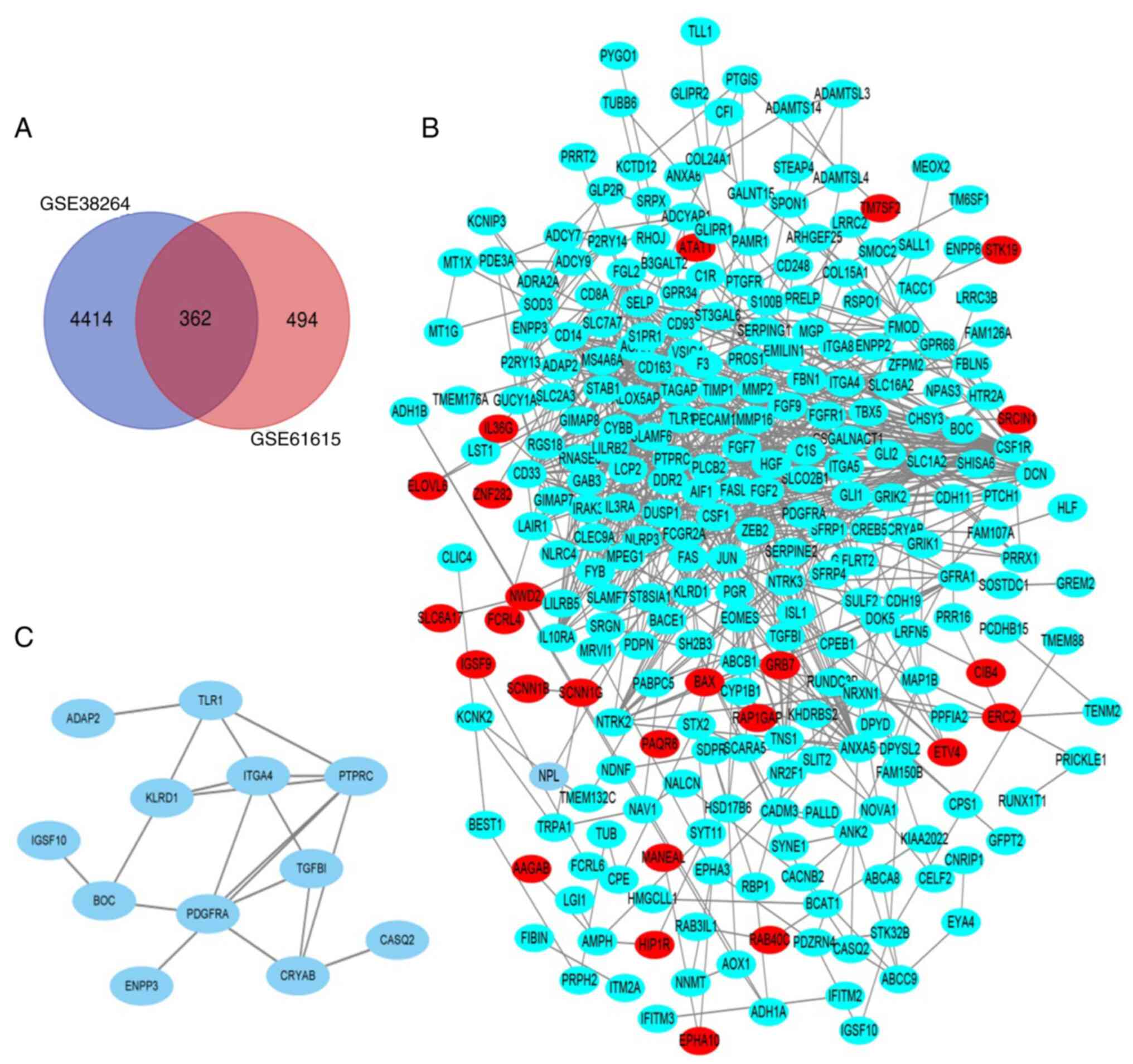
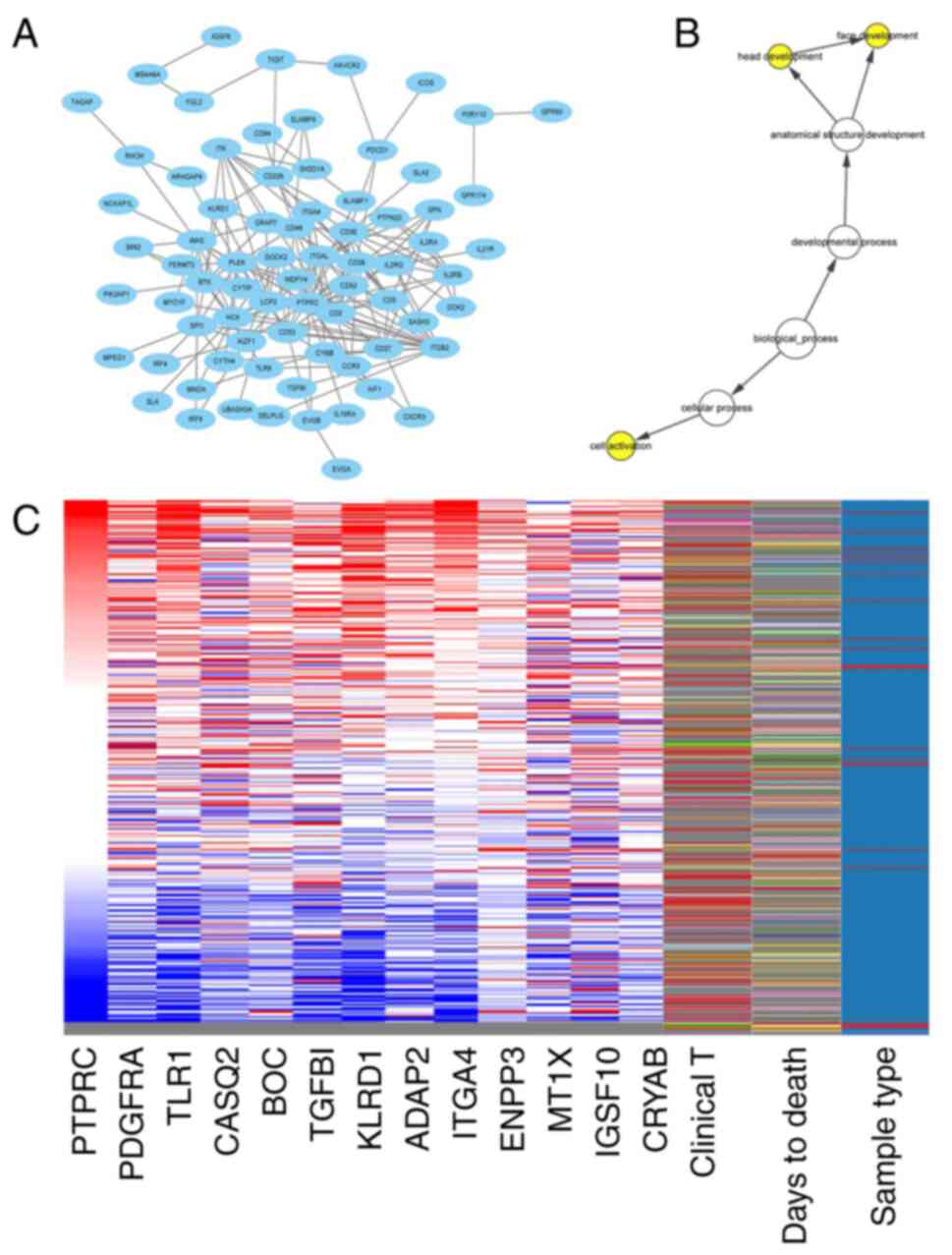
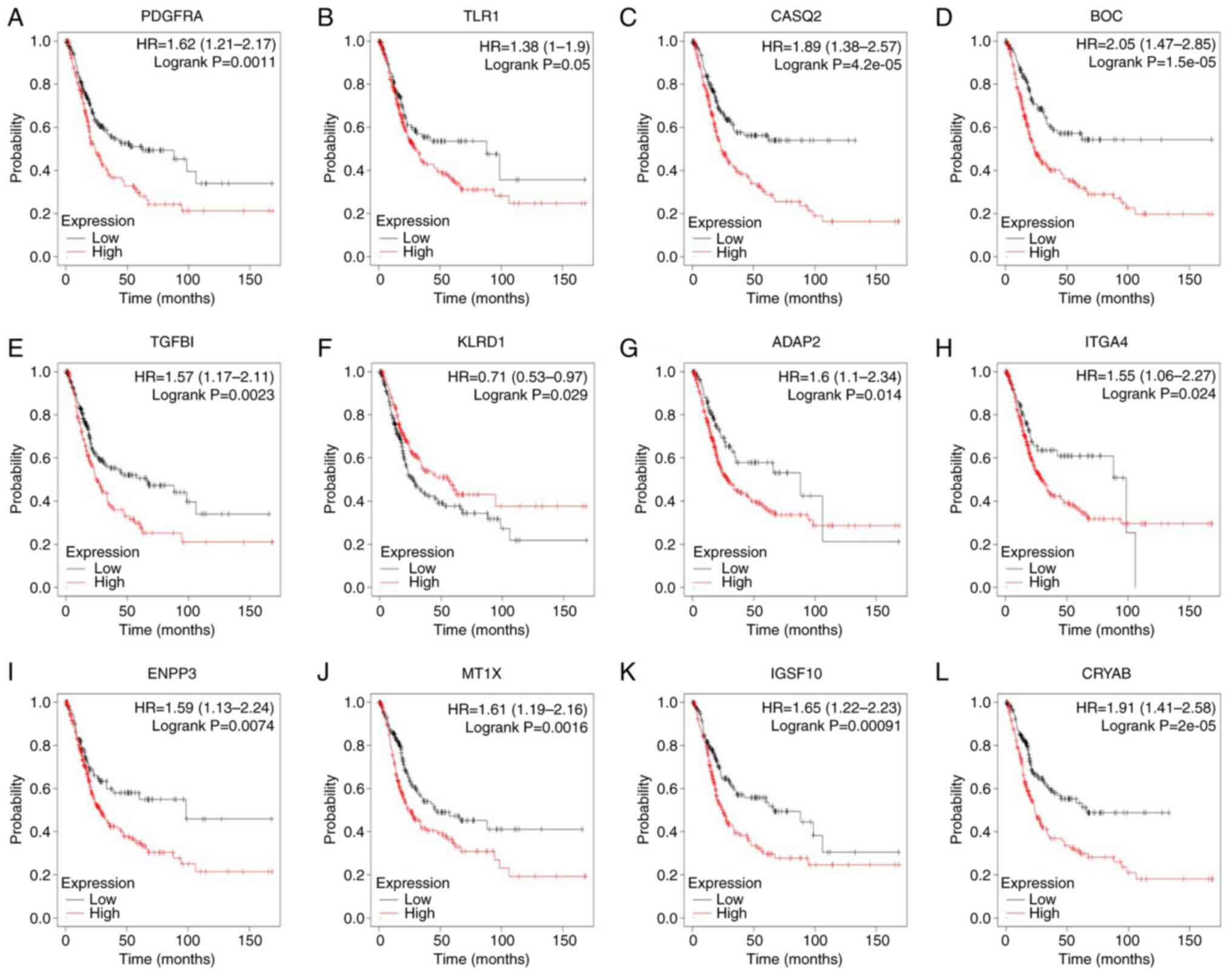
