Introduction
Retinitis pigmentosa (RP; OMIM no. 268000) is a
group of highly heterogeneous but related retinal disorders that
cause progressive vision loss (1-3).
Typically, RP manifests as night blindness in the early stages. As
this disease progresses, the extent of visual field loss becomes
gradually more apparent, with impaired color vision and fundus
degeneration during the advanced stages. The prevalence of RP is
~1/4000 in China (4,5). RP can be classified as syndromic or
non-syndromic. Usher syndrome and Bardet-Biedl syndrome, which also
affect multiple organs, are the most common forms of syndromic RP.
By contrast, non-syndromic RP is typically inherited and can
manifest in an autosomal-recessive (50-60% of cases),
autosomal-dominant (30-40% of cases), X-linked (5-15% of cases) or
mitochondrial manner (6-8).
In the pathophysiology of all types of RP, the majority of mutant
genes reported are expressed exclusively in rod cells. Although a
small number of mutants are specifically expressed in the retinal
pigment epithelium, none are cone-specific (https://sph.uth.edu/retnet/). Despite this, RP can
cause the degeneration of both rod and cone photoreceptors, which
mediate achromatic night vision and high acuity central vision,
respectively (9-14).
One of the reasons for the heterogeneity of RP is
the >80 disease-causing genes that have been identified
(https://sph.uth.edu/retnet/).
Additionally, variations in these genes can cause a wide range of
clinical symptoms that are distinct from typical RP, including
cone-rod dystrophy (CORD), Leber's congenital amaurosis (LCA) and
stationary night blindness. For example, whilst a number of
variants in the cone-rod homeobox (CRX) gene have
been reported to be associated with RP, other variants of
CRX can also trigger CORD and LCA (15-18).
In another example, although the majority of Cytochrome P450
family 4 subfamily V member 2 (CYP4V2) variants are
associated with Bietti crystalline dystrophy (BCD), other variants
in the gene can also cause RP. BCD is an autosomal recessive
chorioretinal degenerative disease that is characterized by
numerous glistening yellow-white crystalline retinal
micro-deposits, progressive atrophy of the retinal pigment
epithelium (RPE) and choroidal sclerosis (19).
In the present study, whole-exome sequencing (WES)
was applied to screen for potential disease-causing variants in a
non-consanguineous Chinese family with autosomal recessive RP. A
novel compound heterozygous variant in CYP4V2 was identified
in a patient with RP.
Materials and methods
Subjects
A Chinese family with RP, including six members with
an affected individual and five unaffected individuals, was
recruited from the Sichuan Provincial People's Hospital (Chengdu,
China). The affected individual (sex, female) was 47 years old when
diagnosed. Additionally, 400 unrelated healthy subjects, including
218 males and 182 females, were recruited from the Sichuan
Provincial People's Hospital (Chengdu, China). The median age of
the healthy controls was 42 (age range, 20-60). The study was
performed in accordance with the tenets of the Declaration of
Helsinki (20) and approved by the
Institutional Review Boards of Sichuan Academy of Medical Sciences
and Sichuan Provincial People's Hospital. Signed informed consent
was obtained from all participants before their inclusion in this
study.
Clinical diagnosis
All participants underwent ophthalmological
examinations. Fundus photography was performed for all members of
the affected individual's family. The clinical data were assessed
by ophthalmologists at Sichuan Academy of Medical Sciences and
Sichuan Provincial People's Hospital.
DNA isolation
Peripheral blood samples were collected in EDTA
tubes from all six members of the family and unrelated normal
controls. Genomic DNA was extracted using a blood DNA extraction
kit according to the manufacturer manual (Tiangen Biotech, Co.,
Ltd.) DNA samples were stored at -20˚C until required.
Whole-exome sequencing (WES)
The DNA from individuals II-1 (the proband), III-1
and III-2 were analyzed by WES with a mean read depth of 100x. The
samples were prepared following the Illumina standard procedure
(Illumina, Inc.). Briefly, ~3 µg genomic DNA was randomly sheared
into small fragments of 150-220 bp using a sonicator (Covaris). The
sheared fragments were purified with reagents supplied with the
AMPure XP system (Beckman Coulter, Inc.). Adapters (Agilent
Technologies, Inc.) were ligated with the polished ends and the
libraries were amplified by PCR. The amplified libraries were
hybridized with biotin-labeled probes in the liquid phase. The DNA
fragments bound to the probes, namely the captured library, were
purified. Then, the library was sequenced on a Illumina HiSeq4000
platform (Illumina, Inc.) and paired-end 150 bp reads were
obtained.
