Introduction
Among the various genetic metabolic disorders,
Angelman syndrome (AS) has attracted considerable attention due to
the abnormal expression of the ubiquitin-protein ligase E3A (UBE3A)
gene (1). In 1965, the British
Doctor Harry Angelman first described AS and named it after his
surname. The frequency of this condition is estimated to be 1 in
15,000 individuals (2). AS is a
maternally inherited neurodevelopmental genetic disease associated
with chromosomal abnormality at the 15q11-q13 genetic region
(3). The loss of the expression of
the maternal allele of the UBE3A gene is typically associated with
the four following mechanisms: Deletion at the 15q11.2-q13 locus,
UBE3A functional loss variation, presence of paternal
parthenogenetic double chromosome or genomic imprinting defect
(4).
The various characteristics of AS are primarily
caused by maternal allele dysfunction of the UBE3A gene and
paternal imprinting (5). UBE3A is
the only gene in the 15q11-q13 region that indicates biased
expression from the maternal allele (6). The expression levels of these genes
are tissue-specific and depend on the origin of the parent. In
normal brain tissues, the maternally inherited UBE3A allele is
actively expressed (7), while the
paternally inherited UBE3A gene is not. The UBE3A gene is located
on the 15q11-q13 locus of chromosome 15. The human UBE3A gene
encodes an E3 ubiquitin ligase, which exhibits three known protein
subtypes (1,8). Little is known regarding the human
subtypes (9). The UBE3A gene plays
a regulatory role on the function of specific monoamine
transmitters, which are associated with the dynamics of synaptic
plasticity. Therefore, UBE3A is considered an important factor
involved in maintaining the normal function of the synapses
(10). Abnormal expression of UBE3A
affects the normal maintenance of the circadian rhythm. The
synaptic function of UBE3A (including neuronal excitability) may be
associated with the balance of the mTOR signals in the developing
neurons (10). Although AS presents
with typical characteristics, genetic diagnosis may be hindered by
the change in disease presentation and the presence of different
molecular mechanisms. Therefore, ~10-15% of patients, which are
suspected to have AS, are not diagnosed at an early stage of the
disease.
In the present study, two sisters of a Chinese
family with clinical features and genetic variations of AS were
tested. The electroencephalograms (EEGs) of the children in this
family were assessed in combination with the expression of the
associated genes in the sisters in order to fully understand AS and
provide additional evidence for its diagnosis.
Materials and methods
Patient consent and approval
This study was approved by the Ethics Committee of
Affiliated Hospital of Jining Medical College (Jining, China). The
parents of the patients provided written informed consent for their
participation. The parents of the children provided written
informed consent for publication of the data and images.
Genetic screening and sample
collection
A total of 5 ml peripheral blood was collected from
both the proband and her sister. A total of 2 ml peripheral blood
was collected from their parents and their grandmother. The blood
samples were sent to Beijing Kangso Medical Inspection for panel
testing (a total of 324 genes). Total exon screening was performed
for the two sisters and copy number variation (CNV) sequencing and
multiplex ligation-dependent probe amplification testing for the
proband.
Genomic DNA extraction
The Qiagen FlexiGene DNA kit (Qiagen GmbH) was used
to extract genomic DNA from blood samples according to the
procedure described by the manufacturer.
DNA library construction
To construct the DNA library, genomic DNA samples
were fragmented into 150-300 bp DNA fragments using an ultrasonic
processor. The adaptors used for both ends of these DNA fragments
were ligated and the cohesive ends of the DNA fragments were
trimmed. Subsequently, the DNA library was amplified using
PrimeSTar HS DNA Polymerase with the following thermocycling
conditions: Initial denaturation at 95˚C for 10 min, followed by 6
cycles of 30 sec at 95˚C, 30 sec at 60˚C and 45 sec at 72˚C. A
final extension was performed at 72˚C for 5 min. The primer
sequences used were as follows: Forward primer,
5'-TGTCAGCTCGCTGGACTCAG-3' and reverse primer,
5'-TTGCAGCCCAAGGAAAACTG-3'. The PCR products were purified using a
nucleic acid purification kit according to the manufacturer's
protocol.
Hybrid capture
The target DNA fragments from the amplified DNA
library were hybridized and captured by specific probes.
Subsequently, the fragments were amplified using the SureSelect
target enrichment system according to the manufacturer's protocol
(Agilent Technologies, Inc.). Finally, A customized gene panel for
inherited metabolic diseases was designed, which consisted of a
total of 324 genes. The DNA fragments were hybridized, isolated and
then amplified using the SureSelect Target Enrichment System
(Herculase ii Fusion enzyme dnTP combo; Agilent Technologies, Inc.)
with the following thermocycling conditions: Initial denaturation
at 98˚C for 2 min; followed by 15 cycles of 30 sec at 98˚C, 30 sec
at 62˚C and 1 min at 72˚C; with a final extension at 72˚C for 10
min.
Sequencing
Single-read sequencing was performed by NextSeq500
(Illumina, Inc.). Raw data were obtained in the FastQ format.
Data analysis
The raw data were transformed into identifiable base
sequences using CASAVA (version 1.8.2; Illumina, inc.). Sequences
were aligned to Grch37 (as known as hg19) using Burrow-Wheeler
aligner version 0.7.15-r1140, and single nucleotide and
deletion/insertion polymorphisms analyses were performed to obtain
mutation information within the targeted regions using GaTK version
3.6. Finally, protein damage analysis was performed to
qualitatively predict the probability of the results using
PolyPhen2 (Version 2; http://genetics.bwh.harvard.edu/pph2/). The mutation
sites, which were obtained, were further validated.
First-generation sequencing
verification
The gene sequences of the aforementioned mutation
sites were obtained from GenBank. The primers were designed by the
website Primer Z (http://genepipe.ncgm.sinica.edu.tw/primerz/primerz4.do)
and subsequently synthesized. The mutation sites were amplified by
PCR and sequenced using first-generation sequencing by Kangso
Medical Inspection (Beijing, Chain). The obtained sequences were
aligned with the previous results and the false positive sites
obtained by next generation sequencing were ruled out.