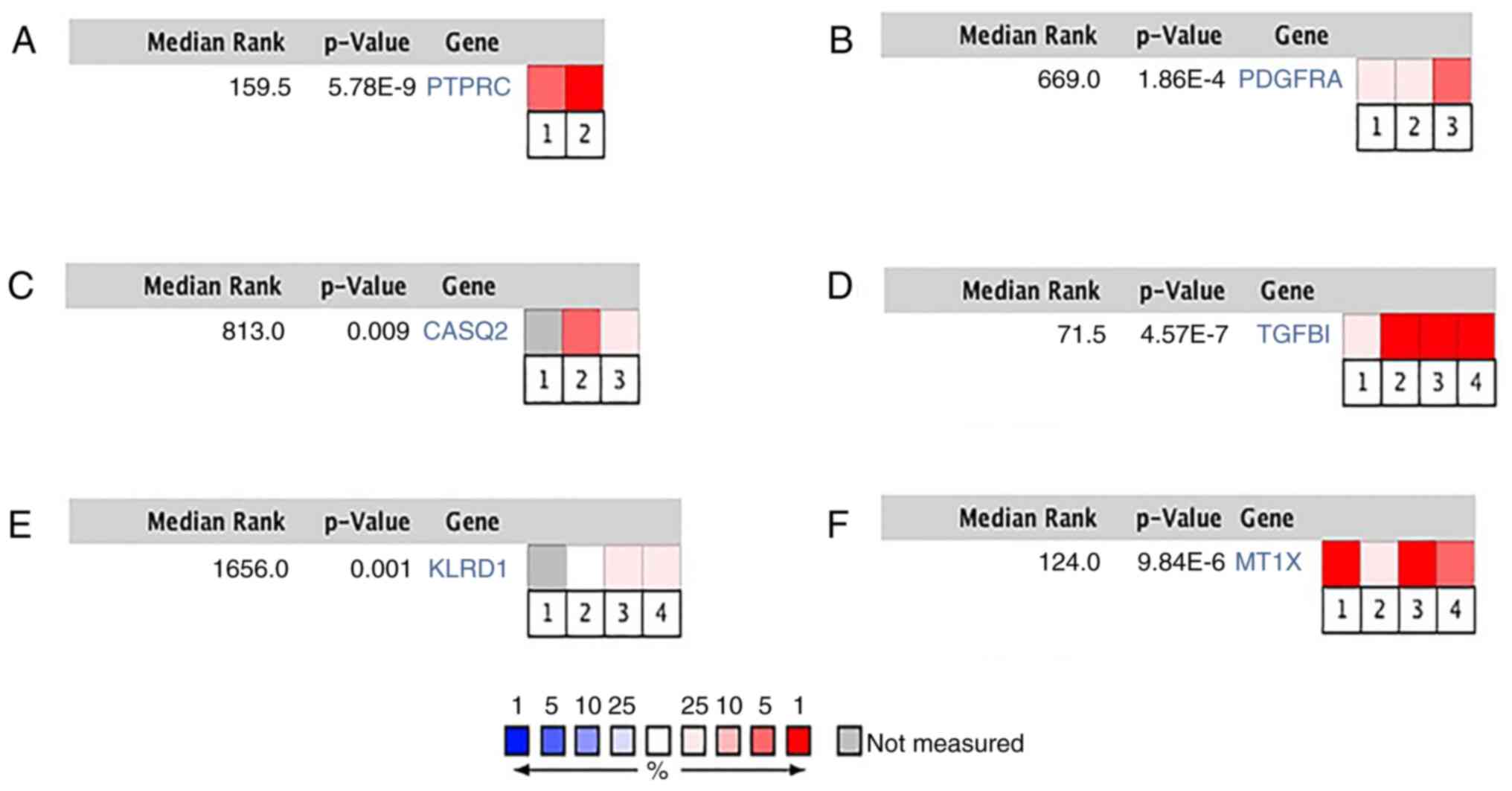
|
1
|
Aghaalikhani N, Rashtchizadeh N, Shadpour
P, Allameh A and Mahmoodi M: Cancer stem cells as a therapeutic
target in bladder cancer. J Cell Physiol. 234:3197–3206.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bhanvadia SK: Bladder cancer survivorship.