Mutation identification and data
analysis
The mutations in CYP4V2 were identified using
the following databases: dbSNP138 (https://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes
Project (http://grch37.ensembl.org/Homo_sapiens/Info/Index),
Exome Aggregation Consortium (https://gnomad.broadinstitute.org/), OMIM (https://www.omim.org/), and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php),
as well as an east Asian population databases (https://blog.nus.edu.sg/sshsphphg/singapore-genome-variation/,
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/GRCh38_positions/,
and the Retnet database (https://sph.uth.edu/Retnet/). Sanger sequencing was
used to verify the identified variants of CYP4V2. All exons
of CYP4V2 were amplified from the genomic DNAs by PCR under
standard conditions (94˚C for 2 min; followed by 30 cycles of 94˚C
for 20 sec, 58˚C for 30 sec, and 72˚C for 60 sec; with a final
extension step of 72˚C for 7 min) followed by sequencing on a 3730
ABI DNA sequencer (Thermo Fisher Scientific, Inc.). Finally, the
sequencing results were analyzed using A plasmid Editor (version
2.0, by M. Wayne Davis at the University of Utah, USA). Online
bioinformatics tools, including Mutation Taster (https://www.mutationtaster.org/), CADD
(https://cadd.gs.washington.edu/),
PROVEAN (http://provean.jcvi.org/index.php), PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/) (21), Panther (http://www.pantherdb.org/tools/)and Sorting Intolerant
from Tolerant (SIFT) (https://sift.bii.a-star.edu.sg/), were used to predict
the potential pathogenic effects of the amino acid substitution in
CYP4V2. The mutant CYP4V2 protein structures were generated
by using SWISS-MODEL (https://swissmodel.expasy.org/).
Results
Clinical characteristics
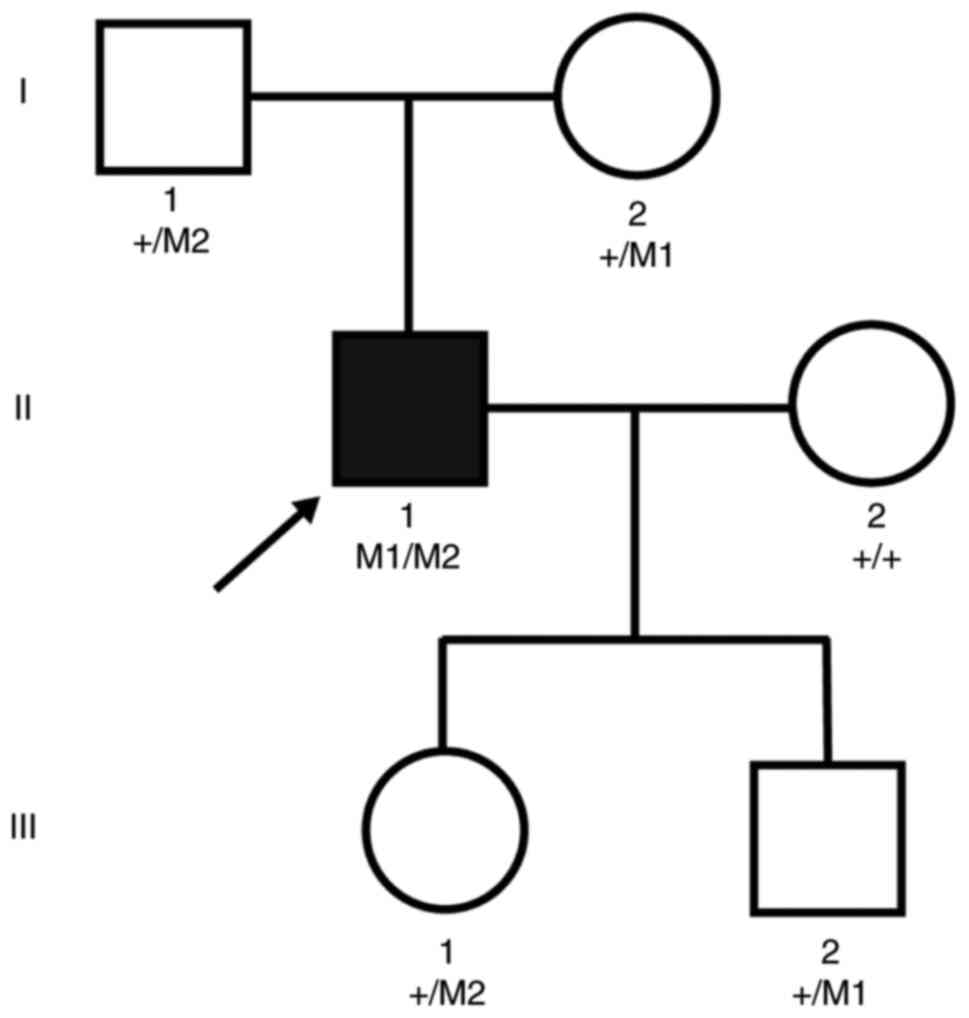
A Chinese family consisting of three generations of
individuals with RP, but no history of consanguineous marriage was
examined in the present study (Fig.
