Introduction
The vascular endothelium is an active monolayer of
cells lining the entire circulatory system, and it is indispensable
for the regulation of vascular tone and in the maintenance of
vascular homeostasis (1).
Endothelial dysfunction is regarded as a key early event in the
development of atherosclerosis and the occurrence of
atherosclerotic complications (2,3).
Notably, endothelial dysfunction is an established predictor of
cardiovascular outcomes (4).
The endothelium regulates the remodeling of the
vessel wall by releasing a large number of vasoactive substances.
Nitric oxide (NO) is an endothelium-derived relaxing factor that is
produced by endothelial NO synthase (eNOS) (5). It protects the vascular system by
acting as an endogenous defense against the development of
atherosclerosis. The reduction in the generation and
bioavailability of NO is accepted as an important factor in the
process of atherosclerosis and thrombosis (6,7).
Endothelin (ET)-1 is a potent vasoconstrictor produced by
endothelial cells that plays a pivotal role in the etiology of
atherosclerotic vascular disease (8). It induces endothelial dysfunction and
contributes to atherosclerosis by stimulating platelet aggregation,
cell adhesion molecule expression and the proliferation of vascular
smooth muscle cells (9,10). A pathophysiological imbalance in NO
and ET-1 is usually referred as endothelial dysfunction (11).
Cilostazol is a selective inhibitor of
phosphodiesterase type III (PDE3) that inhibits platelet
aggregation (12). It is widely
used in patients after receiving percutaneous coronary intervention
(PCI) to reduce the risk of thrombotic events (13,14).
The beneficial effects of cilostazol have been attributed not only
to its antiplatelet functions but also to its actions on the
endothelium. Recent studies suggested that cilostazol could protect
endothelial function by inducing NO production (15-17)
and decreasing ET-1 production (18,19).
As an important member of the MAPK pathway, p38 MAPK
plays an important role in inflammation and stress response. Animal
studies have confirmed that p38/MAPK is closely related to the
expression and secretion of ET-1 (20,21).
It has also been reported that p38/MAPK can regulate the expression
of eNOS (22). A number of studies
have shown that p38/MAPK is one of the targets of cilostazol, which
may explain its endothelial protective effect (23,24). A
study published by Chao et al (25) suggested that cilostazol could
regulate NO production via the p38/MAPK signaling pathway, however,
it did not assess the effects of cilostazol on ET-1. Thus, whether
cilostazol regulates NO and ET-1 via the p38/MAPK signaling pathway
remains to be determined.
Taking the above evidence together, the objective of
this study was to examine the effects of cilostazol on the
regulation of ET-1 and eNOS, as well as its relationship with
p38/MAPK. HUVECs were used in this study as they have proven
invaluable in investigating endothelial damage and repair, and on
the potential impact of atherosclerosis during the early stages and
during atherosclerosis progression (26). Specifically, HUVECs were exposed to
TNF-α and cilostazol with or without specific inhibitors of
p38/MAPK, and the changes in p38/MAPK activity, and in the
expression levels of ET-1 and eNOS were determined.
Materials and methods
Materials
HUVECs (cat. no. 8000) and endothelial cell medium
(ECM, cat. no. #1001) were purchased from ScienCell Research
Laboratories, Inc. Cilostazol (cat. no. PHR1503) and the p38
inhibitor SB203580 were obtained from Sigma-Aldrich; Merck KGaA.
Antibodies were obtained from ProteinTech Group, Inc., Cell
Signaling Technology, Inc., or Zhejiang Kangchen Biotech, Co.,
Ltd.; TRIzol® reagent from Invitrogen; Thermo Fisher
Scientific, Inc.; and TNF-α from Sigma-Aldrich; Merck KGaA. PCR
primers (Integrated DNA Technologies, Inc.); and the
GoTaq® qPCR MasterMix kit was from Promega Corporation.
The study proposal was approved by Zhongshan hospital, Fudan
University.
Cell culture and treatments
HUVECs were cultured at 37˚C in a humidified 5%
CO2 incubator and split in accordance with standard
procedures. When they grew 60-85% confluent, HUVECs were treated
with different concentrations (0, 1, 10 or 50 uM) cilostazol
(25) or TNF-α (10 µg/l) for 24 h
and then collected for further analysis.
SB203580 (10 µM, for 2 h) (27) and TNF-α (for 24 h) (28) were added to cells prior to
incubation with cilostazol. Control HUVECs were pre-treated with
goat serum.
ELISA
ET-1 and eNOS levels in the cell culture supernatant
were quantified by spectrophotometry using ELISA kits according to
the manufacturer's protocol (cat. no. H093 and cat. no. H195,
respectively; both from Nanjing Jiancheng Bioengineering
Institute).