Curr Urol Rep. 19(111)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cumberbatch MGK, Jubber I, Black PC,
Esperto F, Figueroa JD, Kamat AM, Kiemeney L, Lotan Y, Pang K,
Silverman DT, et al: Epidemiology of bladder cancer: A systematic
review and contemporary update of risk factors in 2018. Eur Urol.
74:784–795. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Freedman ND, Silverman DT, Hollenbeck AR,
Schatzkin A and Abnet CC: Association between smoking and risk of
bladder cancer among men and women. JAMA. 306:737–745.
2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ma Y and Li MD: Establishment of a strong
link between smoking and cancer pathogenesis through DNA
methylation analysis. Sci Rep. 7(1811)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Islam MO, Bacchetti T and Ferretti G:
Alterations of antioxidant enzymes and biomarkers of
nitro-oxidative stress in tissues of bladder cancer. Oxid Med Cell
Longev. 2019(2730896)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sawicka E, Lisowska A, Kowal P and Długosz
A: The role of oxidative stress in bladder cancer. Postepy Hig Med
Dosw (Online). 69:744–752. 2015.PubMed/NCBI View Article : Google Scholar : (In Polish).
|
|
8
|
Whongsiri P, Pimratana C, Wijitsettakul U,
Sanpavat A, Jindatip D, Hoffmann MJ, Goering W, Schulz WA and
Boonla C: Oxidative stress and LINE-1 reactivation in bladder
cancer are epigenetically linked through active chromatin
formation. Free Radic Biol Med. 134:419–428. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhang X, Han C and He J: Research progress
of oncogene and tumor suppressor gene in bladder cancer. Panminerva
Med. 57:191–200. 2015.PubMed/NCBI
|
|
10
|
Li L, Lei Q, Zhang S, Kong L and Qin B:
Screening and identification of key biomarkers in hepatocellular
carcinoma: Evidence from bioinformatic analysis. Oncol Rep.
38:2607–2618. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Santos M, Martínez-Fernández M, Dueñas M,
García-Escudero R, Alfaya B, Villacampa F, Saiz-Ladera C, Costa C,
Oteo M, Duarte J, et al: In vivo disruption of an Rb-E2F-Ezh2
signaling loop causes bladder cancer. Cancer Res. 74:6565–6577.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhao F, Lin T, He W, Han J, Zhu D, Hu K,
Li W, Zheng Z, Huang J and Xie W: Knockdown of a novel lincRNA
AATBC suppresses proliferation and induces apoptosis in bladder
cancer. Oncotarget. 6:1064–1078. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID Gene Functional Classification Tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8(R183)2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mougeot JL, Noll BD and Bahrani Mougeot
FK: Sjögren's syndrome X-chromosome dose effect: An epigenetic
perspective. Oral Dis. 25:372–384. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yang G, Chen Q, Xiao J, Zhang H, Wang Z
and Lin X: Identification of genes and analysis of prognostic
values in nonsmoking females with non-small cell lung carcinoma by
bioinformatics analyses. Cancer Manag Res. 10:4287–4295.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–101. 2002.PubMed/NCBI
|
|
18
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Szklarczyk D, Gable AL, Nastou KC, Lyon D,
Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al:
The STRING database in 2021: Customizable protein-protein networks,
and functional characterization of user-uploaded gene/measurement
sets. Nucleic Acids Res. 49(D1):D605–D612. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Doncheva NT, Morris JH, Gorodkin J and
Jensen LJ: Cytoscape StringApp: Network analysis and visualization
of proteomics data. J Proteome Res. 18:623–632. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang J, Zhong J, Chen G, Li M, Wu FX and
Pan Y: ClusterViz: A Cytoscape APP for cluster analysis of
biological network. IEEE/ACM Trans Comput Biol Bioinform.
12:815–822. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu J, Lichtenberg T, Hoadley KA, Poisson
LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee
AV, et al: An integrated TCGA pan-cancer clinical data resource to
drive high-quality survival outcome analytics. Cell.