1). Pedigree analysis suggested a pattern of autosomal
recessive inheritance in this family, which consisted of a member
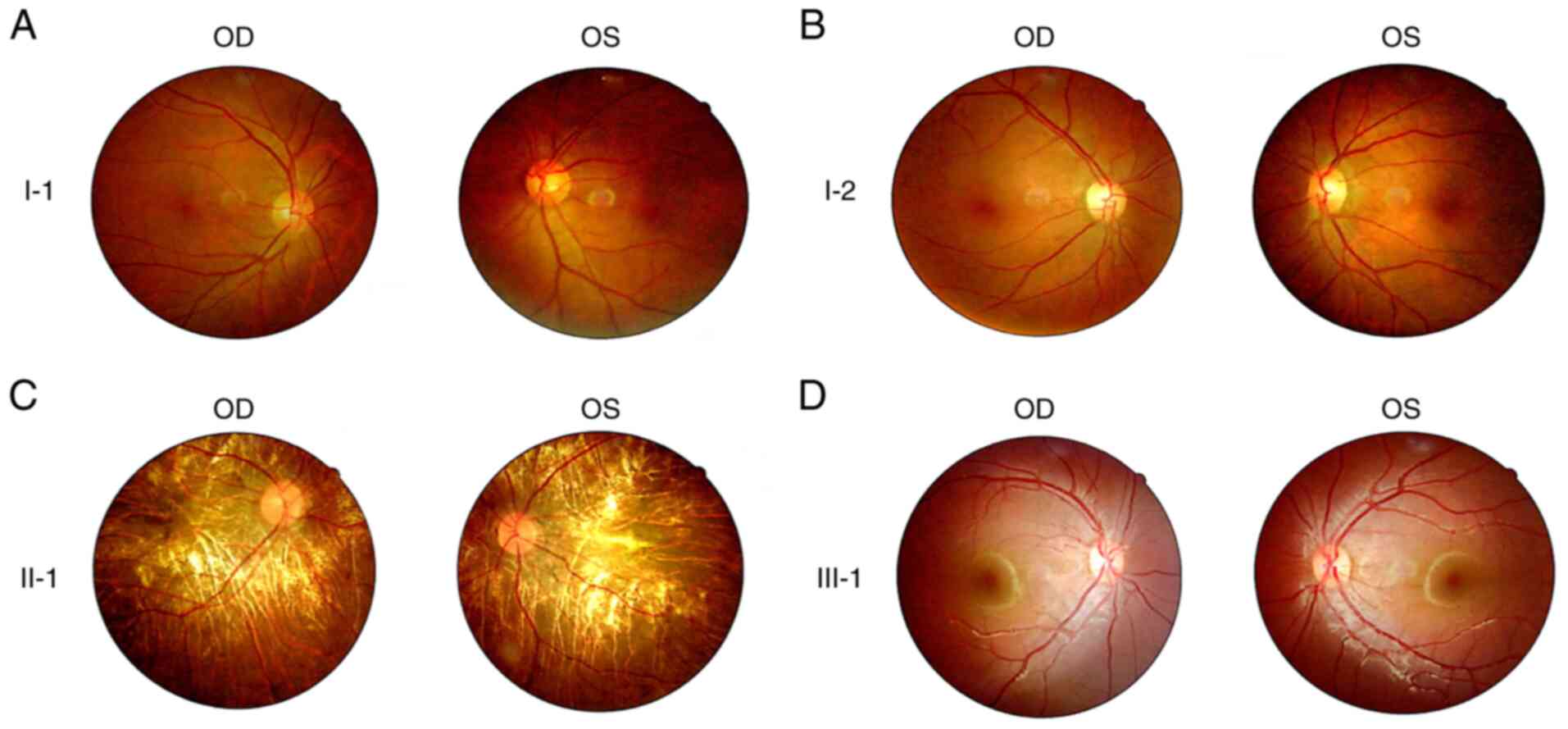
with this disease and five who were unaffected. Fundus examination
revealed that the proband exhibited the characteristic
pathophysiology of RP (22),
including retinal pigmented epithelium atrophy, attenuated blood
vessels, retinal vascular attenuation and a waxy pallor optic disc.
By contrast, fundus photography revealed no abnormalities in the
unaffected individuals (Fig.
2).
WES and data analysis
After WES analysis on the proband (II-1), and
individuals III-1 and III-2, 29,734 variants in the coding regions
and splice junctions were obtained, including 14,298 nonsynonymous
single-nucleotide polymorphisms (SNPs), 14,927 synonymous SNPs, 509
SNPs of miscellaneous types and 744 indels. To screen for the
disease-causative variant in the family with RP, common variants
present in high frequencies in dbSNP138, 1000 Genomes Project,
Exome Aggregation Consortium, OMIM, HGMD and other east Asian
population databases were filtered out. Subsequently, variants
located in introns, 5'untranslated regions (UTRs) and 3'UTRs, in
addition to synonymous SNPs and non-frameshift indels were also
filtered out since they typically do not influence gene function. A
particular focus was placed on possible functional SNPs/indels in
the homozygous or compound heterozygous variants, including
frameshift indels, non-synonymous variants and splicing junction
variants, which are more likely to be pathogenic. These SNP/indels
were filtered further using the criterion that the candidate
variants must be inherited in an autosomal recessive inheritance
manner. Genes affected by these filtered variants were then
compared with the reported genes that have been previously
associated with retinal diseases using the Retnet database
(https://sph.uth.edu/Retnet/; Fig. 3A). As a result, a novel compound
heterozygous variant, c.C958T (p.R320X) and c.G1355A (p.R452H), was
identified in the CYP4V2 gene of the proband, but not in the
other two unaffected family members. The segregation pattern of
this compound variant was consistent with the clinical phenotypical
and genetic profile of the family, suggesting that this is a
candidate disease-causing variant.