Bioinformatics analysis
To investigate the effects of the detected variants,
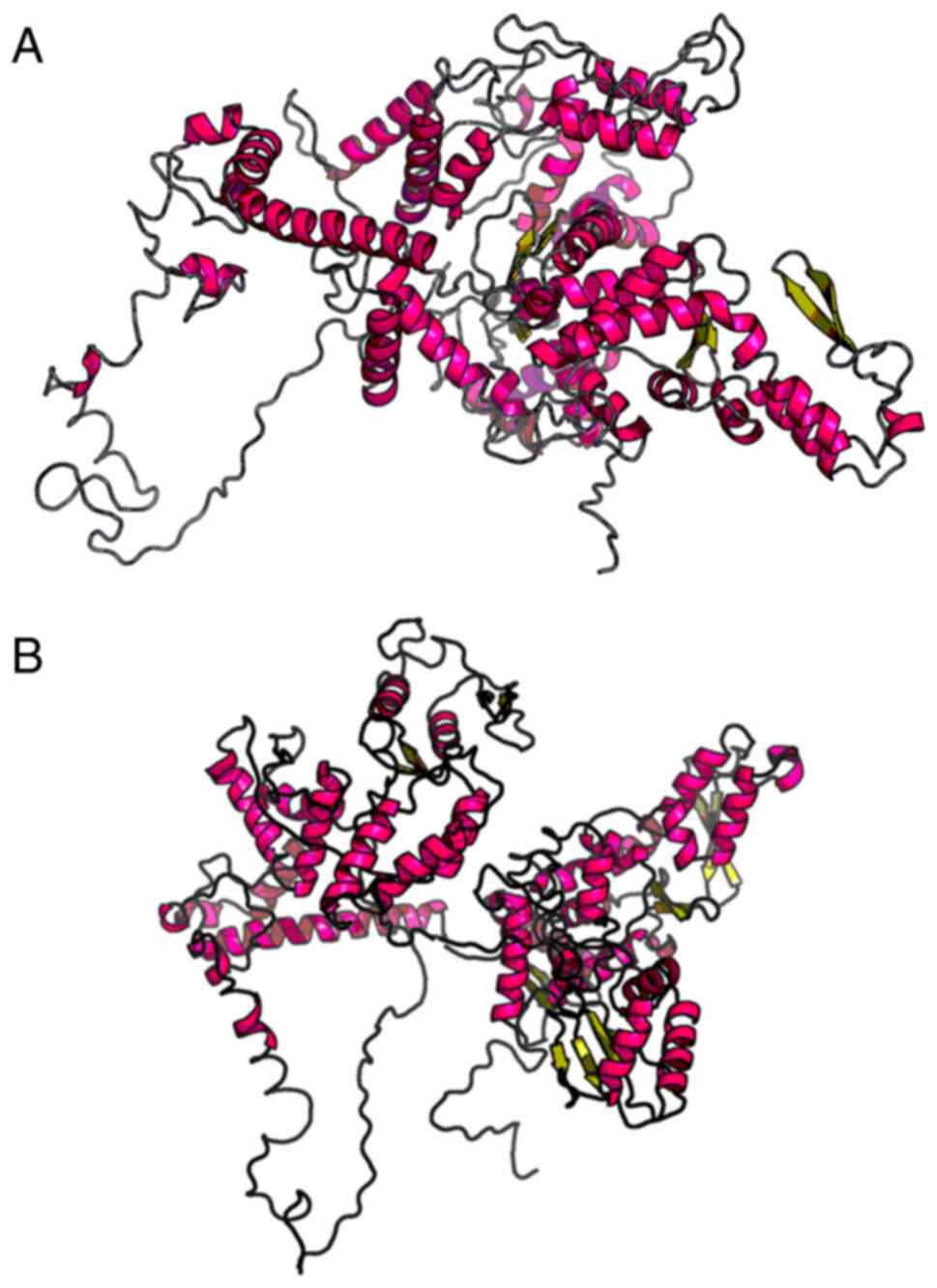
bioinformatics analyses were performed. RaptorX (http://raptorx.uchicago.edu) (11) can predict protein tertiary
structures (5). Following sequence
input, the 3D structure of the protein sequence was predicted from
the protein database (Fig. 1). The
patient's UBE3A gene did not fold completely in its spatial
structure compared with the wild-type gene, thus affecting its
protein function.
Case report
The present study investigated a Chinese family with
five members (Fig. 2). The second
child in the family was a proband, an 8-year-old female who was
delivered via cesarean section at full-term. The proband revealed
no apparent abnormalities at birth, no perinatal problems or
hypotonia, and no apparent abnormality in prenatal examination.
Initially, no apparent difference was noted in diet and sleep
compared with those of healthy infants. The abnormality was
initially discovered when the child was 7-8 months old. She could
not sit alone and could not interact with her parents. The child
was admitted to the Children's Health and Genetics Clinic of the
Affiliated Hospital of Jining Medical University and was initially
diagnosed with ‘stunting’. Due to the similar medical history of
her sister, she did not receive the corresponding examination and
treatment. The child exhibited her first epileptic seizure at the
age of 15 months in the absence of fever. The seizure condition was
unknown and the pediatric patient was treated in the Epilepsy
Clinic of Affiliated Hospital of Jining Medical University with no
apparent abnormality in routine EEG. The dynamic EEG indicated no
epileptic discharge compared with the EEG obtained when the patient
was 16 months and awake. When the patient was asleep, a medium-high
amplitude peak wave and a steeple slow coincidence wave were noted
in the EEG. The central, right parietal, frontal, left middle
temporal and posterior temporal regions were the primary regions
that were monitored. Craniocerebral MRI indicated that the anterior
longitudinal cisterna and the bilateral frontotemporal
extracephalic space were widened. At that time, the patient was
administered oral sodium valproate. The pediatric patient was on
sodium valproate treatment for >2 years and her seizures did not
show significant improvements. Therefore, the family members of the
child terminated drug administration.
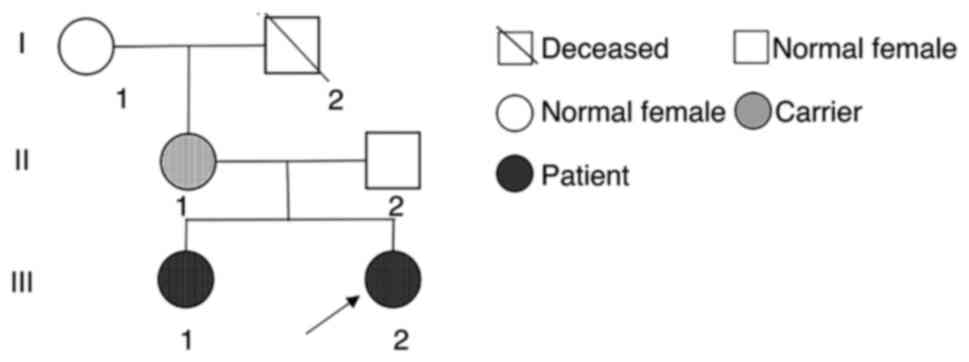 | Figure 2Pedigree chart of the examined family.
The Chinese family investigated consisted of five members: Ⅰ1, the
grandmother, healthy; Ⅰ2, the grandfather, deceased (mutation
status unknown); Ⅱ1, the mother, 41 years old, healthy; Ⅱ2, the
father, 38 years old, healthy; Ⅲ1, the first daughter, 15 years
old, with AS; Ⅲ2, the proband (arrow), 8 years old, with AS. AS,
Angelman Syndrome. |
When the child was 5 years old, she suddenly
exhibited frequent epileptic seizures, including limb paralysis,
falling to the ground and shaking of the limbs. These conditions
were relieved after a few seconds. The number of seizures increased
from 3-5 times a day to >10 times a day, and shock and
stimulation were prone to occur. When the frequency of the
epileptic attacks increased, the child was unable to walk alone
(the child could walk alone prior to the onset of the disease) and
was admitted to Peking University First Hospital. At that time, the
child suffered from poor sleep quality, could not take care of
herself during defecation or urination, and walked unsteadily.
