has also been
reported in other pathologies, such as MS (15), although in the latter,
neurofilaments are the central filaments contributing to
deterioration due to demyelination (16,17).
These proteins have become potential biomarkers, since they are
related to the pathophysiology of the disease, and therefore have a
significant association with diagnosis, treatment and prognosis,
facilitating the diagnosis of individuals at earlier stages of the
disease with less severe symptoms and long-term repercussions.
In this review, we discuss the current body of
literature with regard to the abnormalities of the cytoskeleton and
related proteins that are considered to play critical roles in the
pathology of AD, cerebral ischemia and MS, such as
microtubule-associated protein 2 (MAP2), tau and
neurofilaments.
3. Alzheimer's disease
AD is considered the most prevalent cause of
dementia worldwide (18). AD is
characterized by two proteinopathies that result in a loss of cell
and tissue homeostasis, leading to progressive neuronal
degeneration and loss of cognitive functions in patients (19). Amyloidopathy is a product of the
abnormal cleavage of amyloid precursor protein (APP), which leads
to the accumulation of the Aβ peptide in senile or amyloid plaques,
and tauopathy, which is due to hyperphosphorylation and aggregation
of the tau protein (14). This
protein is normally associated with MTs, but when it is abnormally
phosphorylated, it dissociates and aggregates, leading to the
formation of paired helical filaments (PHFs) and resulting
structures called neurofibrillary tangles (NFTs) (20) (Fig.
1).
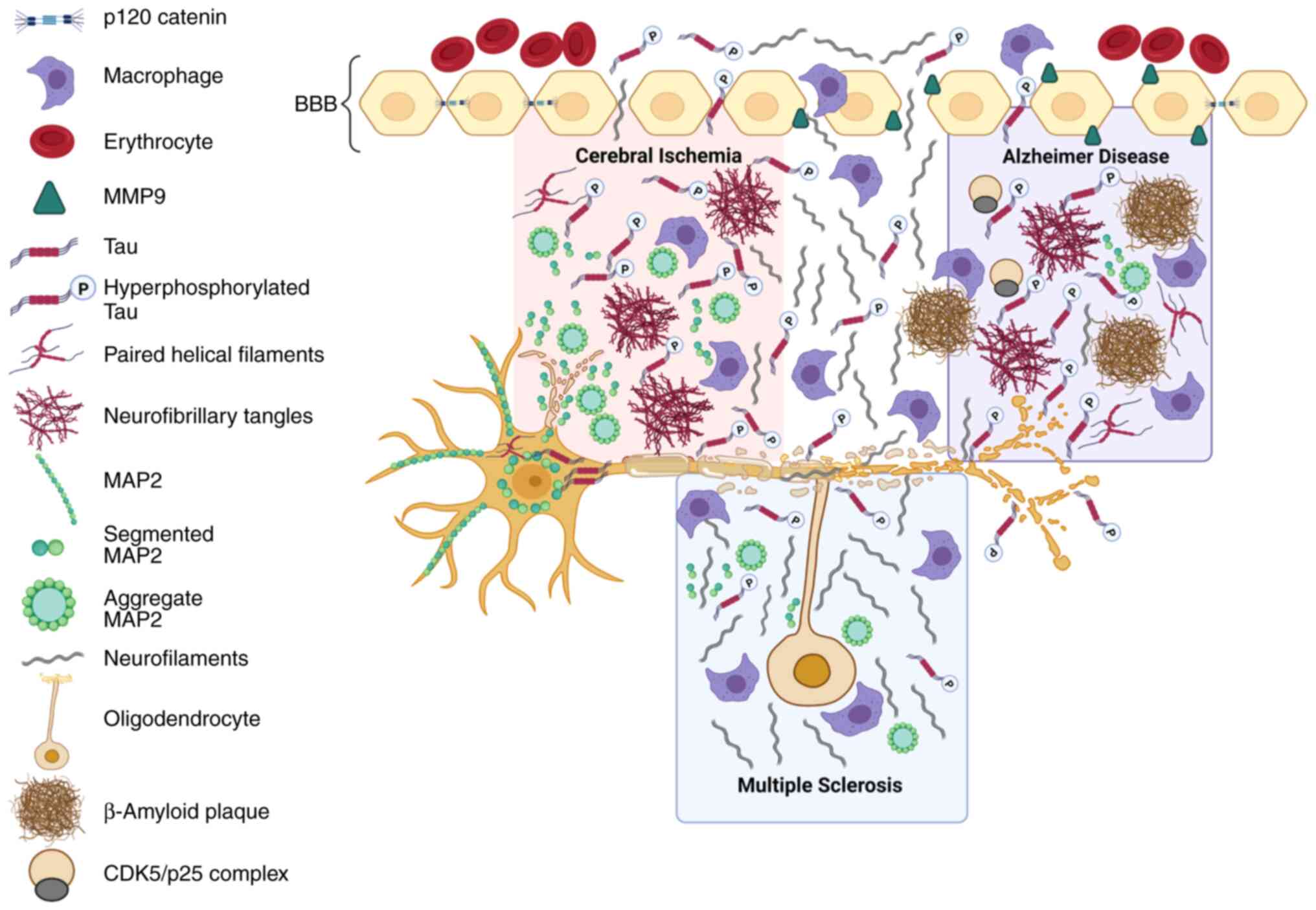 | Figure 1Cytoskeletal alterations in AD,
cerebral ischemia and MS. In patients with AD, cerebral ischemia
and multiple sclerosis, the tau protein is hyperphosphorylated by
different kinases, such as CDK5, which leads to the formation of
NFTs in patients with AD and cerebral ischemia. In individuals with
AD, these tangles are accompanied by the formation of β amyloid
plaques. The MAP2 protein is altered in dendrites, forming
aggregates, and it translocates to the neuronal soma in individuals
with cerebral ischemia. NFs are also a fundamental part of the
neuronal cytoskeleton that are altered in patients with AD,
cerebral ischemia and MS; this deterioration of the cytoskeleton
and other events typical of neurodegeneration generate an immune
response. The disruption of the blood-brain barrier mediated by
MMP9 and the alteration of cell adhesion proteins such as p120
catenin allow these cytoskeletal proteins to be detected in
peripheral blood. AD, Alzheimer's disease; MS, multiple sclerosis;
CDK5, cyclin-dependent kinase; NFT, neurofibrillary tangle; NF,
neurofilament; MMP9, matrix metalloproteinase 9; MAP2,
microtubule-associated protein 2; p-, phospho. |
Tau and MAPs in Alzheimer's
disease
Tauopathy in AD is responsible for a set of
abnormalities that lead to neuronal degeneration, such as defects
in axonal transport and mitochondrial and lysosomal function, among
other functions associated with MTs (21,22).
These abnormalities are due to the overactivation or loss of
inhibition of various kinases and phosphatases that regulate the
binding of MAPs and NFs to the neuronal cytoskeleton and their
functions (23). Under pathological
conditions, the tau protein itself leads to a loss of function,
preventing the binding or shedding of MTs and accumulating in the
cytoplasm along with other MAPs, forming PHFs and subsequently
resulting in extracellular accumulation in NFTs, which leads to
brain deterioration (14,24,25).