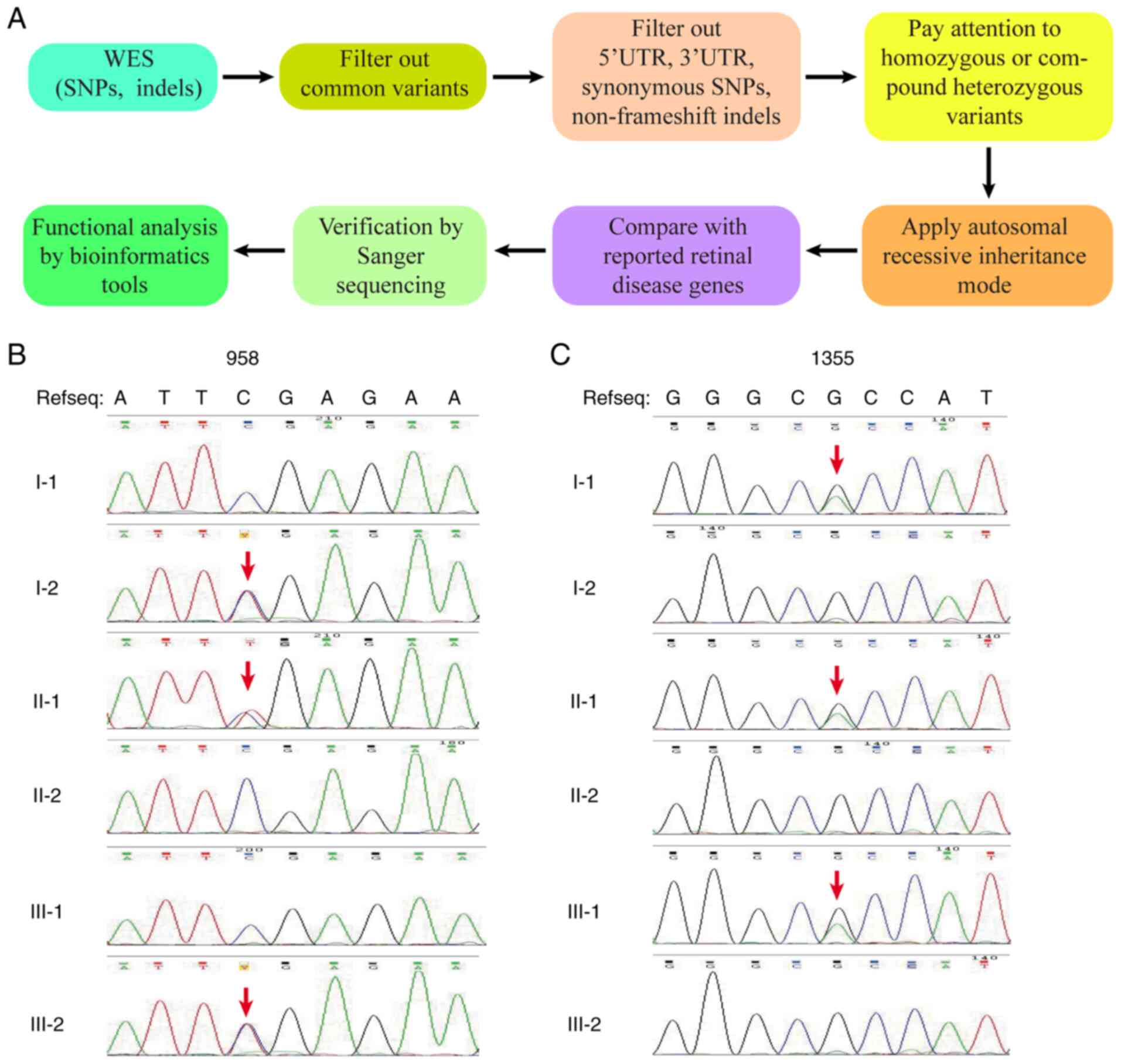
Verification of variants in the CYP4V2
gene
Direct Sanger sequencing confirmed the compound
heterozygous variant in the proband. The proband's father (I-1) and
daughter (III-1) possessed a heterozygous variant of c.G1355A
(p.R452H), his mother (I-2) and son (III-2) were heterozygous
carriers of c.C958T (p.R320X) (Fig.
3B and C). The proband's wife
(II-2) had no variant at either of these two positions. In
addition, neither of the heterozygous compound mutations,
homozygous c.G1355A (p.R452H) nor homozygous c.C958T (p.R320X)
mutations could be detected in 400 ethnically-matched control
samples. However, a heterozygous c.C958T variant was found in 2 of
the 400 controls, whereas a heterozygous c.G1355A variant was found
in 3 of the 400 controls. This compound heterozygous variant
matched the genotype and phenotype of this family. These findings
suggest that the RP symptoms present in the proband can be
attributed to this compound variant in the CYP4V2 gene.