Specialist examination showed intelligence and reaction ability
were low. The child would not take the initiative to speak and
pronounced unconsciously the words ‘mama’ and ‘baba’, could
understand simple sentences and exhibited serious salivation and no
mouth angle skew. The patient also presented with binocular
strabismus, coarse test vision and normal hearing. The pediatric
patient demonstrated the ability to chase an object and had thick
upper limbs, mobility, and a physical examination revealed a lack
of cooperation. The long-range video EEG detection report indicated
that during waking, the full conduction medium, high radiation
frequency (2.5-3.0 Hz rate) and slow composite wave burst were
observed. The normal background was restored for ~5 sec with left
and right synchronization. The high and slow complex wave
distributions were occasionally noted in the left frontal, central,
parietal regions and the left middle-posterior temporal regions. At
the onset stage, the spine rhythm was observed in the bilateral
frontal pole and anterior temporal region at the beginning of the
onset. The spine rhythm was mixed with movement and electromyogenic
pseudo difference in the middle and later stages of the onset,
which lasted for ~15 sec. During the hospitalization, the patient
was treated with nerve pulse fusion therapy and transcranial
magnetic stimulation therapy, which demonstrated aggravation rather
than apparent improvement of the condition. In view of the frequent
seizures, intramuscular injection of diazepam was provided.
However, the seizure intensity nor frequency were not reduced
compared with before diazepam administration. The family refused to
continue the treatment. Oral administration of levetiracetam and
topiramate was provided, which is not a regular regimen.
The facial features of the children exhibited
slightly wider eye spacing, lack of downward sloping eye fissure,
large mouth, thin upper lip, wide mouth, small teeth and wide
spacing, occasionally protruding tongue, serious salivation,
microcephaly and normal skin and hair color. The behaviors of the
proband included the following: Frequent laughing, cheerful mood,
ease of excitement, playing with water, looking at the phone,
tearing paper and being able to identify the WeChat application on
their father's phone. The patient was not obtaining sufficient
sleep. Moderate mental retardation was noted, which resulted in
spelling of the words ‘mom’ and ‘dad’ in an unconscious way. The
female pediatric patient could not speak >2 words, but could
understand simple commands and sentences. She also used gestures to
express her intentions. The upper limb muscle tension was normal.
whereas lower limb muscle tension was decreased. Solitary sitting
was stable and the patient could walk several steps alone, without
pointing her feet. In addition, she failed to walk actively and her
upper arm was flexed when descending, resulting in a slightly
disordered gait. She was uncooperative upon physical examination
and had epileptic seizures ≥10 times a day. Following the induction
of shock stimulation, the patient experienced seizures during which
her limbs were shaking and her eyes rolled up. This state occurred
for no apparent reason and lasted >20 sec prior to returning to
their original condition.
The 15-year-old sister of the proband exhibited
similar clinical manifestations with those of the proband. She had
a natural birth at term, with no apparent abnormalities at birth
and no associated perinatal problems. She demonstrated similar
developmental delays to those of her sister. The initial report of
her condition was at the age of 8 months, when she was found to sit
backwards. At that time, she was admitted to the pediatric health
clinic of Affiliated Hospital of Jining Medical University and was
preliminarily diagnosed as a patient experiencing ‘developmental
delays’. The seizures initially occurred at the age of 1.5 years in
the absence of fever. The specific onset was unknown. The failure
to capture an informative EEG and the insufficient diagnostic
evidence resulted in the lack of the diagnosis of epilepsy.
Therefore, the patient was not treated with the corresponding
treatment required for this condition. At the age of 2, the patient
underwent the electrical evoked potential test. The brainstem
auditory evoked potential test indicated lack of apparent
abnormality. The detection of somatosensory-evoked potential
indicated bilateral cortical potential abnormalities. A medical
practitioner examined the patient when she was 3 years old in
Affiliated Hospital of Jining Medical University due to mental and
motor retardation symptoms. At that time, she could not stand or
walk alone and referred to her mother and father unconsciously. She
was able to walk alone for >10 days when she was 30 months old.
However, she could not walk alone after falling to the ground due
to epilepsy. In addition, she could not control her urination and
her sleep was normal. The muscle strength of both lower limbs was
low, resulting in an inability to stand and walk. The results of
the intelligence test indicated that the speech intelligence
quality was 54 (based on the parent's account as there was no
report card), suggesting moderate mental retardation. At the age of
5, EEG data indicated that a large number of multi-focal low-high
amplitude spinous waves and slow spinous waves were emitted, which
were clustered or continuously distributed in the occipital and
temporal regions in each sleeping stage. The following patterns
were observed: Multi-wide, high to very high amplitude, irregular
slow-spined wave, slow wave inclusions and short-range spined
waves. The patient exhibited increased EEG discharge following
examination when she was awake, asleep and with her eyes closed
(interonset). No significant improvement was noted following oral
administration of valproate for >2 years resulting in the
termination of the drug treatment by the patient's family members.
At the age of 8, a gene deletion was noted at the chromosomal
location 15q11-13, which was methylated. No gene copy number
changes or methylation abnormalities were detected at the 15q11-13
region of the tested samples. The facial features of the patient
included slightly wider eye spacing, lack of downward sloping
palpebral fissure, large mouth, thin upper lip, wide mouth, normal
tooth spacing with protruding tongue, substantial salivation and
microcephaly. No apparent abnormalities were noted in the skin and
hair color of the patient. Her behavioral expression included happy
behavior and excitable mood, which were accompanied by watching TV
and tearing papers. Her sleep time was reduced. Moderate mental
retardation was also noted and the patient unconsciously pronounced
the words ‘dad’, ‘mom’, ‘grandpa’ and ‘grandma’. Furthermore, she
was only able to understand simple commands and sentences, and used
gestures to express her wishes. The upper limb muscle strength was
normal and the lower limb muscle tone was decreased. The patient
could sit alone but could not walk actively, which resulted in a
disordered gait. She could stand and walk with support and flex the
upper arm upon descending. She was uncooperative upon physical
examination. The epileptic seizures occurred at a frequency of
>10 times a day and following their stimulation loss of
consciousness was observed, which was confirmed by visual
observation of the eyes, mouth salivation, extended tongue and limb
shaking. The attack was relieved within 5-10 sec.
The epileptic seizures of the elder sister were
slightly less severe compared with that of the proband. The latter
was an introvert, and the stress associated with contact with a
stranger could trigger an epileptic attack. By contrast, the
patient's sister had an extrovert personality. The two sisters
exhibited unexplained seizures and stimulation of coexisting
seizures in combination with pathological EEG manifestations.
However, the EEG of the sister, which provided the diagnostic data,
appeared subsequently. Since the two sisters did not receive
medical treatment, they did not have regular follow-ups, which
resulted in poor control of the epileptic onset, and this seriously
affected their quality of life. The proband could walk several
steps alone and exhibited mild ataxia. Her sister could not walk
alone and exhibited an abnormal gait when her walk was
assisted.