In mice deficient in the tau protein, decreases in
the numbers of MTs and small caliber axons, muscle spasms and
behavioral deficits have been identified, although MAP2 partially
compensates for the tau deficit (26). In neurons, the tau protein regulates
the establishment of MT dynamics in axons and has been linked to
the formation of cytoplasmic extensions, axonal transport and
protection against compounds that are deleterious to the cell
(13). In vitro models
related to AD, such as glutamate excitotoxicity, generate a
cellular environment that resembles the conditions in the neurons
of a patient (27). This model
shows the disassembly of MTs, the retraction of dendritic and
axonal processes and the hyperphosphorylation and aggregation of
PHFs in the cell soma in the short term. Increased intracellular
calcium levels, abnormal activation of signaling cascades, and loss
of cellular functions trigger cell death signals that represent the
neurodegenerative process of the disease (28).
Studies have described how the inhibition of various
cytoskeletal proteins is directly and indirectly involved in the
disassembly of MTs, the accumulation of the tau protein and
neuronal death (27,29). The tau protein kinases described
here are grouped into three classes: proline-directed protein
kinases (PDPKs), protein kinases (non-PDPKs) and tyrosine protein
kinases (TPKs). Each kinase was assessed based on its structure,
roles, regulation, involvement in tau phosphorylation and
neurodegeneration linked to AD (30,31).
Cyclin Dependent Kinase 5 (CDK5), a kinase related to the tau
protein and its activators p35/p25, cytoskeletal modifying proteins
such as p120 catenin, and enzymes related to the processing of β
amyloid such as BACE1, have been linked to hyperphosphorylation of
tau and neuronal death (Fig. 1).
Silencing of the CDK5 protein, and the spatial and functional
regulation of the activator p35, as well as its cleaved form p25
occupy central nodes in the mechanism regulating cellular signaling
associated with the hyperphosphorylation of tau (23,28,32).
CDK5 regulates NFs and MAPs either by forming protein complexes or
by phosphorylating them directly, as in the case of NF or MAP2,
MAP1b and the tau protein, which, when phosphorylated, induce the
formation and stabilization of MTs (33,34).
In turn, proteins associated with the processing of β amyloid, such
as BACE1, participate in tau hyperphosphorylation as part of the
cellular and molecular machinery that exerts a synergistic effect
on the development of histopathological markers of the disease
(35,36).
One of the main pathways for degradation of
hyperphosphorylated tau is autophagy, which is a selective cellular
catabolic process by which cytoplasmic material is transported from
the cytoskeleton to lysosomes for degradation (37). This mechanism functions at basal
levels in all cells and is required to maintain homeostasis
(38). This process is particularly
important in neurons, since they undergo cell division only at a
low rate, and therefore must survive throughout the lifespan of the
organism through the exchange of proteins and organelles (39). In neurodegenerative diseases such as
AD, the cytoskeleton plays a key role in the degradation of PHFs,
and when cytoskeletal integrity is compromised, PHFs are more
likely to aggregate and accumulate (40). The current body of literature show
an increase in autophagy in the early stages of AD in the 3xTg-AD
model, which loses its effect as the pathological process
progresses (41). The regulation of
CDK5 and BACE1 and the effects on the cytoskeleton restore the
degradation of the transgenically expressed tau protein and promote
the functional recovery of the brain (23,35).
The contribution of other MT-associated proteins,
such as MAP2, MAP1B and MAP1A, in the pathogenesis and progression
of AD may vary as they generate different MT dynamics dependent on
their location in the models described, despite sharing similar
sequences and functions in stabilizing MTs with the tau protein
(42). Unlike the tau protein,
which is abundantly distributed in the axonal compartment, MAP2 is
located exclusively in the somatodendritic compartment, whereas
MAP1B and MAP1A are located in both compartments (43). Likewise, MAP2, MAP1B and MAP1A
undergo hyperphosphorylation but fail to form filaments, as does
the tau protein (44). In turn, a
decrease in MAP2 levels or an increase in the levels of the soluble
protein due to abnormal phosphorylation have been suggested to
trigger neurotoxic processes (22).
These changes are related to the loss of neuronal connectivity
induced by amyloid β, tauopathy and the excitotoxic
microenvironment that compromises the dynamics of dendritic spines
and postsynaptic compartments (22,45).
Neurofilaments in Alzheimer's
disease
NFs are the other type of essential filament
involved in the neurodegeneration process; they are the largest
component of the neuronal cytoskeleton, and together with the tau
protein, they predominate in the nervous system (40). These filaments are composed of a
family of 5 intermediate filaments termed NF heavy (NFH), medium
(NFM) and light (NFL) chains, and α-internexin and peripherin
(46). The assembly of NFs is
essential for the growth and stability of axons in both the central
and peripheral nervous systems (47). Its functions are broad and mainly
mediate the stabilization of the microtubule content and
interactions with organelles such as the mitochondria (48). In AD and other neurodegenerative
processes, NFs have been described as a common biomarker that
reflects the changes in the neuronal cytoskeleton and the
progression of the neurodegenerative process that underlies the
pathology (48).
NFs have been detected in both cerebrospinal fluid
(CSF) and peripheral blood (SP) (49). In a cohort of 1,070 PSEN1 E280A
mutation carriers and 1,074 noncarriers with baseline assessments
and 242 mutation carriers and 262 noncarriers with longitudinal
(6±3 years) measures, ranging in age from 8 to 75 years, plasma NFL
levels increased with age in both groups and began to differentiate
carriers from noncarriers at age 22 (22 years before the estimated
median age of mild cognitive impairment) (50,51).
This biomarker is released in to the CSF and SP as the inflammatory
process and gliosis progress in the white matter, and has been
correlated with cortical and hippocampal atrophy, widening of the
ventricles, memory impairment and mild cognitive impairment
(52,53). Although NFs have proline-directed
phosphorylation sites and are identified in NFTs as one of the
proteins that accumulate with PHFs, their contribution to aggregate
formation has not been clarified (24). Axonal damage associated with MTs and
synaptic compromise are common pathologies such as AD, Down
syndrome, frontotemporal dementia, amyotrophic lateral sclerosis,
dementia with Lewy bodies, progressive supra-nuclear palsy, and
cortico-basal syndrome; along with the aging process, they convert
NFs into biomarkers of CNS and peripheral involvement (54-56).
4. Cerebral ischemia
Cerebral ischemia is a disorder that affects the
brain tissue and is characterized mainly by the disrupted supply of
oxygen and other nutrients, which leads to tissue death (57). This pathology triggers cognitive
impairment or dementia, conditions that include alterations in
learning, memory and functions needed to perform basic activities
of daily life (58). Within the
pathophysiology of the disease, changes in the cytoskeleton that
reflect neuronal damage have been identified (59).