In silico analysis of the variants
identified in CYP4V2
The c.C958T replacement caused the substitution of
arginine with a stop codon (p.R320X), whilst the c.G1355A
replacement caused the substitution of histidine with arginine
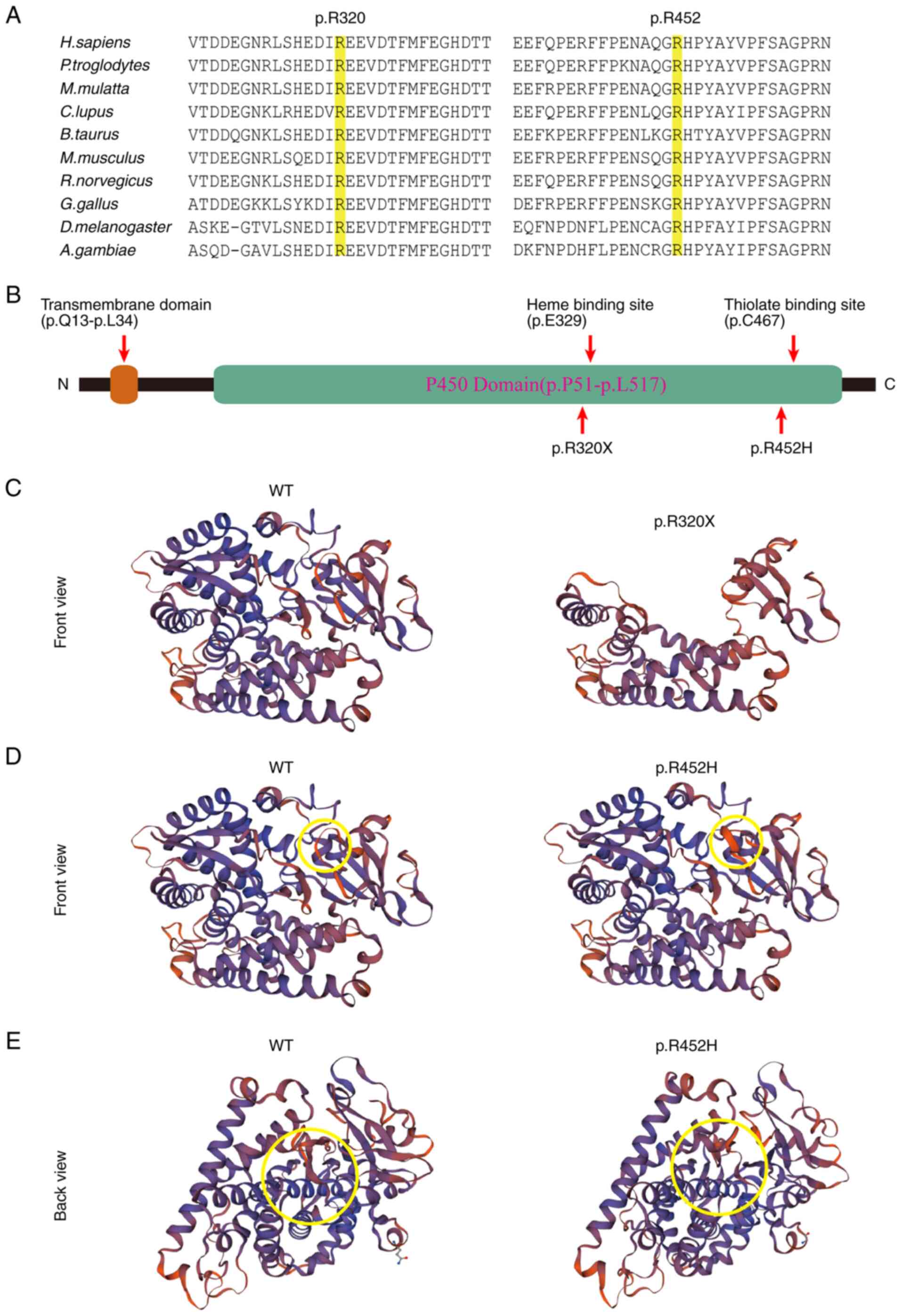
(p.R452H). Amino acid sequence alignment of the CYP4V2 protein
among different species revealed that these two variants are
located in two highly conserved regions (Fig. 4A). Bioinformatics tools Mutation
Taster, CADD, PROVEAN, PolyPhen-2 and SIFT, were used to analyze
the potential impact of p.R452H on the function of CYP4V2. The
results showed that these two variants are potentially pathogenic
(Table I). The p.R320X mutation is
located near the center of the P450 domain. The premature stop
codon at this position results in the loss of the C-terminal
portion of the protein, which accounts for ~33% of the protein. A
model of the CYP4V2 protein structure generated using SWISS-MODEL
showed that both mutants could greatly alter the structure of
CYP4V2, which may affect its function (Fig. 4B-E).
 | Table IBioinformatics analysis of the effect
of p.R320X and p.R452H in CYP4V2. |
Table I
Bioinformatics analysis of the effect
of p.R320X and p.R452H in CYP4V2.
| CYP4V2
Mutation | Mutation
Taster | CADD, RawScore
/Phred | PROVEAN | PolyPhen-2 | SIFT |
|---|
| p.R320X | Disease
causing | 8.551470/43,
Deleterious | -13.84,
Deleterious | N/A | N/A |
| p.R452H | Disease
causing | 4.218231/29.6,
Deleterious | -4.79,
Deleterious | 0.999, Probably
damaging | Damaging |
Discussion
In the present study, a novel compound heterozygous
CYP4V2 variant was discovered in a Chinese family with RP.
To date, 129 variants of CYP4V2 have been reported to be
associated with either BCD, corneal dystrophy, fundus dystrophy or
RP (23-25).
These variants include 82 missense mutations, 20 splicing
substitutions, 17 small deletions, 4 small insertions, 3 small
indels and 3 cross deletions (20-22).
Mutations in the CYP4V2 gene are documented
to primarily result in BCD (26).
BCD was first was described in 1937 by Gian Battista Bietti. The
typical clinical features of BCD include numerous yellow-white
glistening crystalline deposits, progressive night blindness and
constriction of the visual field (27). In the present study, a novel
compound heterozygous CYP4V2 variant that could cause RP and
not BCD was discovered in a Chinese family. The association of
CYP4V2 with RP is not new, as this has been reported
previously (23). However, RP is a
highly heterogenous retinal disease and it is not uncommon that
mutations in the same gene can result in a variety of clinical
manifestations. For example, mutations in RP GTPase regulator have
been reported to cause either RP or cone-dystrophies in different
individuals (28). In addition,
RPE65 mutations can result in LCA and early-onset severe retinal
dystrophy (29,30) or relatively mild phenotypes with
slower rates of progression (29,31).