173:400–416.e11. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chan BKC: Data analysis using R
Programming. Adv Exp Med Biol. 1082:47–122. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Rangwala SH, Kuznetsov A, Ananiev V,
Asztalos A, Borodin E, Evgeniev V, Joukov V, Lotov V, Pannu R,
Rudnev D, et al: Accessing NCBI data using the NCBI Sequence Viewer
and Genome Data Viewer (GDV). Genome Res. 31:159–169.
2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Park J, Lee SI, Shin S, Hong JH, Yoo HM
and Kim JG: Genetic profiling of somatic alterations by Oncomine
Focus Assay in Korean patients with advanced gastric cancer. Oncol
Lett. 20(129)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu D, Mao Y, Chen C, Zhu F, Lu W and Ma
H: Expression patterns and clinical significances of ENO2 in lung
cancer: An analysis based on Oncomine database. Ann Transl Med.
8(639)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhu J, Jin L, Zhang A, Gao P, Dai G, Xu M,
Xu L and Yang D: Coexpression analysis of the EZH2 gene using The
cancer genome atlas and oncomine databases identifies coexpressed
genes involved in biological networks in breast cancer,
glioblastoma, and prostate cancer. Med Sci Monit.
26(e922346)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sahin Y, Yucetas U, Ates HA, Erkan E,
Yucetas E, Temiz MZ, Toktas MG, Kadihasanoglu M and Topkaya BC:
Improving the diagnosis of high grade and stage bladder cancer by
detecting increased urinary calprotectin expression in tumor tissue
and tumor-associated inflammatory response. Investig Clin Urol.
60:343–350. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang Q, Zhang T, Wu J, Wen J, Tao D, Wan T
and Zhu W: Prognosis and risk factors of patients with upper
urinary tract urothelial carcinoma and postoperative recurrence of
bladder cancer in central China. BMC Urol. 19(24)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Rozanec JJ and Secin FP: Epidemiology,
etiology and prevention of bladder cancer. Arch Esp Urol.
73:872–878. 2020.PubMed/NCBI(In Spanish).
|
|
33
|
Koie T, Ohyama C, Makiyama K, Shimazui T,
Miyagawa T, Mizutani K, Tsuchiya T, Kato T and Nakane K: Utility of
robot-assisted radical cystectomy with intracorporeal urinary
diversion for muscle-invasive bladder cancer. Int J Urol.
26:334–340. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Saleh AA, Gohar SF, Hemida AS, Elgharbawy
M and Soliman SE: Evaluation of ASPM and TEF gene expressions as
potential biomarkers for bladder cancer. Biochem Genet. 58:490–507.
2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Adelmann TG, Camerota TC, Ceausu AR,
Cimpean AM, Mazzanti M and Raica M: Chloride intracellular channel
protein 1 (CLIC1) ιs over-expressed in muscle invasive urinary
bladder cancer. Anticancer Res. 40:6879–6884. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li X, Shang D, Shen H, Song J, Hao G and
Tian Y: ZSCAN16 promotes proliferation, migration and invasion of
bladder cancer via regulating NF-kB, AKT, mTOR, P38 and other
genes. Biomed Pharmacother. 126(110066)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Barrows D, Feng L, Carroll TS and Allis
CD: Loss of UTX/KDM6A and the activation of FGFR3 converge to
regulate differentiation gene-expression programs in bladder
cancer. Proc Natl Acad Sci USA. 117:25732–25741. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bolón-Canedo V, Alonso-Betanzos A,
López-de-Ullibarri I and Cao R: Challenges and future trends for
microarray analysis. Methods Mol Biol. 1986:283–293.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lu W, Li N and Liao F: Identification of
key genes and pathways in pancreatic cancer gene expression profile
by integrative analysis. Genes (Basel). 10(612)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ma X, Wang P, Xu G, Yu F and Ma Y:
Integrative genomics analysis of various omics data and networks
identify risk genes and variants vulnerable to childhood-onset
asthma. BMC Med Genomics. 13(123)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ma Y, Huang Y, Zhao S, Yao Y, Zhang Y, Qu
J, Wu N and Su J: Integrative genomics analysis reveals a 21q22.11
locus contributing risk to COVID-19. Hum Mol Genet. 30:1247–1258.