MAP2 and tau in cerebral ischemia
Numerous antibodies have been used in experimental
models to identify the damage and chronological changes in
cytoskeletal proteins, including antibodies against microtubule
proteins such as MAP2, a marker of cytoskeletal disruption; in
fact, studies have documented a correlation of MAP2 expression with
prolonged postischemic periods (60-62).
In animal models, immunohistochemistry and immunofluorescence
techniques revealed the loss or discontinuity of MAP2
immunoreactivity in lesions. The discontinuity reflects the
fragmentation of dendrites. The translocation of this protein to
neuronal cells has also been reported (63-65)
(Fig. 1). Taken together, these
data reveal the changes in dendrites that lead to cell death in
areas of both the ischemic focus and in regions of the ischemic
penumbra, as well as the loss of synaptic plasticity in regions
further from the ischemic focus (65). The loss of MAP2 may contribute to
the initial phase of neuronal dysfunction, and dendritic
degradation may be the first sign of neurodegeneration 1 h after
cerebral ischemia (66). In
patients who have suffered cerebral ischemia, a decrease in MAP2
immunoreactivity has been observed in the motor (area 4), temporal
(area 21), frontal (area 10) and visual (area 17) cortices in the
left hemisphere; additionally, in all the areas studied, the most
significant decrease in MAP2 expression was detected in cortical
layers II-III compared to cortical layers V-VII. The maximum
reduction in MAP2-positive pyramidal neurons was observed in
cortical layers II-III of the motor cortex after 1 year of survival
after cerebral infarction (67).
Based on this finding, markers of the cytoskeleton are sensitive to
the deterioration that occurs long after cerebral infarction.
Another protein involved in microtubule disruption
is the tau protein. The hyperphosphorylation of the tau protein is
associated with the development of dementia in the late
postischemia phase. Its roles in initiating synaptic and cognitive
dysfunction, in addition to neuronal toxicity and
neurodegeneration, have been described (68). Experimental animals subjected to
cerebral ischemia present disruptions in memory and learning
processes accompanied by tau hyperphosphorylation, the formation of
PHFs and changes in the immunoreactivity of the MAP2 protein
(69).
The relationship between the CA1 region of the
hippocampus and the expression of the tau gene has been established
after transient global cerebral ischemia in rats with a survival
period of 2, 7 and 30 days (70).
In the hippocampal CA1 region, the expression of the tau gene
increased to a maximum 3.3-fold change on the second day after
cerebral ischemia. A total of 7 days after ischemic episodes, the
expression ranged from 0.2 to 0.5 times the base value (70). On the 30th day of survival after
ischemic injury, the expression of the tau protein gene decreased
to 0.4 times that of the base value (70). Studies have shown that tau protein
phosphorylation patterns differ depending on the models of cerebral
ischemia. After ischemia and global brain recirculation, the tau
protein is phosphorylated and slowly accumulates (71). Transient focal cerebral ischemia
with 1 day of reperfusion induces local hyperphosphorylation
(regions of injury) of the tau protein (72,73).
Current research indicates that after ischemia, hyperphosphorylated
tau protein accumulates in cortical neuronal cells and is
accompanied by apoptosis (72,74).
Additionally, hyperphosphorylation of tau leads to the formation of
NFTs 24 h postinfarction in regions such as the motor, sensory and
hippocampal cortex. These tangles persist in these same regions for
up to 1 month after infarction, and lead to learning and memory
deficits in the affected animals (69).
In clinical studies, an increase in total tau levels
in human CSF and blood has been reported after brain injury,
including ischemic stroke (75-78).
Measurable tau has been detected in serum within 6 h after the
onset of ischemic symptoms (78).
The concentration may peak after 3-5 days (78) or later (79). Furthermore, a significant
correlation between serum tau levels and the severity of the
clinical deficit or disability evaluated using the Barthel index
(BI) was not observed. However, serum tau levels correlate with
infarct volumes (7-48 ml) and functional results at 90 days
postischemia (78). These findings
are consistent with other studies indicating that the absence of
serum tau during the acute phase (<24 h) of ischemia might
predict good clinical outcomes 90 days after stroke (80). Patients in whom tau was detected in
serum had more severe neurological deficits and poorer functional
outcomes than patients without tau (81). However, other researchers found that
tau protein levels are correlated with neurological deficit (BI)
scores after 48 h. Additionally, serum tau levels did not have a
significant correlation with the etiology of stroke, as represented
by the TOAST criteria (82). A
prospective study revealed that tau levels in both plasma and CSF
were closely related not only to stroke severity assessed using the
National Institutes of Health Stroke Scale but also to long-term
outcomes (83). In particular, the
study of autopsy specimens of the brains from patients with
cerebral infarction showed an increase in tau immunoreactivity and
tau deposition in the ischemic area (73,84).
However, the tau protein has been detected in the serum of ~40% of
patients with stroke (78,79). Some researchers propose that tau
appears in the blood due to disruption of the blood-brain barrier
(BBB). Some factors, such as matrix metalloproteinase 9 (MMP9), may
play a key role in the release of tau into the circulation
(79) (Fig. 1).
In addition to BBB damage after stroke, another
major cause of persistent disability after stroke is neuroaxonal
damage, which is crucial for the functional outcomes and long-term
survival of patients with stroke. Predicting functional outcomes
after ischemic stroke is very important for patients and clinicians
in terms of allocating healthcare resources and optimizing patient
care. Furthermore, the amount of acute neuroaxonal damage reflected
by the infarct nucleus guides patient selection for stroke
therapies, e.g., endovascular thrombectomy beyond the 6-h time
window. To date, only diffusion magnetic resonance imaging (MRI)
and computed tomography (CT) perfusion approaches serve to assess
the core of the infarct; therefore, blood biomarkers are urgently
needed to guide individualized treatments of patients. NFs may be a
suitable candidate for this purpose as they are part of the
neuronal cytoskeleton, are exclusively expressed in neurons
(85), and show encouraging
results, as described further below.
Neurofilaments in cerebral
ischemia
After the occlusion of a cerebral artery and
subsequent neuroaxonal damage, the NF protein is released into the
CSF and to a lesser extent in SP. Ultrasensitive assays with the
single molecule matrix method facilitate the highly sensitive
quantification of NF levels in blood (86). Due to the ease and wide
applicability, research on NFs in various diseases, such as MS and
other neurodegenerative diseases, including cerebral infarction, is
emerging (16). In a study
conducted by Peters et al (87), where they evaluated 503 patients
with small vessel disease (SVD), NFs were associated with the
presence of lacunae and microbleeds and with MRI imaging markers
related to SVD. In addition, NF was associated with gap incidents
during patient follow-up, as well as with future cognitive decline
after adjustment for age, sex, education and depression. The risk
of dementia increased with higher NF levels. Therefore, NFs were
proposed as an emerging blood biomarker for neuroaxonal damage in
various neurological diseases affecting the elderly, including
small vessel neurodegenerative and cerebral disease (88,89).
Thus, cerebrovascular diseases appear to be a major vascular
contributor to dementia (88-90).