One reason for this is that different mutations can mediate
differential impacts on subsequent gene functions. In addition,
another reason may be related to the different genetic backgrounds
in different individuals, who can harbor different genetic
modifiers. For example, a genetic modifier has been previously
identified in patients with Bardet-Biedl syndrome (BBS) (32). A genetic variation in the
coiled-coil domain-containing 28B gene, which encodes a
pericentriolar protein, was found to greatly influence the
phenotype of patients with BBS. Therefore, for any given disease,
the symptoms exhibited are most likely to be the outcomes of
interactions amongst multiple genes. As such, the precise phenotype
in each individual may depend on the nature of mutations and the
genetic modifier profile.
In the present pedigree, the proband (II-1) was
found to be carrying the compound heterozygous variant of c.C958T
and c.G1355A, whereas other family members were found to either
carry none of the variants of interest or one heterozygous variant
of c.C958T and c.G1355A. Furthermore, this compound heterozygous
variant could not be found in 400 unrelated healthy Chinese control
individuals or in any of the public databases probed, including
HGMD (http://www.hgmd.org/), 1000 Genome
project (http://www.internationalgenome.org/) and the NHLBI
Exome Sequencing Project (ESP) Exome Variant Server (http://evs.gs.washington.edu/EVS/). Both c.C958T
and c.G1355A in the CYP4V2 gene were predicted to damage the
function of the CYP4V2 protein according to PolyPhen-2. Therefore,
the c.C958T and c.G1355A variants are likely to be putative
pathogenic mutations. The homozygous c.C958T (pR320X) or c.G1355A
(p.R452H) mutation in the CYP4V2 gene has been previously
associated with BCD, supporting the notion that this compound
heterozygous c.C958T (pR320X) and c.G1355A (p.R452H) variant can
cause retinal diseases (24,33,34).
CYP4V2 is also known as BCD or CYP4AH1, and belongs
to a subfamily within the cytochrome P450 superfamily.
CYP4V2 is located on chromosome 4q35 and contains 11 exons,
such that the CYP4V2 protein is ubiquitously expressed. In the eye,
it is predominantly expressed in RPE cells with lower expression
levels in the cornea. CYP4V2 is one of 57 functional human enzymes
in the cytochrome P450 superfamily of heme-containing monooxygenase
enzymes (35). Specifically, the
CYP4V2 protein catalyzes the omega-3 hydroxylation of
poly-unsaturated fatty acids (PUFAs), such as eicosapentaenoic acid
and docosahexaenoic acid (36).
PUFAs are widely distributed throughout the retina and are
essential components of retinal rod outer segment membranes
(37).
Structurally, CYP4V2 encodes a protein
containing 525 amino acid residues. As a member of the cytochrome
P450 family, the CYP4V2 protein requires a cysteine
thiolate-coordinated Fe(II) heme complex to activate the bound
molecular oxygen in proximity to the cysteine thiolate (38). Therefore, the heme-binding and
thiolate ligand-binding sites are indispensable for CYP4V2
function. The pR320X mutant causes the premature termination of the
polypeptide and truncation of ~33% of the protein at the
C-terminus, which includes the heme-binding site (at E329) and
thiolate ligand-binding site (at C467). This may severely impair
enzymatic activity. Therefore, the p.R320X mutation was considered
to be in the loss-of-function category. The p.R452H mutant is a
missense mutation occurring at a position close to the thiolate
ligand-binding site within the conserved P450 domain. The p.R452H
mutant was predicted to alter protein conformation drastically,
which may disrupt the formation of the cysteine thiolate-Fe(III)
heme complex, thereby compromising the enzymatic function of
CYP4V2. As a result, p.R452H was considered to be either a
loss-of-function mutation or a hypomorphic mutation. To conclude,
this compound variant may severely impair the activity of CYP4V2,
which is required for normal retinal function.
This compound variant of CYP2V4 in this
family described in the present study is different from one that
was previously reported (c.802-8_810del17insGC) (23). The variants of c.802-8_810del17insGC
and c.1091-2A>G were found to disrupt the splicing acceptors of
exon 7 and 9, respectively. In turn, they were predicted to cause
the in-frame deletion of exon 7 (encoding 62 amino acids) and exon
9 (encoding 45 amino acids) (26,27,39,40).
Both variants were predicted to cause the deletion of a significant
portion of the peptide sequence in the key P450 domain. This is
particularly the case in the c.802-8_810del17insGC variant, which
spans the heme binding site and is critical for the protein
activity.
In summary, a novel compound heterozygous mutation
of c.C958T and c.G1355A in the CYP4V2 gene was identified in
a Chinese family with RP using WES. The present study not only
confirmed WES to be a powerful method for screening for causative
mutations for RP, but also expanded the spectrum of disease-causing
variants in the CYP4V2 gene, which will facilitate the
further molecular screening of genetic variants that can cause
RP.
Acknowledgements
Not applicable.