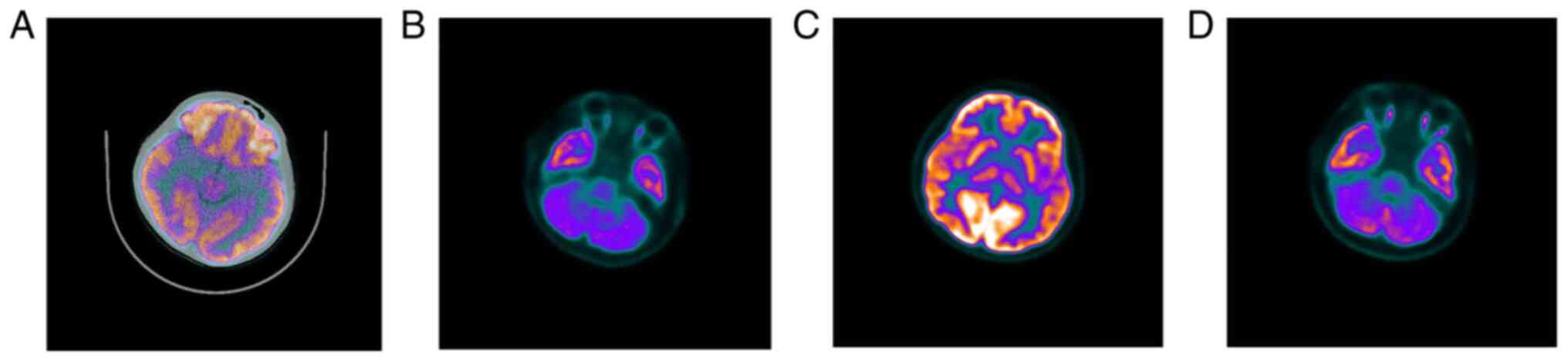
In September 2020, both of the sisters underwent
positron emission computed tomography (PET) and video-EEG in the
brain. Due to the uncooperative nature of the sister of the
proband, only the proband underwent cranial MRI.
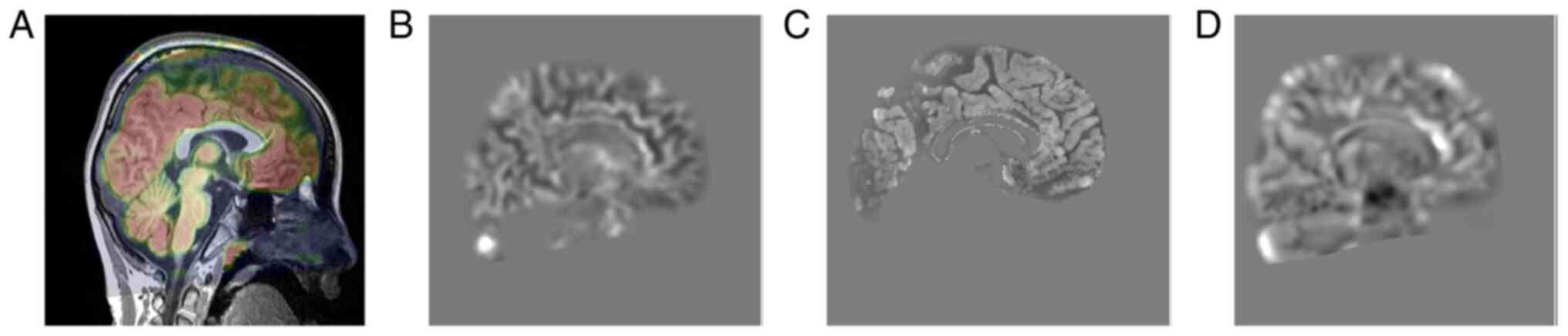
The cerebral MRI (Fig.
3) results of the 8-year-old proband were combined with the
PET/CT findings (Fig. 4) of the
proband's 15-year-old sister. The results indicated lack of
apparent abnormalities in the occipital lobe.