In 2019, Timo Uphaus and colleagues were the first
to show that NF is a valuable biomarker for functional independence
at 90 days after ischemic stroke and predicts long-term
cardiovascular outcomes (85).
Accordingly, NF may be useful in selecting patients at high risk of
future cardiovascular events. The superiority of NF over other
existing biomarkers was also shown with respect to its predictive
value for functional outcomes and cardiovascular survival after
ischemic stroke (85). However,
studies examining larger patient cohorts are required to confirm
the results and to change current clinical practice, since NF may
be used in clinical practice as a screening biomarker to select
patients at high risk of accident occurrence, recurrent
cerebrovascular disease and those more susceptible to death
following cerebral ischemia.
5. Multiple sclerosis
MS is a chronic demyelinating disease of the central
nervous system (CNS). Although its etiology remains unknown, immune
cells are generally accepted to invade the CNS, where demyelination
occurs. This process is attributed to the migration of autoreactive
T lymphocytes from the periphery toward the CNS with the capacity
to cross the BBB and progressively compromise brain function
(91) (Fig. 1). The clinical manifestation of MS
represents the final stage of a process that involves inflammation,
demyelination, remyelination and the depletion of oligodendrocytes,
astrocytes and neurons along with axonal degeneration (91).
Tau and MAP2 in multiple
sclerosis
In the affected part of the CNS, constitutively
expressed proteins in the axon, such as tau protein and NFs, can be
detected using antibodies (Fig. 1).
CSF tau protein levels are associated with axonal injury, are
elevated in patients with MS and correlate with the progression of
disability (75,92). In serum samples from patients with
MS collected before and after autologous hematopoietic stem cell
transplantation, tau protein levels were increased 3 months after
transplantation; this observation may reflect brain atrophy induced
by chemotherapy-mediated toxicity (88). In a case-controlled study of 30
healthy women and 30 women with MS, the levels of the total tau
protein in serum and saliva were analyzed. Total tau protein was
level in the serum of patients with MS compared with the control
group; therefore, tau represents a potential biomarker for MS.
However, the levels of tau protein in saliva was not a suitable
biomarker for the detection of MS (93). Some MS studies have sought to
investigate the mechanisms of remyelination by analyzing the MAP2
protein levels in brain lesions affected by MS using
immunocytochemistry and a series of specific monoclonal antibodies.
MAP2 expression was increased in the brains of patients with MS
(94). MAP2 was expressed in the
regenerating oligodendrocytes associated with demyelinated lesions,
with the highest levels detected in regions with extensive
remyelination. Using electron microscopy, MAP2 was shown to be
located in oligodendrocytes involved in remyelination, as evidenced
by the extension of its process and association with finely
myelinated (remyelinated) axons (94).
Neurofilaments in multiple
sclerosis
NF, a cytoskeletal protein that has been most
closely associated with axonal deterioration in MS, has been used
as a biomarker for axonal degeneration and may be used to predict
the neurological outcomes of patients (48,86).
Following axonal damage in the CNS, NF proteins released into the
CSF reflect the degree of axonal damage and neuronal death. Since
neurofilaments are located in the cytoplasm of neurons, all
diseases that lead to neuronal and axonal damage potentially
increase the levels of these proteins in the CSF (86). In fact, NFs have been evaluated both
in clinical studies and in in vivo models that simulate the
degenerative processes of MS, where the most commonly used model is
the experimental autoimmune encephalitis (EAE) model. In this
model, the mean concentration of NFL is higher in the supernatant
and brain pellet of rats with all EAE subtypes compared to samples
collected from controls (95).
Furthermore, NFM and NFH levels change in the later phases of the
disease. Therefore, the NFL biomarker may reflect acute axonal
damage mediated by inflammatory mechanisms, and may have prognostic
value for the evaluation of MS, while NFM and NFH may be better
indicators of nerve regeneration after acute inflammation (95).
In humans, NFs were used for the first time as
markers of neuronal damage in a study of 12 patients with
amyotrophic lateral sclerosis and 11 patients with AD (96). Subsequently, CSF NF levels were
higher in 60 patients with relapsing-remitting multiple sclerosis
than in the control subjects (97),
suggesting that these proteins may serve as a biomarker of the
disease activity of MS.
NFs have been associated with the progression of
disability in patients with MS (74), and according to studies comparing
the serum and CSF levels of NFs, they are strongly correlated
(83). An ultrasensitive technique
called single molecule arrays (SIMOA) has been developed recently
to detect NF levels in blood, enabling, for the first time, the
detection of NF levels in serum. Compared to detection using ELISA
or ECL-based assays, SIMOA has >25-fold higher analytical
sensitivity (SIMOA: 0.62 pg/ml, ECL: 15.6 pg/ml, ELISA: 78, 0
pg/ml) (98).
NFL is associated with axonal damage, while NFH is
associated with the progression of disability (87,99).
In some of the population based studies, NFL levels were shown to
be associated with an increased risk of progressive phenotypes
(16), and they have also been
useful in diagnosing the pathology; when NFL levels were compared
between patients with MS and controls, where the values between the
two groups were different, they were higher in patients with MS
(100).
A recent meta-analysis found that patients with MS
present with axonal injury (101),
which may explain the neurodegeneration caused by the disease and
its effect on NF levels, especially in the early stages of the
disease. The cause of axonal loss is not yet known, but a
destructive process directed against components of the axonal
cytoskeleton appears to contribute to the progression of the
disability (99). Serum NF levels
also correlate with MRI activity, degree of disability and rate of
brain atrophy (102,103). Furthermore, NFs are also suitable
as a prognostic biomarker for the conversion of clinically isolated
syndrome (CIS) to MS (104,105).
A recent study showed the prognostic importance of serum NF levels
in the conversion of isolated radiological syndrome to CIS
(106).
The serum concentration of NFL, which does not
require a lumbar puncture, appears to correlate with several
clinical and magnetic tomographic features of MS (107,108). Therefore, it may conceivably be
established as a prognostic biomarker in clinical practice in the
future.
6. Conclusions
Cytoskeletal damage, including disruptions to
microtubule stability, NFs and axonal transport, have been
characterized in several unrelated neurodegenerative conditions.
Although no direct link has been established between the different
pathologies addressed in this review, common cytoskeletal players
have been shown to contribute to deterioration, suggesting that
defects in the organization of the cytoskeleton are a common
feature that contributes to neurodegeneration. Findings from the
in vitro and in vivo experimental models discussed in
this review show that the tau protein, MAP2 and NFs are essential
for maintaining the stability of the cytoskeleton, and that their
dysregulation triggered by anoxia (cerebral ischemia), the Aβ
peptide (AD) and demyelination (MS) leads to the disassembly of
MAP2 and NFs, hyperphosphorylation and tau accumulation. These
proteins are subsequently released into the SP after the disruption
of the BBB through the actions of metalloproteases such as MMP9 or
the post-traumatic inflammatory response (Fig. 1). The latter suggests that
cytoskeletal components detected in the SP are promising protein
targets that may facilitate the diagnosis and progression of
cerebral ischemia, AD and MS in a practical and safe manner, as
reported in several clinical studies discussed in the review.