Funding
Funding: This study was funded by the National Precision
Medicine Project (grant no. 2016YFC0905200), the National Natural
Science Foundation of China (grant nos. 81570882, 81770935 and
81800830), a grant from the Department of Science and Technology of
Sichuan Province, China (grant nos. 2020YJ0445 and 2020ZYD035) and
the Key Research and Development and Promotion Project (Science and
Technology) program of Henan Province (grant no. 192102310077).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TZ, TW, FZ and SD performed the experiments and
analyzed the data. BG and HZ analyzed the data and supervised the
project. TZ and HZ wrote the manuscript. All authors read and
approved the final manuscript. TZ, BG and HZ confirmed the
authenticity of all the raw data.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Boards of Sichuan Provincial People's Hospital (Chengdu, China).
Written informed consent was obtained from all participants or
parents of children prior to their inclusion in the present
study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Ali MU, Rahman MSU, Cao J and Yuan PX:
Genetic characterization and disease mechanism of retinitis
pigmentosa; current scenario. 3 Biotech. 7(251)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pearlman JT: Mathematical models of
retinitis pigmentosa: A study of the rate of progress in the
different genetic forms. Trans Am Ophthalmol Soc. 77:643–656.
1979.PubMed/NCBI
|
|
3
|
Ferrari S, Di Iorio E, Barbaro V, Ponzin
D, Sorrentino FS and Parmeggiani F: Retinitis pigmentosa: Genes and
disease mechanisms. Curr Genomics. 12:238–249. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chen X, Liu X, Sheng X, Gao X, Zhang X, Li
Z, Li H, Liu Y, Rong W, Zhao K and Zhao C: Targeted next-generation
sequencing reveals novel EYS mutations in Chinese families with
autosomal recessive retinitis pigmentosa. Sci Rep.
5(8927)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Xiao X, Cao Y, Zhang Z, Xu Y, Zheng Y,
Chen LJ, Pang CP and Chen H: Novel mutations in PRPF31 causing
retinitis pigmentosa identified using whole-exome sequencing.
Invest Ophthalmol Vis Sci. 58:6342–6350. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Grondahl J: Estimation of prognosis and
prevalence of retinitis pigmentosa and Usher syndrome in Norway.
Clin Genet. 31:255–264. 1987.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Paloma E, Martínez-Mir A, García-Sandoval
B, Ayuso C, Vilageliu L, Gonzàlez-Duarte R and Balcells S: Novel
homozygous mutation in the alpha subunit of the rod cGMP gated
channel (CNGA1) in two Spanish sibs affected with autosomal
recessive retinitis pigmentosa. J Med Genet. 39(E66)2002.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bunker CH, Berson EL, Bromley WC, Hayes RP
and Roderick TH: Prevalence of retinitis pigmentosa in maine. Am J
Ophthalmol. 97:357–365. 1984.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xu J, Morris LM, Michalakis S, Biel M,
Fliesler SJ, Sherry DM and Ding XQ: CNGA3 deficiency affects cone
synaptic terminal structure and function and leads to secondary rod
dysfunction and degeneration. Invest Ophthalmol Vis Sci.
53:1117–1129. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yang RB, Robinson SW, Xiong WH, Yau KW,
Birch DG and Garbers DL: Disruption of a retinal guanylyl cyclase
gene leads to cone-specific dystrophy and paradoxical rod behavior.
J Neurosci. 19:5889–5897. 1999.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yang Z, Peachey NS, Moshfeghi DM,
Thirumalaichary S, Chorich L, Shugart YY, Fan K and Zhang K:
Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol
Genet. 11:605–611. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yu DY, Cringle S, Valter K, Walsh N, Lee D
and Stone J: Photoreceptor death, trophic factor expression,
retinal oxygen status, and photoreceptor function in the P23H rat.
Invest Ophthalmol Vis Sci. 45:2013–2019. 2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Michaelides M, Hardcastle AJ, Hunt DM and
Moore AT: Progressive cone and cone-rod dystrophies: Phenotypes and
underlying molecular genetic basis. Surv Ophthalmol. 51:232–258.
2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Narayan DS, Wood JP, Chidlow G and Casson
RJ: A review of the mechanisms of cone degeneration in retinitis
pigmentosa. Acta Ophthalmol. 94:748–754. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
den Hollander AI, Roepman R, Koenekoop RK
and Cremers FP: Leber congenital amaurosis: genes, proteins and
disease mechanisms. Prog Retin Eye Res. 27:391–419. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Huang L, Xiao X, Li S, Jia X, Wang P, Guo
X and Zhang Q: CRX variants in cone-rod dystrophy and mutation
overview. Biochem Biophys Res Commun. 426:498–503. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Nichols LL II, Alur RP, Boobalan E,
Sergeev YV, Caruso RC, Stone EM, Swaroop A, Johnson MA and Brooks
BP: Two novel CRX mutant proteins causing autosomal dominant Leber
congenital amaurosis interact differently with NRL. Hum Mutat.
31:E1472–E1483. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Walia S, Fishman GA, Jacobson SG, Aleman
TS, Koenekoop RK, Traboulsi EI, Weleber RG, Pennesi ME, Heon E,
Drack A, et al: Visual acuity in patients with Leber's congenital
amaurosis and early childhood-onset retinitis pigmentosa.
Ophthalmology. 117:1190–1198. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chung JK, Shin JH, Jeon BR, Ki CS and Park
TK: Optical coherence tomographic findings of crystal deposits in
the lens and cornea in Bietti crystalline corneoretinopathy
associated with mutation in the CYP4V2 gene. Jpn J Ophthalmol.