Reverse transcription-quantitative
(RT-q)PCR
Total mRNA was extracted from HUVECs using
TRIzol® reagent. Reverse transcription was performed
using a Prime Script RT reagent kit (Takara Bio, Inc.) according to
the manufacturer's protocols. The mRNA levels of ET-1 and eNOS were
measured by qPCR using an iCycler real-time system (Bio-Rad
Laboratories, Inc.). The thermocycling conditions were as follows:
An initial pre-denaturation step for 10 sec at 95˚C; followed by 45
cycles of denaturation at 95˚C for 10 sec, annealing at 60˚C for 20
sec and extension at 70˚C for 30 sec; with a final extension step
at 70˚C for 5 min.
The sequences of the primers used for amplification
were: ET-1 forward, 5'-AAGGCAACAGACCGTGAAAAT-3' and reverse,
5'-CGACCTGGTTTGTCTTAGGTG-3'; eNOS forward,
5'-TGATGGCGAAGCGAGTGAAG-3' and reverse,
5'-ACTCATCCATACACAGGACCC-3'; and GAPDH forward,
5'-AGAAGGCTGGGGCTCATTTG-3' and reverse,
5'-AGGGGCCATCCACAGTCTTC-3'.
Western blotting
HUVECs were washed three times with PBS and lysed
with ice-cold RIPA buffer (Beyotime Institute of Biotechnology)
supplemented with protease inhibitor cocktail (Thermo Fisher
Scientific, Inc.). A BCA protein assay kit (Thermo Fisher
Scientific, Inc.) was used to determine the protein concentrations
in the lysates. Equivalent amounts (30 µg) of total protein were
resolved by SDS-PAGE (10% tris-glycine gels) and immunoblotted with
primary antibodies at 4˚C overnight (29) [anti-ET-1 (1:1,000, cat. no.
67008-1-lg,ProteinTech Group, Inc.), anti-eNOS (dilution 1:1,000,
cat. no. 27120-1AP, ProteinTech Group, Inc.), anti-p38/p-p38
(1:1,000, cat. nos. 4511 and 8690, respectively, Cell Signaling
Technology, Inc.) and anti-GAPDH (1:5,000, cat. no. 5174, Cell
Signaling Technology, Inc.)]. After washing three times, the
horseradish peroxidase-conjugated secondary antibody (1:5,000, cat.
no. KC-RB-035; Zhejiang Kangchen Biotech Co., Ltd.) was added and
incubated for 1 h at room temperature.
Statistical analysis
All experiments were performed at least three times.
The representative results and corresponding quantification data of
one repeat is shown for each experiment. Data are presented as the
mean ± SEM and were compared using a Student's t-test or a one-way
ANOVA with a post-hoc Tukey's test using GraphPad Prism version
5.01 (GraphPad software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of cilostazol on the levels of
ET-1 and eNOS in HUVECs
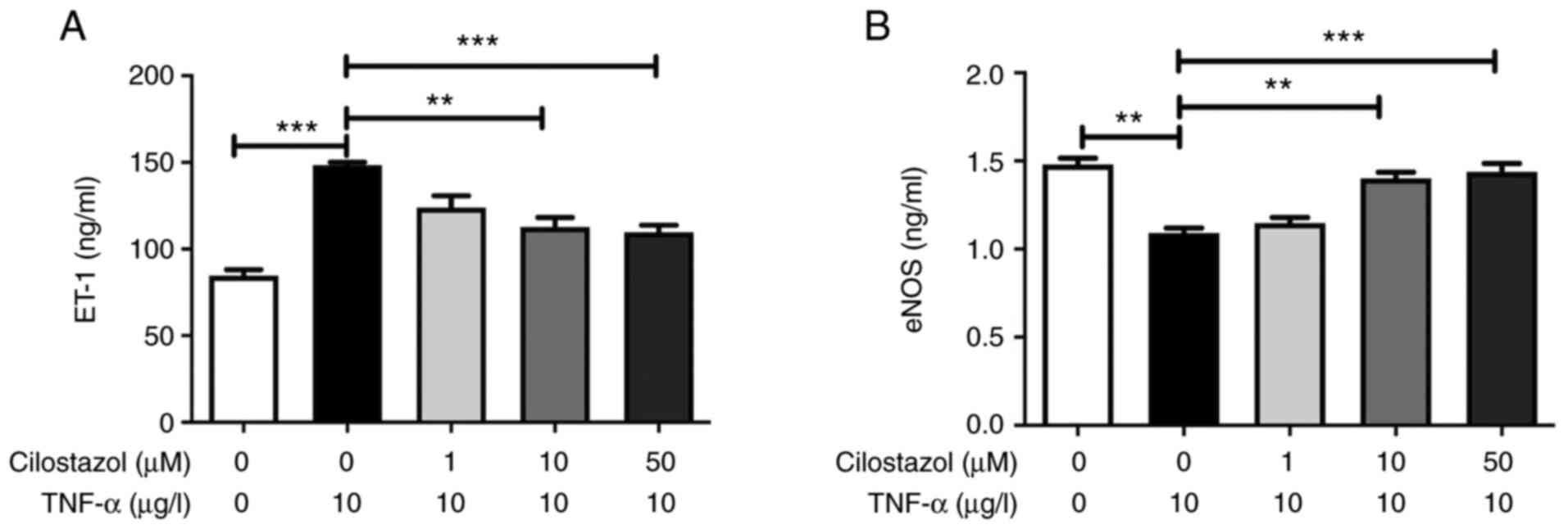
Compared with the untreated cells, TNF-α markedly
increased the levels of ET-1, and decreased the levels of eNOS in
the HUVECs culture supernatant. Cilostazol (1, 10 and 50 µM)
administration significantly decreased the levels of ET-1, and
increased the levels of eNOS in HUVECs compared with the
TNF-α-induced cells (Fig. 1).