However, more studies are required to correlate the clinical
manifestations presented by patients with these pathologies.
Acknowledgements
Not applicable.
Funding
Funding: We would like to thank Remington University Corporation
for the financial support.
Availability of data and materials
Not applicable.
Authors' contributions
JAGV, JFCA, JFZB, KAR and AAN wrote and revised the
current review. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fletcher DA and Mullins RD: Cell mechanics
and the cytoskeleton. Nature. 463:485–492. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rottner K, Faix J, Bogdan S, Linder S and
Kerkhoff E: Actin assembly mechanisms at a glance. J Cell Sci.
130:3427–3435. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jay D, García EJ, Lara JE, Medina MA and
de la Luz Ibarra M: Determination of a cAMP-dependent protein
kinase phosphorylation site in the C-terminal region of human
endothelial actin-binding protein. Arch Biochem Biophys. 377:80–84.
2000.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Liem RKH and Messing A: Dysfunctions of
neuronal and glial intermediate filaments in disease. J Clin
Invest. 119:1814–1824. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Da Silva JS and Dotti CG: Breaking the
neuronal sphere: Regulation of the actin cytoskeleton in
neuritogenesis. Nat Rev Neurosci. 3:694–704. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Medana IM and Esiri MM: Axonal damage: A
key predictor of outcome in human CNS diseases. Brain. 126:515–530.
2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Goodson HV and Jonasson EM: Microtubules
and microtubule-associated proteins. Cold Spring Harb Perspect
Biol. 10(a022608)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jellinger KA: Cell death mechanisms in
neurodegeneration. J Cell Mol Med. 5:1–17. 2001.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Muñoz-Lasso DC, Romá-Mateo C, Pallardó FV
and Gonzalez-Cabo P: Much more than a scaffold: Cytoskeletal
proteins in neurological disorders. Cells. 9(E358)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Henriques AG, Müller T, Oliveira JM, Cova
M, da Cruz e Silva CB and da Cruz E Silva OA: Altered protein
phosphorylation as a resource for potential AD biomarkers. Sci Rep.
6(30319)2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mietelska-Porowska A, Wasik U, Goras M,
Filipek A and Niewiadomska G: Tau protein modifications and
interactions: Their role in function and dysfunction. Int J Mol
Sci. 15:4671–4713. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
McMurray CT: Neurodegeneration: Diseases
of the cytoskeleton? Cell Death Differ. 7:861–865. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Guo T, Noble W and Hanger DP: Roles of tau
protein in health and disease. Acta Neuropathol. 133:665–704.
2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kosik KS, Joachim CL and Selkoe DJ:
Microtubule-associated protein tau (tau) is a major antigenic
component of paired helical filaments in Alzheimer disease. Acta
Neuropathol. 83:4044–4048. 1986.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Islas-Hernandez A, Aguilar-Talamantes HS,
Bertado-Cortes B, Mejia-delCastillo GJ, Carrera-Pineda R,
Cuevas-Garcia CF and Garcia-delaTorre P: BDNF and Tau as biomarkers
of severity in multiple sclerosis. Biomark Med. 12:717–726.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Siller N, Kuhle J, Muthuraman M, Barro C,
Uphaus T, Groppa S, Kappos L, Zipp F and Bittner S: Serum
neurofilament light chain is a biomarker of acute and chronic
neuronal damage in early multiple sclerosis. Mult Scler.
25:678–686. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zetterberg H: Plasma Neurofilament light
in progressive multiple sclerosis. Acta Neurol Scand. 141:14–15.
2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
GBD 2017 Disease and Injury Incidence and
Prevalence Collaborators. Global, Regional, and national incidence,
prevalence, and years lived with disability for 354 Diseases and
Injuries for 195 countries and territories, 1990-2017: A systematic
analysis for the Global Burden of Disease Study 2017. Lancet.
392:1789–1858. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sanchez JS, Hanseeuw BJ, Lopera F,
Sperling RA, Baena A, Bocanegra Y, Aguillon D, Guzmán-Vélez E,
Pardilla-Delgado E, Ramirez-Gomez L, et al: Longitudinal amyloid
and tau accumulation in autosomal dominant Alzheimer's disease:
Findings from the Colombia-Boston (COLBOS) biomarker study.
Alzheimers Res Ther. 13(27)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Busche MA and Hyman BT: Synergy between
amyloid-β and tau in Alzheimer's disease. Nat Neurosci.
23:1183–1193. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Drummond E, Pires G, MacMurray C, Askenazi
M, Nayak S, Bourdon M, Safar J, Ueberheide B and Wisniewski T:
Phosphorylated tau interactome in the human Alzheimer's disease
brain. Brain. 143:2803–2817. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kandimalla R, Manczak M, Yin X, Wang R and
Reddy PH: Hippocampal phosphorylated tau induced cognitive decline,
dendritic spine loss and mitochondrial abnormalities in a mouse
model of Alzheimer's disease. Hum Mol Genet. 27:30–40.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Castro-Alvarez JF, Uribe-Arias A, Raigoza
DM and Cardona-Gómez GP: Cyclin-dependent kinase 5, a node protein
in diminished tauopathy: A systems biology approach. Front Aging
Neurosci. 6(232)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Rudrabhatla P, Jaffe H and Pant HC: Direct
evidence of phosphorylated neuronal intermediate filament proteins
in neurofibrillary tangles (NFTs): Phosphoproteomics of Alzheimer's
NFTs. FASEB J. 25:3896–3905. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Richetin K, Steullet P, Pachoud M, Perbet
R, Parietti E, Maheswaran M, Eddarkaoui S, Bégard S, Pythoud C, Rey
M, et al: Tau accumulation in astrocytes of the dentate gyrus
induces neuronal dysfunction and memory deficits in Alzheimer's
disease. Nat Neurosci. 23:1567–1579. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ,
Alaverdyan M, Kiosea NC, Nazari S, Chen PP, Nothias F, et al: Loss
of MAP function leads to hippocampal synapse loss and deficits in
the Morris water maze with aging. J Neurosci. 34:7124–7136.
2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lopez-Tobón A, Cepeda-Prado E and
Cardona-Gómez GP: Decrease of tau hyperphosphorylation by 17β
estradiol requires sphingosine kinase in a glutamate toxicity
model. Neurochem Res. 34:2206–2214. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Posada-Duque RA, Ramirez O, Härtel S,
Inestrosa NC, Bodaleo F, González-Billault C, Kirkwood A and
Cardona-Gómez GP: CDK5 downregulation enhances synaptic plasticity.
Cell Mol Life Sci. 74:153–172. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Uribe-Arias A, Posada-Duque RA,
González-Billault C, Villegas A, Lopera F and Cardona-Gómez GP:
p120-catenin is necessary for neuroprotection induced by CDK5
silencing in models of Alzheimer's disease. J Neurochem.