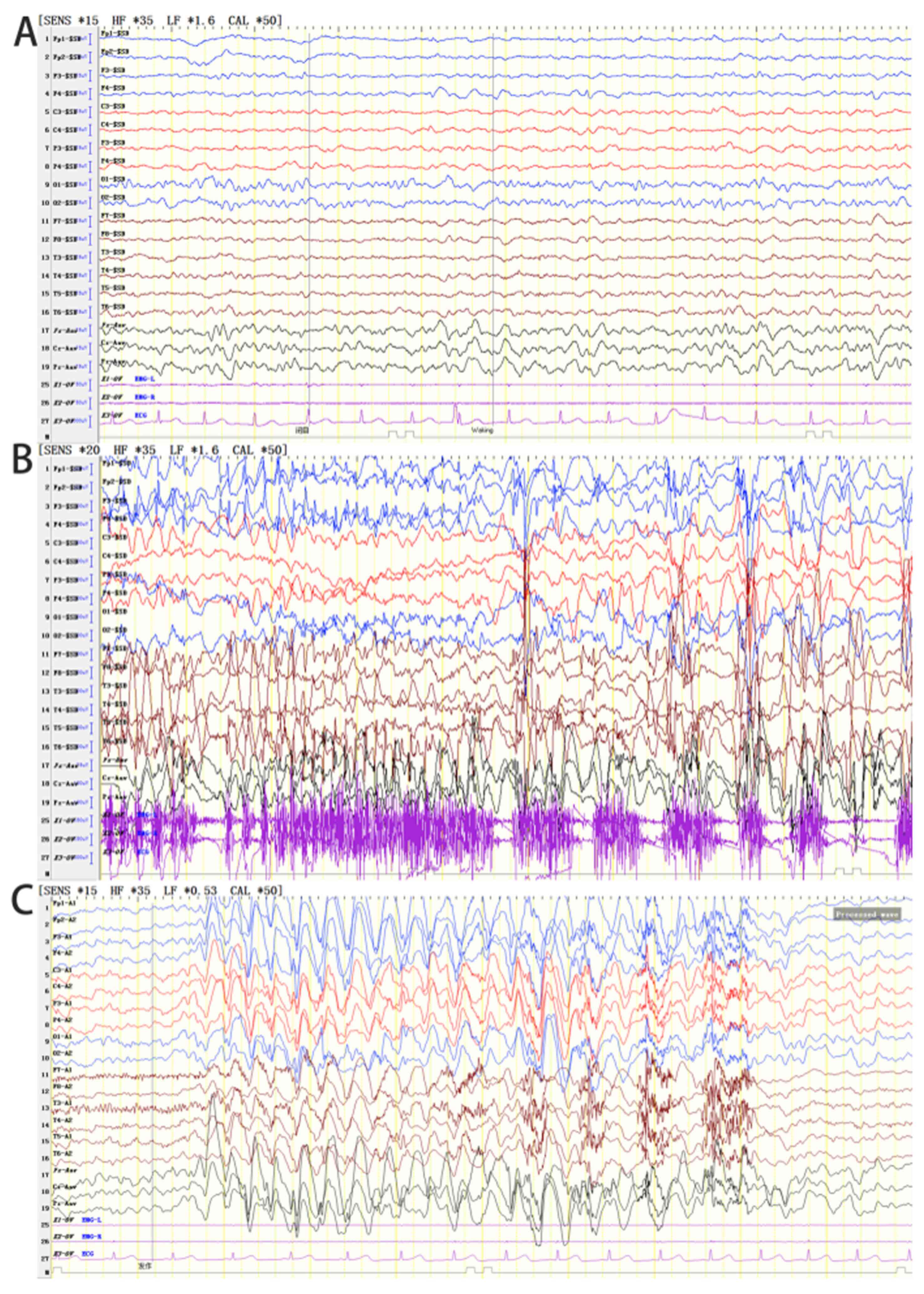
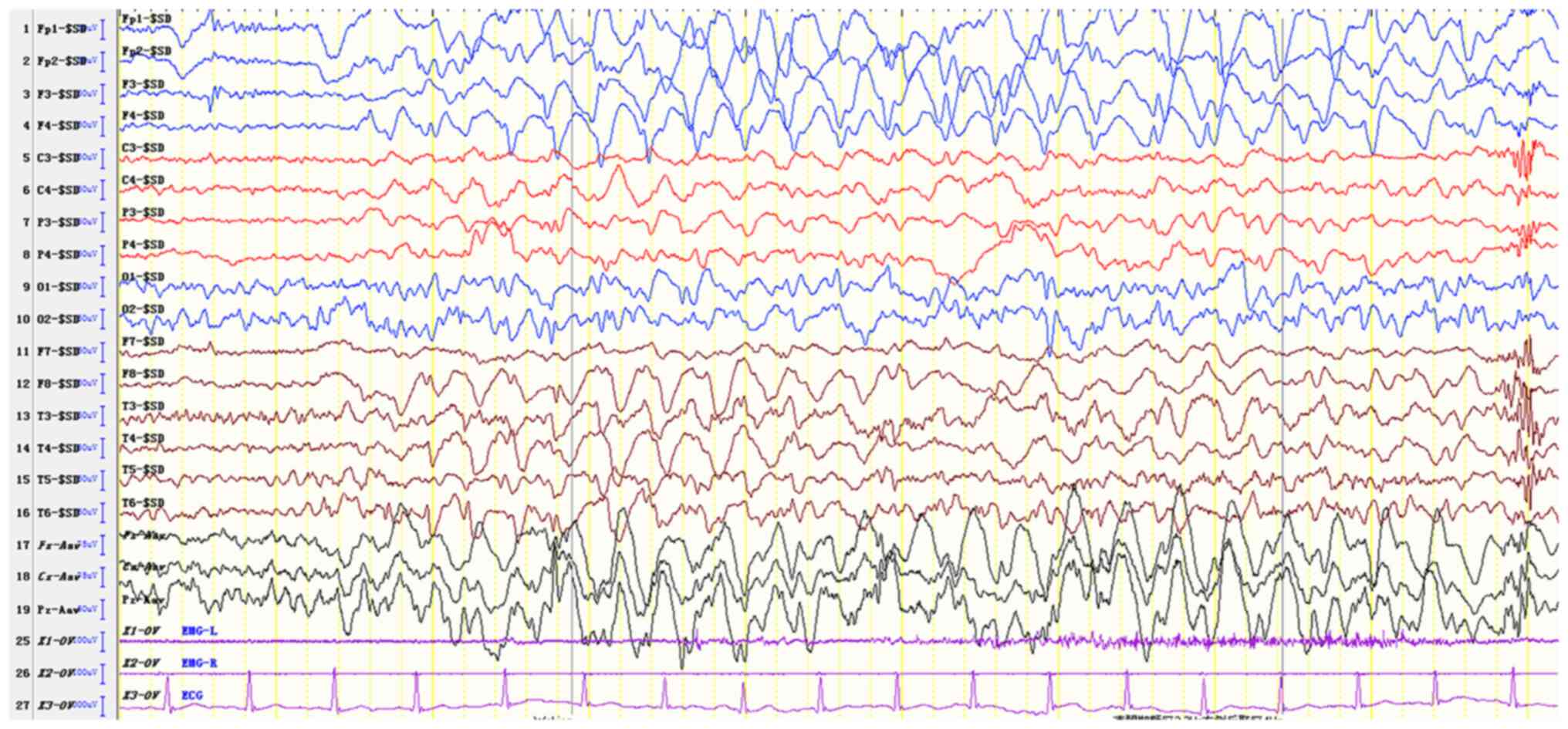
The EEG results of the proband suggested an abnormal
EEG for a child of that age. The full conduction was more frequent
when a sharp slow wave was noted with a frequency range of 2.5-3.5
Hz, or during the rhythmical release of the slow wave.
Occasionally, the posterior head exhibited a 4-6 Hz rhythm and was
accompanied by a multi-focal sharp wave (apparent in the left and
right occipital areas) and comprehensive tonic-clonic and atypical
absence seizure patterns (Fig. 5).