Effect of cilostazol on the mRNA
expression levels of ET-1 and eNOS in HUVECs
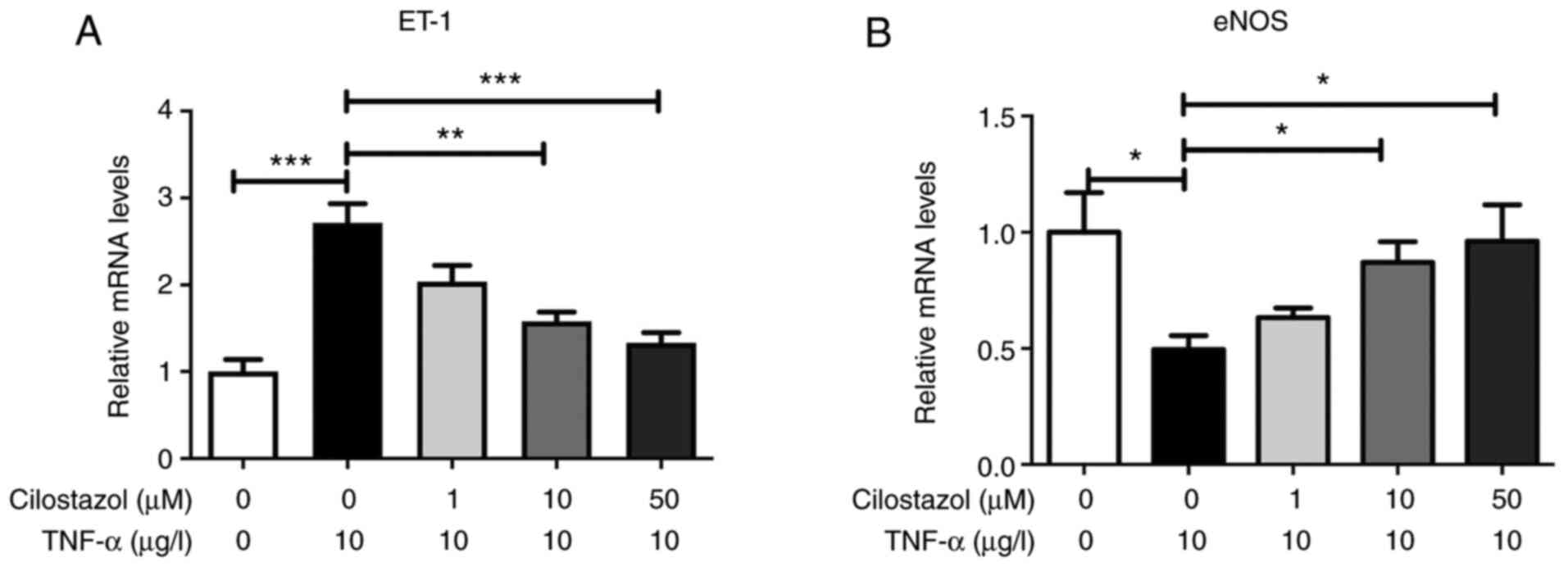
To determine whether ET-1 and eNOS expression in
HUVECs was affected by cilostazol at the mRNA level, the HUVECs
were incubated overnight with TNF-α and then treated with different
concentrations of cilostazol (1, 10 or 50 µM). Total RNA was
isolated from HUVECs and subjected to RT-qPCR analysis. As shown in
Fig. 2, HUVECs exposed to
cilostazol exhibited upregulated eNOS mRNA levels and downregulated
ET-1 mRNA levels in a dose-dependent manner (P<0.05).