138:624–639. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Martin L, Latypova X, Wilson CM,
Magnaudeix A, Perrin ML, Yardin C and Terro F: Tau protein kinases:
Involvement in Alzheimer's disease. Ageing Res Rev. 12:289–309.
2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Reimer L, Betzer C, Kofoed RH, Volbracht
C, Fog K, Kurhade C, Nilsson E, Överby AK and Jensen PH: PKR kinase
directly regulates tau expression and Alzheimer's disease-related
tau phosphorylation. Brain Pathol. 31:103–119. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Posada-Duque RA, López-Tobón A, Piedrahita
D, González-Billault C and Cardona-Gómez GP: p35 and Rac1 underlie
the neuroprotection and cognitive improvement induced by CDK5
silencing. J Neurochem. 134:354–370. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zheng YL, Li BS, Kanungo J, Kesavapany S,
Amin N, Grant P and Pant HC: Cdk5 modulation of mitogen-activated
protein kinase signaling regulates neuronal survival. Mol Biol
Cell. 18:404–413. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cicero S and Herrup K: Cyclin-dependent
kinase 5 is essential for neuronal cell cycle arrest and
differentiation. J Neurosci. 25:9658–9668. 2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Piedrahita D, Castro-Alvarez JF, Boudreau
RL, Villegas-Lanau A, Kosik KS, Gallego-Gomez JC and Cardona-Gómez
GP: β-Secretase 1's targeting reduces hyperphosphorilated tau,
implying autophagy actors in 3xTg-AD mice. Front Cell Neurosci.
9(498)2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Choi SH, Kim YH, Hebisch M, Sliwinski C,
Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, et al: A
three-dimensional human neural cell culture model of Alzheimer's
disease. Nature. 515:274–278. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Choi AMK, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662.
2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Komatsu M, Qing JW, Holstein GR, Friedrich
VL Jr, Iwata J, Kominami E, Chait BT, Tanaka K and Yue Z: Essential
role for autophagy protein Atg7 in the maintenance of axonal
homeostasis and the prevention of axonal degeneration. Proc Natl
Acad Sci USA. 104:14489–14494. 2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Komatsu M, Waguri S, Chiba T, Murata S,
Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E and
Tanaka K: Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature. 441:880–884. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kast DJ and Dominguez R: The
cytoskeleton-autophagy connection. Curr Biol. 27:R318–R326.
2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Villamil Ortiz JG and Cardona Gomez GP:
Comparative analysis of autophagy and tauopathy related markers in
cerebral ischemia and Alzheimer's disease animal models. Front
Aging Neurosci. 7(84)2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mohan R and John A: Microtubule-associated
proteins as direct crosslinkers of actin filaments and
microtubules. IUBMB Life. 67:395–403. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xie C, Soeda Y, Shinzaki Y, In Y, Tomoo K,
Ihara Y and Miyasaka T: Identification of key amino acids
responsible for the distinct aggregation properties of
microtubule-associated protein 2 and tau. J Neurochem. 135:19–26.
2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Xie C, Miyasaka T, Yoshimura S, Hatsuta H,
Yoshina S, Kage-Nakadai E, Mitani S, Murayama S and Ihara Y: The
homologous carboxyl-terminal domains of microtubule-associated
protein 2 and Tau induce neuronal dysfunction and have differential
fates in the evolution of neurofibrillary tangles. PLoS One.
9(e89796)2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Takahashi RH, Capetillo-Zarate E, Lin MT,
Milner TA and Gouras GK: Accumulation of Intraneuronal β-Amyloid 42
peptides is associated with early changes in microtubule-associated
protein 2 in neurites and synapses. PLoS One.
8(e51965)2013.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yuan A, Sasaki T, Kumar A, Peterhoff CM,
Rao MV, Liem RK, Julien JP and Nixon RA: Peripherin is a subunit of
peripheral nerve neurofilaments: Implications for differential
vulnerability of CNS and peripheral nervous system axons. J
Neurosci. 32:8501–8508. 2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yuan A, Sershen H, Veeranna Basavarajappa
BS, Kumar A, Hashim A, Berg M, Lee JH, Sato Y, Rao MV, et al:
Neurofilament subunits are integral components of synapses and
modulate neurotransmission and behavior in vivo. Mol Psychiatry.
20:986–994. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gafson AR, Barthélemy NR, Bomont P, Carare
RO, Durham HD, Julien JP, Kuhle J, Leppert D, Nixon RA, Weller RO,
et al: Neurofilaments: Neurobiological foundations for biomarker
applications. Brain. 143:1975–1998. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Barry DM, Stevenson W, Bober BG, Wiese PJ,
Dale JM, Barry GS, Byers NS, Strope JD, Chang R, Schulz DJ, et al:
Expansion of Neurofilament Medium C terminus increases axonal
diameter independent of increases in conduction velocity or myelin
thickness. J Neurosci. 32:6209–6219. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Guzmán-Vélez E, Zetterberg H, Fox-Fuller
JT, Vila-Castelar C, Sanchez JS, Baena A, Garcia-Ospina G, Aguillon
D, Pardilla-Delgado E, Gatchel JR, et al: Associations between
plasma neurofilament light, in vivo brain pathology, and cognition
in non-demented individuals with autosomal-dominant Alzheimer's
disease. Alzheimers Dement. 17:813–821. 2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Quiroz YT, Zetterberg H, Reiman EM, Chen
Y, Su Y, Fox-Fuller JT, Garcia G, Villegas A, Sepulveda-Falla D,
Villada M, et al: Plasma neurofilament light chain in the
presenilin 1 E280A autosomal dominant Alzheimer's disease kindred:
A cross-sectional and longitudinal cohort study. Lancet Neurol.
19:513–521. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Rajan KB, Aggarwal NT, McAninch EA, Weuve
J, Barnes LL, Wilson RS, DeCarli C and Evans DA: Remote blood
biomarkers of longitudinal cognitive outcomes in a population
study. Ann Neurol. 88:1065–1076. 2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Walsh P, Sudre CH, Fiford CM, Ryan NS,
Lashley T, Frost C and Barnes J: ADNI Investigators. The
age-dependent associations of white matter hyperintensities and
neurofilament light in early- and late-stage Alzheimer's disease.
Neurobiol Aging. 97:10–17. 2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Delaby C, Alcolea D, Carmona-Iragui M,
Illán-Gala I, Morenas-Rodríguez E, Barroeta I, Altuna M, Estellés
T, Santos-Santos M, Turon-Sans J, et al: Differential levels of
Neurofilament Light protein in cerebrospinal fluid in patients with
a wide range of neurodegenerative disorders. Sci Rep.
10(9161)2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Idland AV, Sala-Llonch R, Borza T, Watne
LO, Wyller TB, Brækhus A, Zetterberg H, Blennow K, Walhovd KB and
Fjell AM: CSF neurofilament light levels predict hippocampal
atrophy in cognitively healthy older adults. Neurobiol Aging.