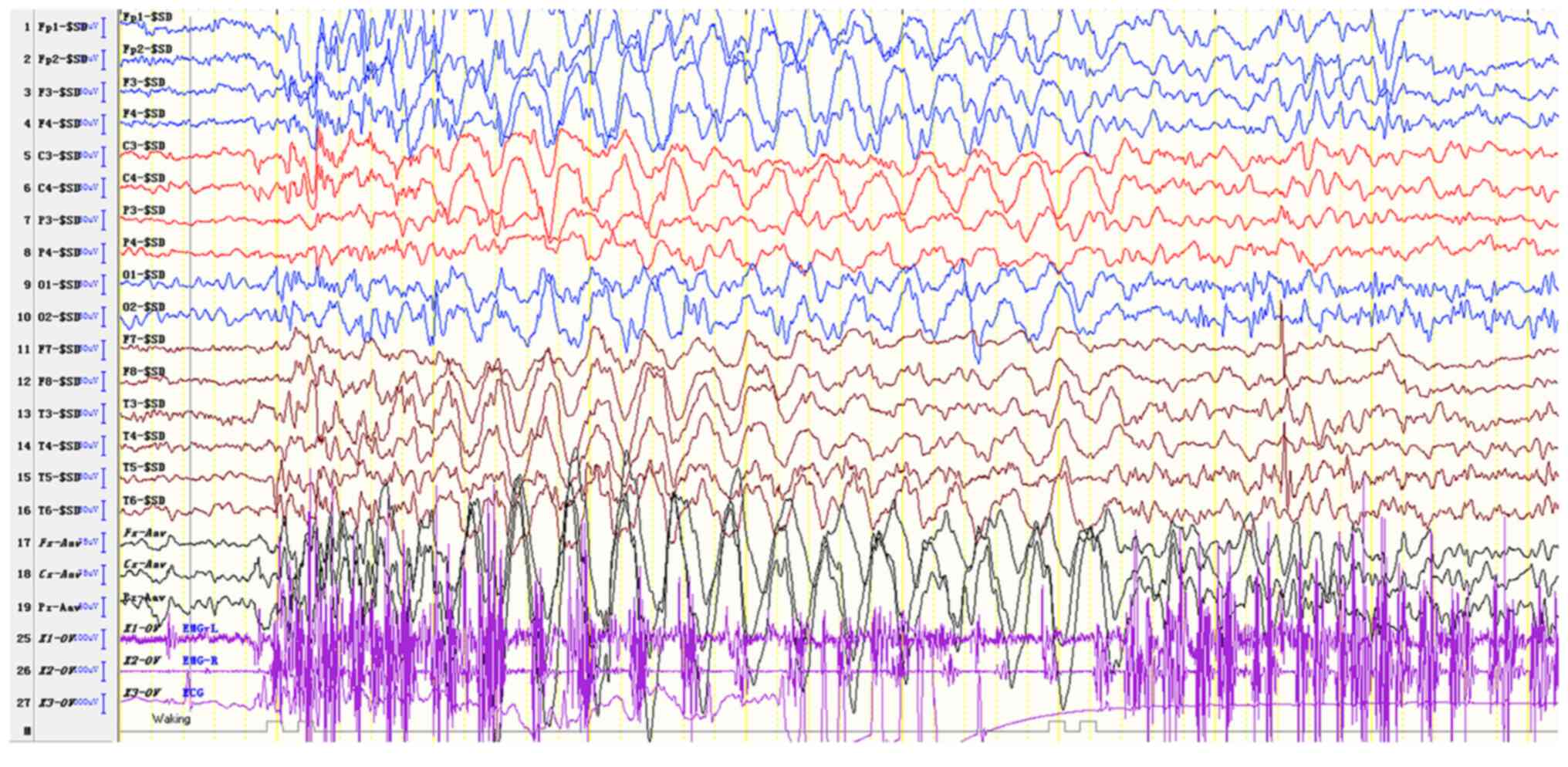
The EEG results of the proband's sister suggested abnormal EEG in
the pediatric patients examined. The EEG activity was mainly
distributed in the bilateral frontal region with a frequency of
2.5-3.5 Hz and was associated with a high amplitude cusp slow
composite wave or slow wave rhythmic release, which occurred more
frequently (Fig. 6). A sharper slow
wave emission was noted in the occipital region. A limited number
of slow sharp waves with a frequency of 4 Hz were noted in the
right posterior temporal region. Several ankylosing-atypical
absence seizures were noted, which were accompanied with myoclonic
seizures (Fig. 7). The sleep cycle
of the proband's sister was disturbed and therefore she did not
sleep during video EEG.
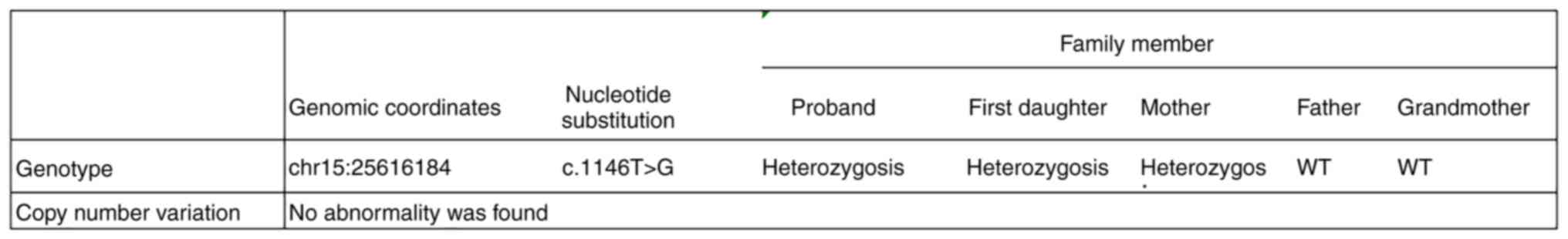
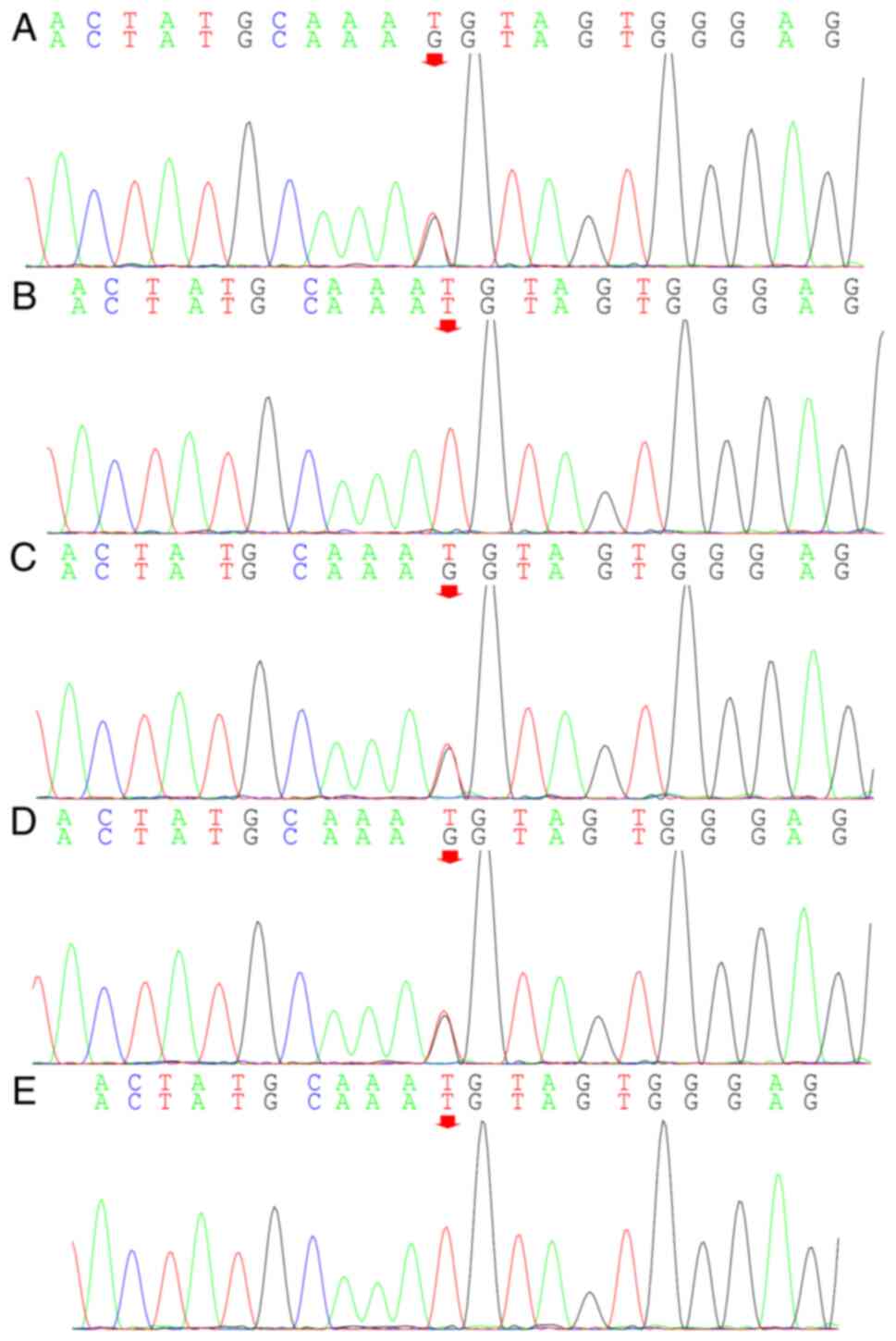
The UBE3A gene of the proband and his sister was
found to possess a C.1146T>G (p.Asn382Lys) mutation. This
mutation has not been previously reported in the HGMD Pro, PubMed
and Clinvar databases, and, to the best of our knowledge, had not
been previously reported in the literature. The Exome Aggregation
Consortium, ESP6500, 1000 Genomes, 1000 Genomes and 1000 Genomes
databases were not included. SIFT (Version 2; https://sift.bii.a-star.edu.sg/), Polyphen2
(Version 2; http://genetics.bwh.harvard.edu/pph2/) and
MutationTaster software were used to predict the protein damage and
the results were classified as follows: Harmful, benign and
possibly harmful. The mutation was inherited from the mother as
determined by family source verification. The missense mutation
resulted in a lysine residue replacing an asparagine residue at
position 382 (p.Asn382Lys). No large fragments of mutations were
noted in the associated genes and no methylation abnormalities were
found. The 25-bp repeat mutation was not found at the shear
mutation site of exon 1 of the small nuclear ribonucleoprotein
polypeptide N gene in the subjects examined. The suspected CNV was
not found following analysis. To determine the pathogenicity of the
locus, the proband's grandmother was tested by genetic analysis and
she was found to be free of the mutation (Fig. 8). The discovery of the UBE3A
mutation was consistent with the clinical manifestations of the
patients, the diagnosis of AS and it's familial transmission.
Discussion
AS is an autosomal dominant genetic disease
secondary to maternal imprinting, which is primarily characterized
by dysfunction of the maternal allele of the UBE3A gene (5). UBE3A encodes the E3 ubiquitin ligase,
and its deletion or replication leads to various neurodevelopmental
disorders. However, how changes in the copy number of ubiquitin
ligase genes affect brain development remains unclear (10). UBE3A has six functional domains as
follows: The homologous to the E6-AP carboxyl terminus domain
(12), the E6 binding domain, the
p53 binding domain and three nuclear receptor interaction and
activation domains (13). The
expression of UBE3A is widespread in various tissues, including the
human fetal brain and adult frontal cortex. The UBE3A gene is
primarily expressed by maternal imprinting, but not by paternal
methylation (10). UBE3A is the
only gene that demonstrates partial expression from maternal
alleles in the 15q11-q13 region (6). The UBE3A gene encodes two proteins
with known functions, of which one is an E6-AP ubiquitin-protein
ligase, and this catalyzes the combination of ubiquitin and
substrate proteins, which are indispensable in the process of
protein catabolism. The other protein is a steroid hormone receptor
coactivator (1). The synaptic
function of UBE3A (including neuronal excitability) may be
associated with the balance of mTOR signaling in the developing
neurons (10). Although the
cellular functions of UBE3A are incompletely understood, it is
known that it plays a role in neurodevelopmental disorders
(1).
The novel heterozygous mutation C.1146T> G (p.Asn
382Lys) of the UBE3A gene found in this family was considered the