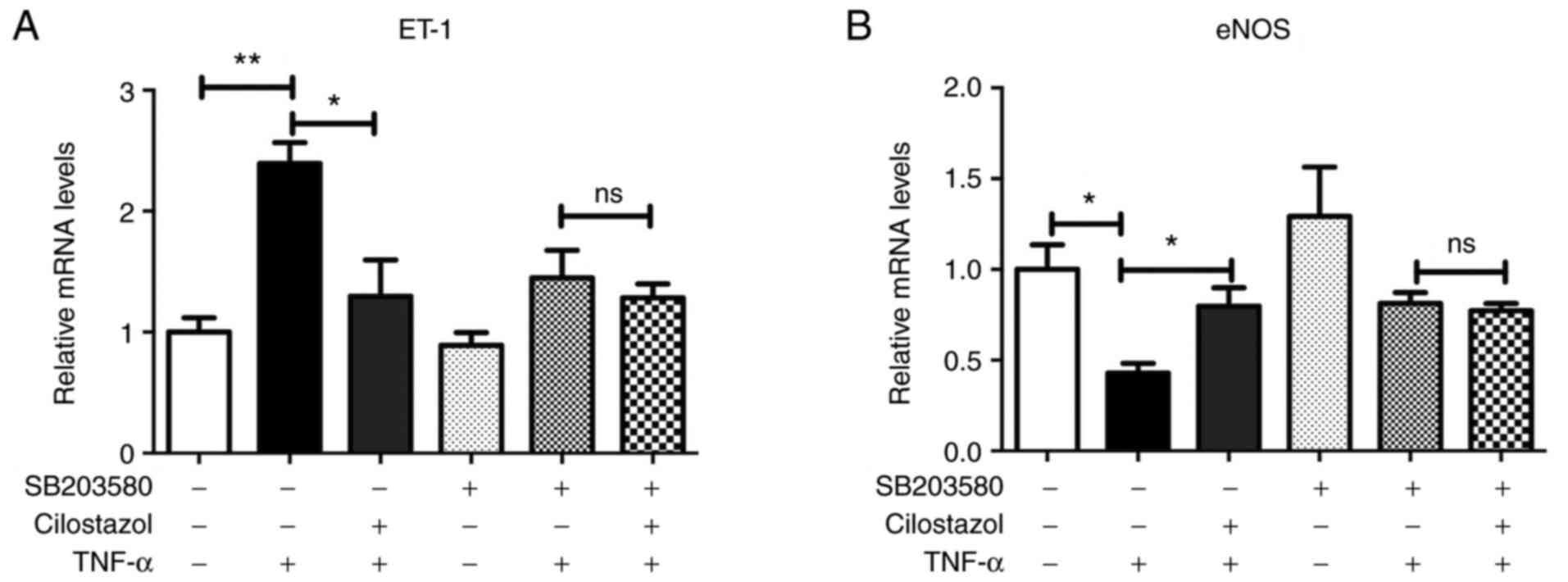
HUVECs were pre-treated with a p38 inhibitor
(SB203580-10 µM) for 2 h prior to incubation with cilostazol. The
increase in ET-1 mRNA expression levels were fully abolished in the
presence of this p38 inhibitor indicating the involvement of this
kinase in cilostazol-induced regulation of ET-1 in HUVECs
(P<0.05; Fig. 3A). Additionally,
the effects of cilostazol on eNOS mRNA were also abolished by
SB203580 (Fig. 3B).
Effect of cilostazol on p-p38 MAPK
expression in HUVECs
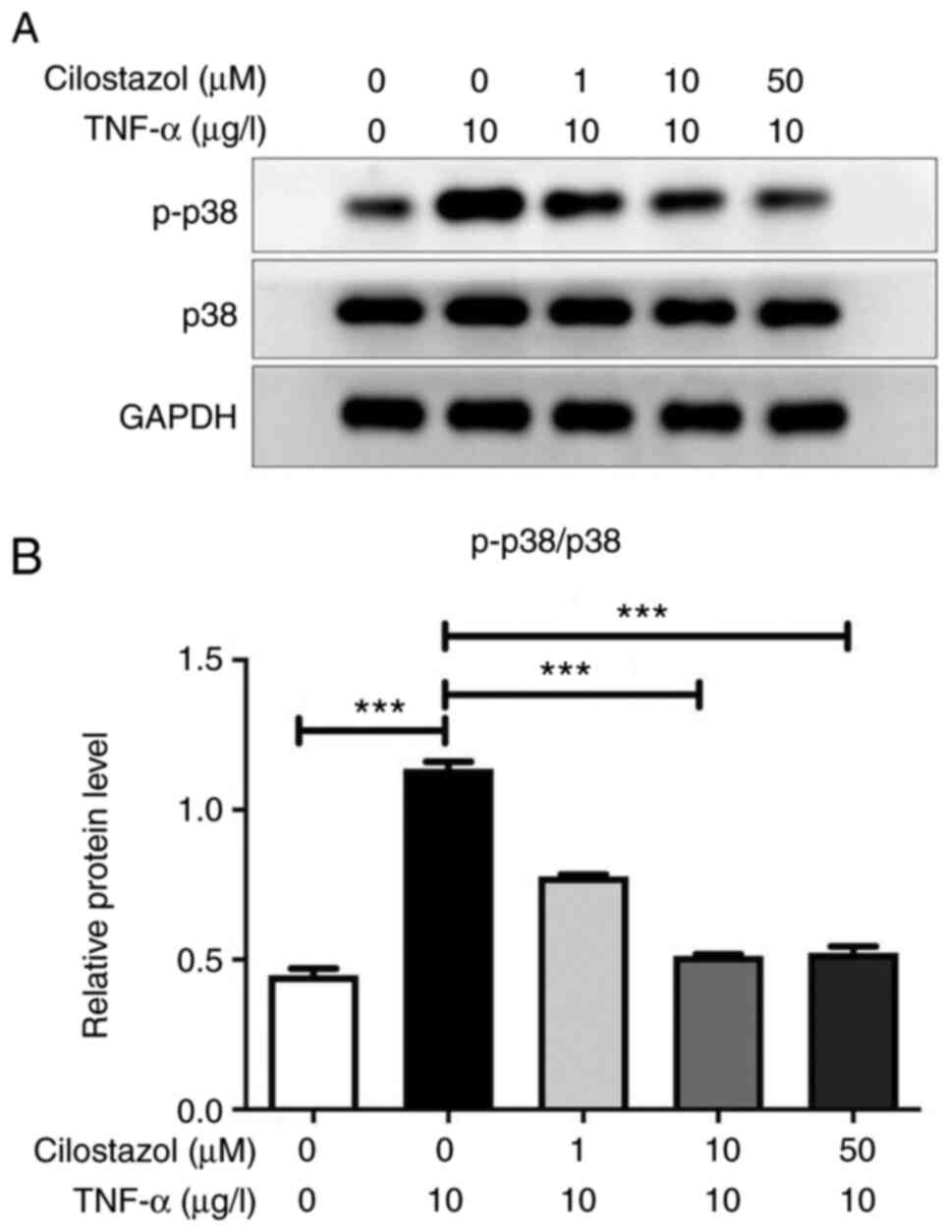
Lysates from HUVECs treated with different
cilostazol concentrations were analyzed by western blotting
utilizing specific antibodies against the activated (i.e.