49:138–144. 2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Henson RL, Doran E, Christian BT, Handen
BL, Klunk WE, Lai F, Lee JH, Rosas HD, Schupf N, Zaman SH, et al:
Cerebrospinal fluid biomarkers of Alzheimer's disease in a cohort
of adults with Down syndrome. Alzheimers Dement (Amst).
12(e12057)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Sveinsson OA, Kjartansson O and
Valdimarsson EM: Cerebral ischemia/infarction-epidemiology, causes
and symptoms. Laeknabladid. 100:271–279. 2014.PubMed/NCBI View Article : Google Scholar : (In
Icelandic).
|
|
58
|
Cao L, Tan L, Wang HF, Jiang T, Zhu XC and
Yu JT: Cerebral Microinfarcts and dementia: A systematic review and
metaanalysis. Curr Alzheimer Res. 14:802–808. 2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Pluta R, Januszewski S and Czuczwar SJ:
Brain ischemia as a prelude to Alzheimer's disease. Front Aging
Neurosci. 13(636653)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Yoshimi K, Takeda M, Nishimura T, Kudo T,
Nakamura Y, Tada K and Iwata N: An immunohistochemical study of
MAP2 and clathrin in gerbil hippocampus after cerebral ischemia.
Brain Res. 560:149–158. 1991.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Vanicky I, Balchen T and Diemer NH:
Alterations in MAP2 immunostainability after prolonged complete
brain ischaemia in the rat. Neuroreport. 7:161–164. 1995.PubMed/NCBI
|
|
62
|
Mages B, Fuhs T, Aleithe S, Blietz A,
Hobusch C, Härtig W, Schob S, Krueger M and Michalski D: The
Cytoskeletal Elements MAP2 and NF-L show substantial alterations in
different stroke models while elevated serum levels highlight
especially MAP2 as a sensitive biomarker in stroke patients. Mol
Neurobiol. 58:4051–4069. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Johanna GV, Fredy CA, David VC, Natalia
MV, Angel CR and Patricia CG: Rac1 activity changes are associated
with neuronal pathology and spatial memory long-term recovery after
global cerebral ischemia. Neurochem Int. 57:762–773.
2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Gutiérrez-Vargas JA, Moreno H and
Cardona-Gómez GP: Targeting CDK5 post-stroke provides long-term
neuroprotection and rescues synaptic plasticity. J Cereb Blood Flow
Metab. 37:2208–2223. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Pérez-Corredor PA, Gutiérrez-Vargas JA,
Ciro-Ramírez L, Balcazar N and Cardona-Gómez GP: High fructose
diet-induced obesity worsens post-ischemic brain injury in the
hippocampus of female rats. Nutr Neurosci: Mar 2, 2020 (Epub ahead
of print).
|
|
66
|
Dawson DA and Hallenbeck JM: Acute focal
ischemia-induced alterations in MAP2 immunostaining: Description of
temporal changes and utilization as a marker for volumetric
assessment of acute brain injury. J Cereb Blood Flow Metab.
16:170–174. 1996.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Akulinin VA and Dahlstrom A: Quantitative
analysis of MAP2 immunoreactivity in human neocortex of three
patients surviving after brain ischemia. Neurochem Res. 28:373–378.
2003.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Pluta R, Ułamek-Kozioł M, Januszewski S
and Czuczwar SJ: Tau protein dysfunction after brain ischemia. J
Alzheimers Dis. 66:429–437. 2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Gutiérrez-Vargas JA, Múnera A and
Cardona-Gómez GP: CDK5 knockdown prevents hippocampal degeneration
and cognitive dysfunction produced by cerebral ischemia. J Cereb
Blood Flow Metab. 35:1937–1949. 2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Pluta R, Bogucka-Kocka A, Ułamek-Kozioł M,
Bogucki J, Januszewski S, Kocki J and Czuczwar SJ: Ischemic tau
protein gene induction as an additional key factor driving
development of Alzheimer's phenotype changes in CA1 area of
hippocampus in an ischemic model of Alzheimer's disease. Pharmacol
Rep. 70:881–884. 2018.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Mailliot C, Podevin-Dimster V, Rosenthal
RE, Sergeant N, Delacourte A, Fiskum G and Buée L: Rapid tau
protein dephosphorylation and differential rephosphorylation during
cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb
Blood Flow Metab. 20:543–549. 2000.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Wen Y, Yang S, Liu R and Simpkins JW:
Transient cerebral ischemia induces site-specific
hyperphosphorylation of tau protein. Brain Res. 1022:30–38.
2004.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Uchihara T, Nakamura A, Arai T, Ikeda K
and Tsuchiya K: Microglial tau undergoes
phosphorylation-independent modification after ischemia. Glia.
45:180–187. 2004.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Fujii H, Takahashi T, Mukai T, Tanaka S,
Hosomi N, Maruyama H, Sakai N and Matsumoto M: Modifications of tau
protein after cerebral ischemia and reperfusion in rats are similar
to those occurring in Alzheimer's disease-Hyperphosphorylation and
cleavage of 4- and 3-repeat tau. J Cereb Blood Flow Metab.
37:2441–2457. 2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Shiiya N, Kunihara T, Miyatake T,
Matsuzaki K and Yasuda K: Tau protein in the cerebrospinal fluid is
a marker of brain injury after aortic surgery. Ann Thorac Surg.
77:2034–2038. 2004.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Hesse C, Rosengren L, Andreasen N,
Davidsson P, Vanderstichele H, Vanmechelen E and Blennow K:
Transient increase in total tau but not phospho-tau in human
cerebrospinal fluid after acute stroke. Neurosci Lett. 297:187–190.
2001.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Onatsu J, Vanninen R, JÄkÄlÄ P, Mustonen
P, Pulkki K, Korhonen M, Hedman M, HÖglund K, Blennow K, Zetterberg
H, et al: Tau, S100B and NSE as blood biomarkers in acute
cerebrovascular events. In Vivo. 34:2577–2586. 2020.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Bitsch A, Horn C, Kemmling Y, Seipelt M,
Hellenbrand U, Stiefel M, Ciesielczyk B, Cepek L, Bahn E, Ratzka P,
et al: Serum tau protein level as a marker of axonal damage in
acute ischemic stroke. Eur Neurol. 47:45–51. 2002.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Kurzepa J, Bielewicz J, Grabarska A,
Stelmasiak Z, Stryjecka-Zimmer M and Bartosik-Psujek H: Matrix
metalloproteinase-9 contributes to the increase of tau protein in
serum during acute ischemic stroke. J Clin Neurosci. 17:997–999.
2010.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Lasek-Bal A, Jedrzejowska-Szypulka H,
Rozycka J, Bal W, Kowalczyk A, Holecki M, Dulawa J and
Lewin-Kowalik J: The presence of Tau protein in blood as a
potential prognostic factor in stroke patients. J Physiol
Pharmacol. 67:691–696. 2016.PubMed/NCBI
|
|
81
|
Bielewicz J, Kurzepa J, Czekajska-Chehab
E, Stelmasiak Z and Bartosik-Psujek H: Does serum Tau protein
predict the outcome of patients with ischemic stroke? J Mol
Neurosci. 43:241–245. 2011.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Wunderlich MT, Lins H, Skalej M, Wallesch
CW and Goertler M: Neuron-specific enolase and tau protein as
neurobiochemical markers of neuronal damage are related to early
clinical course and long-term outcome in acute ischemic stroke.