cause of the phenotype of these two children with AS. The missense
mutation C.1146T>G resulted in asparagine replacement by lysine
at position 382 (p.Asn382Lys), which affected the normal expression
of UBE3A (Figs. 8 and 9) and reduced ubiquitin ligase synthesis.
The mother of the patient carried the same mutation, but did not
exhibit the symptoms of AS. Following verification of the mother's
pedigree, this locus was not inherited from the grandmother of the
patient. Since the grandfather of the proband had passed away, it
was impossible to assess whether this locus was inherited from the
grandfather of the proband or whether it originated as a novel
mutation in the mother. It could only be assessed by referral to
the clinical evidence of the proband. Since the locus of the
proband was inherited from the mother, the pathogenic
characteristics of AS were caused by to the UBE3A gene mutation.
The clinical manifestations of the 2 patients reported were highly
consistent with AS, suggesting that the comprehensively inferred
locus was the pathogenic cause of the proband and her sister. Since
a heterozygous mutation of UBE3A was identified by genetic testing
of the proband's mother, it was considered that the two mutations
were inherited by the sisters via maternal imprinting.
The imaging results of the two sisters indicated a
slightly blurred and increased signal intensity of periventricular
white matter on T2 weighted and FLAIR image sequences (14). The data were consistent with the
results from Harding et al (15) and Castro-Gago et al (16), suggesting that certain infants with
AS may exhibit delayed myelination. However, the imaging findings
do not usually exhibit substantial specificity, suggesting that
this type of imaging examination may not be sufficient for
diagnosis of AS.
In contrast to these findings, video EEG examination
of children without sedatives usually produce more reliable data,
which can be used to establish a more accurate clinical suspicion
index prior to the final molecular biology-based diagnosis
(14).
It has been previously reported that 80% of patients
with AS may demonstrate characteristic EEG seizures and epileptic
discharges with notch δ and rhythmic θ activity (14). The seizures usually begin prior to
the age of 3 and last until adulthood (15). The EEG results of the two siblings
were consistent with the EEG results noted in the literature
(16).
AS is characterized by severe stunting and
dyskinesia, including ataxia and motor spasms (17). Noticeable behavioral activities
include a cheery manner, excitement, frequent smiling, unexplained
laughter, and no or limited use of words (18). Other common features of AS include
dystonia, tongue protrusion, an abnormal sleep-wake cycle,
decreased sleep demand and salivation (19).
The clinical manifestations of the pediatric
patients with the 15q11-q13 mutation are similar to those of the
clinical phenotype of patients with large deletions in the
chromosomal 15q11.2 region, and to those with single diploid and
imprint deletion. The clinical phenotype of the former includes
severe mental retardation, early epilepsy, microcephaly and severe
ataxia, whereas the clinical phenotype of the latter is less severe
and is characterized by a lower incidence of epilepsy, microcephaly
and special facial deformity, light ataxia and optimal cognitive
ability (3).
In a study conducted by Valente et al
(20) it was reported that sodium
valproic acid could improve the seizures of 19 patients who
received monotherapy or multidrug therapy, notably when combined
with clonazepam or phenobarbital. In the present study, the
seizures of the two sisters were not significantly controlled
following treatment with sodium valproic acid. Moreover, the
proband had taken levetiracetam combined with topiramate, but this
did not control the seizures. At present, lamotrigine combined with
sodium valproate is used for the treatment of seizures and this has
resulted in a slight reduction in their frequency. However, this
effect may not be directly observed.