phosphorylated) form of p38. Compared with the untreated cells,
TNF-α markedly increased the p-p38 MAPK levels in HUVECs. Treatment
of HUVECs with several different concentrations of cilostazol
resulted in a decrease in p-p38 levels in a dose-dependent manner
(Fig. 4).
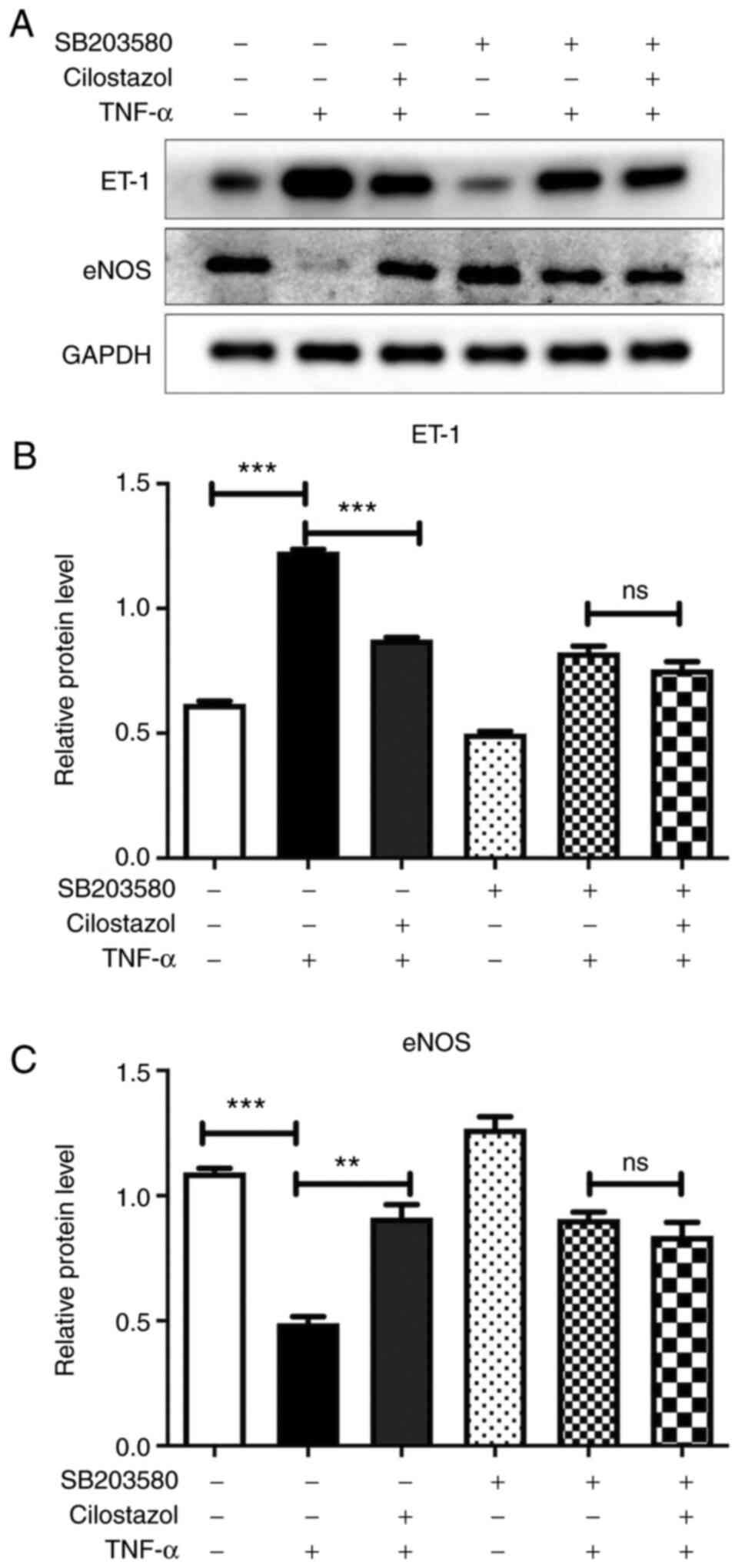
Effect of cilostazol on the protein
expressions levels of ET-1 and eNOS in HUVECs
To further validate the above findings, lysates from
HUVECs exposed to p38 inhibitor (SB203580) and cilostazol were
analyzed by western blotting. Compared with the untreated cells,
TNF-α markedly increased the levels of and ET-1, and decreased the
levels of eNOS in HUVECs. Cilostazol administration significantly
decreased the protein expression levels of ET-1, and increased the
protein expression levels of eNOS in HUVECs. These effects of
cilostazol on ET-1 and eNOS were also abolished by SB203580
treatment (Fig. 5).