57:447–450. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Association WM: World Medical Association
Declaration of Helsinki: Ethical principles for medical research
involving human subjects. JAMA. 310:2191–2194. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sorrentino FS, Gallenga CE, Bonifazzi C
and Perri P: A challenge to the striking genotypic heterogeneity of
retinitis pigmentosa: A better understanding of the pathophysiology
using the newest genetic strategies. Eye (Lond). 30:1542–1548.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang Y, Guo L, Cai SP, Dai M, Yang Q, Yu
W, Yan N, Zhou X, Fu J, Guo X, et al: Exome sequencing identifies
compound heterozygous mutations in CYP4V2 in a pedigree with
retinitis pigmentosa. PLoS One. 7(e33673)2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xiao X, Mai G, Li S, Guo X and Zhang Q:
Identification of CYP4V2 mutation in 21 families and overview of
mutation spectrum in Bietti crystalline corneoretinal dystrophy.
Biochem Biophys Res Commun. 409:181–186. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yin H, Jin C, Fang X, Miao Q, Zhao Y, Chen
Z, Su Z, Ye P, Wang Y and Yin J: Molecular analysis and phenotypic
study in 14 Chinese families with Bietti crystalline dystrophy.
PLoS One. 9(e94960)2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lin J, Nishiguchi KM, Nakamura M, Dryja
TP, Berson EL and Miyake Y: Recessive mutations in the CYP4V2 gene
in East Asian and Middle Eastern patients with Bietti crystalline
corneoretinal dystrophy. J Med Genet. 42(e38)2005.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li A, Jiao X, Munier FL, Schorderet DF,
Yao W, Iwata F, Hayakawa M, Kanai A, Shy Chen M, Alan Lewis R, et
al: Bietti crystalline corneoretinal dystrophy is caused by
mutations in the novel gene CYP4V2. Am J Hum Genet. 74:817–826.
2004.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Branham K, Othman M, Brumm M, Karoukis AJ,
Atmaca-Sonmez P, Yashar BM, Schwartz SB, Stover NB, Trzupek K,
Wheaton D, et al: Mutations in RPGR and RP2 account for 15% of
males with simplex retinal degenerative disease. Invest Ophthalmol
Vis Sci. 53:8232–8237. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kumaran N, Moore AT, Weleber RG and
Michaelides M: Leber congenital amaurosis/early-onset severe
retinal dystrophy: Clinical features, molecular genetics and
therapeutic interventions. Br J Ophthalmol. 101:1147–1154.
2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Thompson DA, Gyurus P, Fleischer LL,
Bingham EL, McHenry CL, Apfelstedt-Sylla E, Zrenner E, Lorenz B,
Richards JE, Jacobson SG, et al: Genetics and phenotypes of RPE65
mutations in inherited retinal degeneration. Invest Ophthalmol Vis
Sci. 41:4293–4299. 2000.PubMed/NCBI
|
|
31
|
Hull S, Holder GE, Robson AG, Mukherjee R,
Michaelides M, Webster AR and Moore AT: Preserved visual function
in retinal dystrophy due to hypomorphic RPE65 mutations. Br J
Ophthalmol. 100:1499–1505. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Badano JL, Leitch CC, Ansley SJ,
May-Simera H, Lawson S, Lewis RA, Beales PL, Dietz HC, Fisher S and
Katsanis N: Dissection of epistasis in oligogenic Bardet-Biedl
syndrome. Nature. 439:326–330. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jiao X, Li A, Jin ZB, Wang X, Iannaccone
A, Traboulsi EI, Gorin MB, Simonelli F and Hejtmancik JF:
Identification and population history of CYP4V2 mutations in
patients with Bietti crystalline corneoretinal dystrophy. Eur J Hum
Genet. 25:461–471. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Meng XH, Guo H, Xu HW, Li QY, Jin X, Bai
Y, Li SY and Yin ZQ: Identification of novel CYP4V2 gene mutations
in 92 Chinese families with Bietti's crystalline corneoretinal
dystrophy. Mol Vis. 20:1806–1814. 2014.PubMed/NCBI
|
|
35
|
Lockhart CM, Nakano M, Rettie AE and Kelly
EJ: Generation and characterization of a murine model of Bietti
crystalline dystrophy. Invest Ophthalmol Vis Sci. 55:5572–5581.
2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kelly EJ, Nakano M, Rohatgi P,
Yarov-Yarovoy V and Rettie AE: Finding homes for orphan cytochrome
P450s: CYP4V2 and CYP4F22 in disease states. Mol Interv.
11:124–132. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Giusto NM, Pasquare SJ, Salvador GA,
Castagnet PI, Roque ME and Ilincheta de Boschero MG: Lipid
metabolism in vertebrate retinal rod outer segments. Prog Lipid
Res. 39:315–391. 2000.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shimizu T: Binding of cysteine thiolate to
the Fe(III) heme complex is critical for the function of heme
sensor proteins. J Inorg Biochem. 108:171–177. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Shan M, Dong B, Zhao X, Wang J, Li G, Yang
Y and Li Y: Novel mutations in the CYP4V2 gene associated with
Bietti crystalline corneoretinal dystrophy. Mol Vis. 11:738–743.
2005.PubMed/NCBI
|
|
40
|
Jin ZB, Ito S, Saito Y, Inoue Y, Yanagi Y
and Nao IN: Clinical and molecular findings in three Japanese
patients with crystalline retinopathy. Jpn J Ophthalmol.
50:426–431. 2006.PubMed/NCBI View Article : Google Scholar
|