Clin Neurol Neurosurg. 108:558–563. 2006.PubMed/NCBI View Article : Google Scholar
|
|
83
|
De Vos A, Bjerke M, Brouns R, De Roeck N,
Jacobs D, Van den Abbeele L, Guldolf K, Zetterberg H, Blennow K,
Engelborghs S and Vanmechelen E: Neurogranin and tau in
cerebrospinal fluid and plasma of patients with acute ischemic
stroke. BMC Neurol. 17(170)2017.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Irving EA, Nicoll J, Graham DI and Dewar
D: Increased tau immunoreactivity in oligodendrocytes following
human stroke and head injury. Neurosci Lett. 213:189–192.
1996.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Uphaus T, Bittner S, Gröschel S, Steffen
F, Muthuraman M, Wasser K, Weber-Krüger M, Zipp F, Wachter R and
Gröschel K: NfL (Neurofilament Light Chain) levels as a predictive
marker for long-term outcome after ischemic stroke. Stroke.
50:3077–3084. 2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Khalil M, Teunissen CE, Otto M, Piehl F,
Sormani MP, Gattringer T, Barro C, Kappos L, Comabella M, Fazekas
F, et al: Neurofilaments as biomarkers in neurological disorders.
Nat Rev Neurol. 14:577–589. 2018.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Peters N, van Leijsen E, Tuladhar AM,
Barro C, Konieczny MJ, Ewers M, Lyrer P, Engelter ST, Kuhle J,
Duering M and de Leeuw FE: Serum Neurofilament light Chain is
associated with incident Lacunes in progressive cerebral small
vessel disease. J Stroke. 22:369–376. 2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Duering M, Konieczny MJ, Tiedt S, Baykara
E, Tuladhar AM, Leijsen EV, Lyrer P, Engelter ST, Gesierich B,
Achmüller M, et al: Serum Neurofilament Light Chain levels are
related to small vessel disease burden. J Stroke. 20:228–238.
2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Paolini Paoletti F, Simoni S, Parnetti L
and Gaetani L: The contribution of small vessel disease to
neurodegeneration: Focus on Alzheimer's disease, Parkinson's
disease and multiple sclerosis. Int J Mol Sci.
22(4958)2021.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Knopman DS: Cerebrovascular disease and
dementia. Br J Radiol. 80 (Suppl 2):S121–S127. 2007.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Dendrou CA, Fugger L and Friese MA:
Immunopathology of multiple sclerosis. Nat Rev Immunol. 15:545–558.
2015.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Virgilio E, Vecchio D, Crespi I, Serino R,
Cantello R, Dianzani U and Comi C: Cerebrospinal Tau levels as a
predictor of early disability in multiple sclerosis. Mult Scler
Relat Disord. 56(103231)2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Mirzaii-Dizgah MH, Mirzaii-Dizgah MR and
Mirzaii-Dizgah I: Serum and saliva total tau protein as a marker
for relapsing-remitting multiple sclerosis. Med Hypotheses.
135(109476)2020.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Shafit-Zagardo B, Kress Y, Zhao ML and Lee
SC: A novel microtubule-associated protein-2 expressed in
oligodendrocytes in multiple sclerosis lesions. J Neurochem.
73:2531–2537. 1999.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Wang P, Jiang LL, Wang C, Zhu Z and Lai C:
Neurofilament protein as a potential biomarker of axonal
degeneration in experimental autoimmune encephalomyelitis. Neurol
India. 68:364–367. 2020.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Rosengren LE, Karlsson JE, Karlsson JO,
Persson LI and Wikkelsø C: Patients with amyotrophic lateral
sclerosis and other neurodegenerative diseases have increased
levels of neurofilament protein in CSF. J Neurochem. 67:2013–2018.
1996.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Lycke JN, Karlsson JE, Andersen O and
Rosengren LE: Neurofilament protein in cerebrospinal fluid: A
potential marker of activity in multiple sclerosis. J Neurol
Neurosurg Psychiatry. 64:402–404. 1998.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Kuhle J, Barro C, Andreasson U, Derfuss T,
Lindberg R, Sandelius Å, Liman V, Norgren N, Blennow K and
Zetterberg H: Comparison of three analytical platforms for
quantification of the neurofilament light chain in blood samples:
ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem
Lab Med. 54:1655–1661. 2016.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Varhaug KN, Torkildsen Ø, Myhr KM and
Vedeler CA: Neurofilament light Chain as a biomarker in multiple
sclerosis. Front Neurol. 10(338)2019.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Norgren N, Rosengren L and Stigbrand T:
Elevated neurofilament levels in neurological diseases. Brain Res.
987:25–31. 2003.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Cai L and Huang J: Neurofilament light
chain as a biological marker for multiple sclerosis: A
meta-analysis study. Neuropsychiatr Dis Treat. 14:2241–2254.
2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Disanto G, Barro C, Benkert P, Naegelin Y,
Schädelin S, Giardiello A, Zecca C, Blennow K, Zetterberg H,
Leppert D, et al: Serum Neurofilament light: A biomarker of
neuronal damage in multiple sclerosis. Ann Neurol. 81:857–870.
2017.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Barro C, Benkert P, Disanto G, Tsagkas C,
Amann M, Naegelin Y, Leppert D, Gobbi C, Granziera C, Yaldizli Ö,
et al: Serum neurofilament as a predictor of disease worsening and
brain and spinal cord atrophy in multiple sclerosis. Brain.
141:2382–2391. 2018.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Arrambide G, Espejo C, Eixarch H, Villar
LM, Alvarez-Cermeño JC, Picón C, Kuhle J, Disanto G, Kappos L,
Sastre-Garriga J, et al: Neurofilament light chain level is a weak
risk factor for the development of MS. Neurology. 87:1076–1084.
2016.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Comabella M and Montalban X: Body fluid
biomarkers in multiple sclerosis. Lancet Neurol. 13:113–126.
2014.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Matute-Blanch C, Villar LM,
Álvarez-Cermeño JC, Rejdak K, Evdoshenko E, Makshakov G, Nazarov V,
Lapin S, Midaglia L, Vidal-Jordana A, et al: Neurofilament light
chain and oligoclonal bands are prognostic biomarkers in
radiologically isolated syndrome. Brain. 141:1085–1093.
2018.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Khalil M: Are neurofilaments valuable
biomarkers for long-term disease prognostication in MS? Mult Scler.
24:1270–1271. 2018.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Giovannoni G: Peripheral blood
neurofilament light chain levels: The neurologist's C-reactive
protein? Brain. 141:2235–2237. 2018.PubMed/NCBI View Article : Google Scholar
|