Discussion
Endothelial dysfunction increases the vulnerability
of plaque formation, triggers plaque rupture, and promotes
thrombosis. It is considered a key precursor in the initiation,
progression, and complication of coronary atherosclerotic heart
disease (30,31). Given its role in the development of
coronary artery and cerebrovascular diseases, endothelial
dysfunction may be an attractive target in the efforts to optimize
individualized therapeutic strategies to reduce cardiovascular
morbidity and mortality rates (32).
Endothelial cells can release mediators and
transcription factors to regulate important functions such as the
tension of blood vessels and the growth of smooth muscle cells.
Among those endothelial active factors, the relaxation factor NO
and the contraction factor ET-1 are two leading active substances
(5,33). Endothelial dysfunction is in part
characterized by enhanced ET-1 and diminished eNOS expression
(34). The balance between ET-1 and
eNOS expression maintains the normal function of endothelial cells
in terms of cellular integrity.
Cilostazol is a novel type of antiplatelet drug,
which inhibits platelets by selectively inhibiting PDE3, and
functions to expand blood vessels, inhibiting intimal hyperplasia
and protecting the endothelium. Unlike other antiplatelet agents,
cilostazol not only exhibits antiplatelet properties, but also
appears to have beneficial effects on endothelial function
(34-37).
A number of studies suggested that cilostazol can induce NO
production (15-17).
Furthermore, it has been reported that cilostazol can decrease ET-1
production (18,19).
The inflammatory cytokine TNF-α plays a pivotal role
in the disruption of macrovascular and microvascular circulation.
It is suggested that TNF-α can induce the synthesis and release of
ET-1(38). It also accelerates the
degradation of eNOS mRNA, decreases the activity of the gene
promoter and downregulates the expression of eNOS (39). In this study, TNF-α and HUVECs were
used to study the effects of cilostazol on the expression of ET-1
and eNOS. The results showed that cilostazol reduced the expression
of ET-1 and increased the expression of eNOS in a dose-dependent
manner. HUVECs exposed to TNF-α exhibited increased ET-1 levels and
ET-1 mRNA expression. This suggested that TNF-α-mediated increases
in ET-1 expression may be achieved through regulation of ET-1 gene
transcription. After incubating with cilostazol, the expression of
ET-1 was significantly reduced. Similarly, cilostazol upregulated
the mRNA and protein expression levels of eNOS.
The MAPKs are a family of kinases that transduce
signals from the cell membrane to the nucleus in response to a wide
range of stimuli, including stress and injury. The MAPK pathway
regulates cell proliferation, mitosis, transformation, apoptosis
and other biological activities by affecting gene transcription and
regulation. p38/MAPK is an important member of the MAPK family.
There is a close relationship between the p38/MAPK signaling
pathway and vascular remodeling. Recent studies have shown that the
p38/MAPK signaling pathway is closely related to the regulation of
ET-1 and eNOS expression (20,22).
It has also been suggested that p38/MAPK is one of the targets of
cilostazol, which may explain the beneficial effects of cilostazol
in slowing down the progression of atherosclerosis (23,24).
To explore the effects of cilostazol on the p38
singling pathway in HUVECs, the intracellular signaling pathways
involved in eNOS and ET-1 expression were examined. HUVECs were
treated with the p38 inhibitor SB203580 prior to cilostazol
treatment. The observed upregulation of eNOS and ET-1 mRNA levels
was completely abolished upon p38 inhibitor treatment, indicating
the involvement of p38/MAPK and the associated signaling cascades
in cilostazol-induced regulation of eNOS and ET-1 in HUVECs.
Western blotting analysis of lysates from HUVECs exposed to
cilostazol further validated this finding. These results suggested
that the regulation of NO and ET-1 in HUVECs by cilostazol may be
mediated through the p38MAPK signaling pathway.