In conclusion, studies have shown that UBE3A is a
multifunctional protein, which has important nuclear and
cytoplasmic regulatory functions, and affects proteasome function,
Wnt signaling, circadian rhythm, imprinted gene network and
chromatin. The synaptic function of UBE3A interacts with light GABA
mTOR signals, which is the most critical signal amongst GABAergic
neurons (10). The two pediatric
patients examined in the present study exhibited AS and carried
novel UBE3A mutations that had not been previously reported.
Moreover, the identification of specific pathogenic genes can aid
genetic counseling and prenatal testing for families and may be
used for early diagnosis of AS in pediatrics. The latter process
can improve the control of epilepsy in children and reduce the
incidence of brain injury. These pediatric patients must receive
specialized education at an early stage in order to improve their
quality of life.
Acknowledgements
We would like to thank Dr Xi-Bin Hu and Dr Yu Kong
(Affiliated Hospital of Jining Medical University, Jining, China)
for their assistance in collecting the clinical data; Dr Zeng-xian
Zhang (Beijing Kangso Medical inspection for Technical Support,
Beijing, China); Dr Yi-dan Liu (Peking University, Peking, China),
Dr Rui-han Liu (Jining Medical University, Jining, China) and Ms
Meng-yu Ma (Shandong University, Shandong, China) for their
assistance in drafting the manuscript.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Shandong Province (grant no ZR2019MH060), the
Teaching Case Database of Professional Degree Postgraduates in
Shandong Province (grant no. SDYAL19213), the Key Research and
Development Plan of Jining City (grant no. 2019SMNS019) and the
Research Fund for Lin He's Academician Workstation of New Medicine
and Clinical Translation in Jining Medical University (grant no.
JYHL2018FMS10).
Availability of date and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
QXK and QBL designed the study. RHL, GFS, LY, QLZ,
SYW, and CL collected the clinical data. CL analyzed the data and
wrote the manuscript. All authors have read and approved the final
manuscript. QXK, QBL, GFS and RHL confirm the authenticity of all
the raw data.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Affiliated Hospital of Jining Medical College (Jining, China). The
parents of the patients provided written informed consent for their
participation.
Patient consent for publication
The parents of the children provided written
informed consent for publication of the data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sirois CL, Bloom JE, Fink JJ, Gorka D,
Keller S, Germain ND, Levine ES and Chamberlain SJ: Abundance and
localization of human UBE3A protein isoforms. Hum Mol Genet.
29:3021–3031. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yang X: Towards an understanding of
angelman syndrome in mice studies. J Neurosci Res. 98:1162–1173.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ranasinghe JC, Chandradasa D, Fernando S,
Kodithuwakku U, Mandawala DE and Dissanayake VH: Angelman syndrome
presenting with a rare seizure type in a patient with 15q11.2
deletion: A case report. J Med Case Rep. 9(142)2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Curtis M, Baribeau D, Walker S, Carter M,
Costain G, Lamoureux S, Liston E, Marshall CR, Reuter MS, Snell M,
et al: A novel intronic variant in UBE3A identified by genome
sequencing in a patient with an atypical presentation of Angelman
syndrome. Am J Med Genet A. 182:2145–2151. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bonello D, Camilleri F and Calleja-Agius
J: Angelman syndrome: Identification and management. Neonatal Netw.
36:142–151. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gu B, Carstens KE, Judson MC, Dalton KA,
Rougié M, Clark EP, Dudek SM and Philpot BD: Ube3a reinstatement
mitigates epileptogenesis in Angelman syndrome model mice. J Clin
Invest. 129:163–168. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Scheffner M, Huibregtse JM, Vierstra RD
and Howley PM: The HPV-16 E6 and E6-AP complex functions as a
ubiquitin-protein ligase in the ubiquitination of p53. Cell.
75:495–505. 1993.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yamamoto Y, Huibregtse JM and Howley PM:
The human E6-AP gene (UBE3A) encodes three potential protein
isoforms generated by differential splicing. Genomics. 41:263–266.
1997.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Valluy J, Bicker S, Aksoy-Aksel A,
Lackinger M, Sumer S, Fiore R, Wüst T, Seffer D, Metge F, Dieterich
C, et al: A coding-independent function of an alternative Ube3a
transcript during neuronal development. Nat Neurosci. 18:666–673.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Lopez SJ, Segal DJ and LaSalle JM: UBE3A:
An E3 ubiquitin ligase with genome-wide impact in
neurodevelopmental disease. Front Mol Neurosci.
11(476)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
D'Angelo R, Donato L, Venza I, Scimone C,
Aragona P and Sidoti A: Possible protective role of the ABCA4 gene
c.1268A>G missense variant in Stargardt disease and syndromic
retinitis pigmentosa in a Sicilian family: Preliminary data. Int J
Mol Med. 39:1011–1020. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Buiting K: Prader-Willi syndrome and
Angelman syndrome. Am J Med Genet C Semin Med Genet. 154C:365–376.
2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ciarlone SL, Grieco JC, D'Agostino DP and
Weeber EJ: Ketone ester supplementation attenuates seizure
activity, and improves behavior and hippocampal synaptic plasticity
in an Angelman syndrome mouse model. Neurobiol Dis. 96:38–46.
2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Leyser M, de Castro Diniz Gonsalvez M,
Vianna PE, Fernandes PA, Carvalho RS, Vasconcelos MM and Nascimento
OJ: Scrutinizing brain magnetic resonance imaging patterns in
Angelman syndrome. Neurol India. 64:228–232. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Harting I, Seitz A, Rating D, Sartor K,
Zschocke J, Janssen B, Ebinger F and Wolf NI: Abnormal myelination
in Angelman syndrome. Eur J Paediatr Neurol. l13:271–276.
2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Castro-Gago M, Gómez-Lado C, Eirís-Puñal J
and Rodríguez-Mugico VM: Abnormal myelination in Angelman syndrome.
Eur J Paediatr Neuro. 14(292)2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Thibert RL, Larson AM, Hsieh DT, Raby AR
and Thiele EA: Neurologic manifestations of Angelman syndrome.
Pediatr Neurol. 48:271–279. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Boyd SG, Harden A and Patton MA: The EEG
in early diagnosis of the Angelman (happy puppet) syndrome. Eur J
Pediatr. 147:508–513. 1988.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tan WH, Bird LM, Thibert RL and Williams
CA: If not Angelman, what is it? A review of Angelman-like
syndromes. Am J Med Genet A. 164A:975–992. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Valente KD, Koiffmann CP, Fridman C,
Varella M, Kok F, Andrade JQ, Grossmann RM and Marques-Dias MJ:
Epilepsy in patients with angelman syndrome caused by deletion of
the chromosome 15q11-13. Arch Neurol. 63:122–128. 2006.PubMed/NCBI View Article : Google Scholar
|