In summary, we found that cilostazol regulates ET-1
and eNOS production by suppressing the p38/MAPK signal pathway in
TNF-α-stimulated HUVECs, and this may contribute to the protective
effect of cilostazol on the endothelium. Further experiments are
required to confirm these results.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Natural Science
Foundation of China (grant no. 81803630).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YX and QL designed the study. YX, ZW and XL
performed the experiments. YX analyzed the data and wrote the
manuscript. All authors have read and approved the final
manuscript. YX, XL and ZW confirmed the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reriani MK, Flammer AJ, Jama A, Lerman LO
and Lerman A: Novel functional risk factors for the prediction of
cardiovascular events in vulnerable patients following acute
coronary syndrome. Circ J. 76:778–783. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
el-Tamimi H, Mansour M, Wargovich TJ, Hill
JA, Kerensky RA, Conti CR and Pepine CJ: Constrictor and dilator
responses to intracoronary acetylcholine in adjacent segments of
the same coronary artery in patients with coronary artery disease.
Endothelial function revisited. Circulation. 89:45–51.
1994.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Huang PH, Leu HB, Chen JW, Wu TC, Lu TM,
Yu-An Ding P and Lin SJ: Decreased heparin cofactor II activity is
associated with impaired endothelial function determined by
brachial ultrasonography and predicts cardiovascular events. Int J
Cardiol. 114:152–158. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Martin BJ and Anderson TJ: Risk prediction
in cardiovascular disease: The prognostic significance of
endothelial dysfunction. Can J Cardiol. 25 (Suppl A):15A–20A.
2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li H, Horke S and Förstermann U: Vascular
oxidative stress, nitric oxide and atherosclerosis.
Atherosclerosis. 237:208–219. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tousoulis D, Kampoli AM, Tentolouris C,
Papageorgiou N and Stefanadis C: The role of nitric oxide on
endothelial function. Curr Vasc Pharmacol. 10:4–18. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sogo N, Magid KS, Shaw CA, Webb DJ and
Megson IL: Inhibition of human platelet aggregation by nitric oxide
donor drugs: relative contribution of cGMP-independent mechanisms.
Biochem Biophys Res Commun. 279:412–419. 2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Miyauchi T and Masaki T: Pathophysiology
of endothelin in the cardiovascular system. Annu Rev Physiol.
61:391–415. 1999.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lüscher TF and Barton M: Endothelins and
endothelin receptor antagonists: Therapeutic considerations for a
novel class of cardiovascular drugs. Circulation. 102:2434–2440.
2000.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rajendran P, Rengarajan T, Thangavel J,
Nishigaki Y, Sakthisekaran D, Sethi G and Nishigaki I: The vascular
endothelium and human diseases. Int J Biol Sci. 9:1057–1069.
2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cone J, Wang S, Tandon N, Fong M, Sun B,
Sakurai K, Yoshitake M, Kambayashi J and Liu Y: Comparison of the
effects of cilostazol and milrinone on intracellular cAMP levels
and cellular function in platelets and cardiac cells. J Cardiovasc
Pharmacol. 34:497–504. 1999.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Guerra E, Byrne RA and Kastrati A:
Pharmacological inhibition of coronary restenosis: Systemic and
local approaches. Expert Opin Pharmacother. 15:2155–2171.
2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jang JS, Jin HY, Seo JS, Yang TH, Kim DK,
Kim DS, Kim DK, Seol SH, Kim DI, Cho KI, et al: A meta-analysis of
randomized controlled trials appraising the efficacy and safety of
cilostazol after coronary artery stent implantation. Cardiology.
122:133–143. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hashimoto A, Miyakoda G, Hirose Y and Mori
T: Activation of endothelial nitric oxide synthase by cilostazol
via a cAMP/protein kinase A- and phosphatidylinositol
3-kinase/Akt-dependent mechanism. Atherosclerosis. 189:350–357.
2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ito H, Hashimoto A, Matsumoto Y, Yao H and
Miyakoda G: Cilostazol, a phosphodiesterase inhibitor, attenuates
photothrombotic focal ischemic brain injury in hypertensive rats. J
Cereb Blood Flow Metab. 30:343–351. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bai Y, Muqier Murakami H, Iwasa M, Sumi S,
Yamada Y, Ushikoshi H, Aoyama T, Nishigaki K, Takemura G, et al:
Cilostazol protects the heart against ischaemia reperfusion injury
in a rabbit model of myocardial infarction: focus on adenosine,
nitric oxide and mitochondrial ATP-sensitive potassium channels.
Clin Exp Pharmacol Physiol. 38:658–665. 2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Pelletier S, Dubé J, Villeneuve A, Gobeil
F Jr, Bernier SG, Battistini B, Guillemette G and Sirois P:
Adenosine induces cyclic-AMP formation and inhibits endothelin-1
production/secretion in guinea-pig tracheal epithelial cells
through A(2B) adenosine receptors. Br J Pharmacol. 129:243–250.
2000.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shima A, Maki T, Mimura N, Yamashita H,
Emoto N, Yoshifuji H and Takahashi R: A case of reversible cerebral
vasoconstriction syndrome associated with anti-phospholipid
antibody syndrome and systemic lupus erythematosus.
eNeurologicalSci. 24(100351)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Armstead WM, Bohman LE, Riley J, Yarovoi
S, Higazi AA and Cines DB: tPA-S(481)A prevents impairment of
cerebrovascular autoregulation by endogenous tPA after traumatic
brain injury by upregulating p38 MAPK and inhibiting ET-1. J
Neurotrauma. 30:1898–1907. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jiang Y, Zeng Y, Huang X, Qin Y, Luo W,
Xiang S, Sooranna SR and Pinhu L: Nur77 attenuates endothelin-1
expression via downregulation of NF-κB and p38 MAPK in A549 cells
and in an ARDS rat model. Am J Physiol Lung Cell Mol Physiol.
311:L1023–Ll035. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Solone X, Wells B and Chrestensen C: MAP
kinases mediate regulation of eNOS through phosphorylation of
different sites. Biochem Mol Biol. 33(S1)(478.10)2019.
|
|
23
|
Lee KM, Lee HJ, Kim MK, Kim HS, Jung GS,
Hur SH, Kim HT, Cho WH, Kim JG, Kim BW, et al: Cilostazol inhibits
high glucose- and angiotensin II-induced type 1 plasminogen
activator inhibitor expression in artery wall and neointimal region
after vascular injury. Atherosclerosis. 207:391–398.
2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lim JH, Woo JS and Shin YW: Cilostazol
protects endothelial cells against lipopolysaccharide-induced
apoptosis through ERK1/2- and P38 MAPK-dependent pathways. Korean J
Intern Med. 24:113–122. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chao TH, Tseng SY, Li YH, Liu PY, Cho CL,
Shi GY, Wu HL and Chen JH: A novel vasculo-angiogenic effect of
cilostazol mediated by cross-talk between multiple signalling
pathways including the ERK/p38 MAPK signalling transduction
cascade. Clin Sci (Lond). 123:147–159. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Medina-Leyte DJ, Domínguez-Pérez M,
Mercado I, Villarreal-Molina MT and Jacobo-Albavera L: Use of human
umbilical vein endothelial cells (HUVEC) as a model to study
cardiovascular disease: A review. Appl Sci. 10(938)2020.
|
|
27
|
Xiong T, Zhang Z, Zheng R, Huang J and Guo
L: N-acetyl cysteine inhibits lipopolysaccharide-induced apoptosis
of human umbilical vein endothelial cells via the p38MAPK signaling
pathway. Mol Med Rep. 20:2945–2953. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cho HY, Park CM, Kim MJ, Chinzorig R, Cho
CW and Song YS: Comparative effect of genistein and daidzein on the
expression of MCP-1, eNOS, and cell adhesion molecules in
TNF-α-stimulated HUVECs. Nutr Res Pract. 5:381–388. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kong LJ, Liu XQ, Xue Y, Gao W and Lv QZ:
Muramyl dipeptide induces reactive oxygen species generation
through the NOD2/COX-2/NOX4 signaling pathway in human umbilical
vein endothelial cells. J Cardiovasc Pharmacol. 71:352–358.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Halcox JP, Schenke WH, Zalos G, Mincemoyer
R, Prasad A, Waclawiw MA, Nour KR and Quyyumi AA: Prognostic value
of coronary vascular endothelial dysfunction. Circulation.
106:653–658. 2002.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Widlansky ME, Gokce N, Keaney JF Jr and
Vita JA: The clinical implications of endothelial dysfunction. J Am
Coll Cardiol. 42:1149–1160. 2003.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: A marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chua BH, Chua CC, Diglio CA and Siu BB:
Regulation of endothelin-1 mRNA by angiotensin II in rat heart
endothelial cells. Biochim Biophys Acta. 1178:201–206.
1993.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Madden JA: Role of the vascular
endothelium and plaque in acute ischemic stroke. Neurology. 79
(Suppl 1):S58–S62. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Goto S: Cilostazol: Potential mechanism of
action for antithrombotic effects accompanied by a low rate of
bleeding. Atheroscler. (Suppl 6):3–11. 2005.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kim KY, Shin HK, Choi JM and Hong KW:
Inhibition of lipopolysaccharide-induced apoptosis by cilostazol in
human umbilical vein endothelial cells. J Pharmacol Exp Ther.
300:709–715. 2002.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kawanabe Y, Takahashi M, Jin X,
Abdul-Majeed S, Nauli AM, Sari Y and Nauli SM: Cilostazol prevents
endothelin-induced smooth muscle constriction and proliferation.
PLoS One. 7(e44476)2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hohlfeld T, Klemm P, Thiemermann C, Warner
TD, Schrör K and Vane JR: The contribution of tumour necrosis
factor-alpha and endothelin-1 to the increase of coronary
resistance in hearts from rats treated with endotoxin. Br J
Pharmacol. 116:3309–3315. 1995.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang H, Park Y, Wu J, Chen Xp, Lee S,
Yang J, Dellsperger KC and Zhang C: Role of TNF-alpha in vascular
dysfunction. Clin Sci (Lond). 116:219–230. 2009.PubMed/NCBI View Article : Google Scholar
|