2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pal A, Haliti P, Dharmadhikari B, Qi W and
Patra P: Manipulating extracellular matrix organizations and
parameters to control local cancer invasion. IEEE/ACM Trans Comput
Biol Bioinform. 18:2566–2576. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Schaeffer J, Tannahill D, Cioni JM,
Rowlands D and Keynes R: Identification of the extracellular matrix
protein Fibulin-2 as a regulator of spinal nerve organization. Dev
Biol. 442:101–114. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Jones CE, Hammer AM, Cho Y, Sizemore GM,
Cukierman E, Yee LD, Ghadiali SN, Ostrowski MC and Leight JL:
Stromal PTEN regulates extracellular matrix organization in the
mammary gland. Neoplasia. 21:132–145. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Ling L, Tan SK, Goh TH, Cheung E, Nurcombe
V, van Wijnen AJ and Cool SM: Targeting the heparin-binding domain
of fibroblast growth factor receptor 1 as a potential cancer
therapy. Mol Cancer. 14(136)2015.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ngernyuang N, Yan W, Schwartz LM, Oh D,
Liu YB, Chen H and Shao R: A heparin binding motif rich in arginine
and lysine is the functional domain of YKL-40. Neoplasia.
20:182–192. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lauritzen SP, Boye TL and Nylandsted J:
Annexins are instrumental for efficient plasma membrane repair in
cancer cells. Semin Cell Dev Biol. 45:32–38. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhu J, Xu M, Gao M, Zhang Z, Xu Y, Xia T
and Liu S: Graphene oxide induced perturbation to plasma membrane
and cytoskeletal meshwork sensitize cancer cells to
chemotherapeutic agents. ACS Nano. 11:2637–2651. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Peters AA, Milevskiy MJ, Lee WC, Curry MC,
Smart CE, Saunus JM, Reid L, da Silva L, Marcial DL, Dray E, et al:
The calcium pump plasma membrane Ca(2+)-ATPase 2 (PMCA2) regulates
breast cancer cell proliferation and sensitivity to doxorubicin.
Sci Rep. 6(25505)2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Tan L, Cho KJ, Neupane P, Capon RJ and
Hancock JF: An oxanthroquinone derivative that disrupts RAS plasma
membrane localization inhibits cancer cell growth. J Biol Chem.
293:13696–13706. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Jones RL, Serrano C, von Mehren M, George
S, Heinrich MC, Kang YK, Schöffski P, Cassier PA, Mir O, Chawla SP,
et al: Avapritinib in unresectable or metastatic PDGFRA
D842V-mutant gastrointestinal stromal tumours: Long-term efficacy
and safety data from the NAVIGATOR phase I trial. Eur J Cancer.
145:132–142. 2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ranjbaran R, Abbasi M, Rafiei Dehbidi G,
Seyyedi N, Behzad-Behbahani A and Sharifzadeh S: Phosflow
assessment of PDGFRA phosphorylation state: A guide for tyrosine
kinase inhibitor targeted therapy in hypereosinophilia patients.
Cytometry A. 99:784–792. 2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Jašek K, Váňová B, Grendár M, Štanclová A,
Szépe P, Hornáková A, Holubeková V, Plank L and Lasabová Z: BRAF
mutations in KIT/PDGFRA positive gastrointestinal stromal tumours
(GISTs): Is their frequency underestimated? Pathol Res Pract.
216(153171)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Tao X, Cheng L, Li Y, Ci H, Xu J, Wu S and
Tao Y: Expression of CRYAB with the angiogenesis and poor prognosis
for human gastric cancer. Medicine (Baltimore).
98(e17799)2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Qin H, Ni Y, Tong J, Zhao J, Zhou X, Cai
W, Liang J and Yao X: Elevated expression of CRYAB predicts
unfavorable prognosis in non-small cell lung cancer. Med Oncol.
31(142)2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Thompson JA, Motzer RJ, Molina AM,
Choueiri TK, Heath EI, Redman BG, Sangha RS, Ernst DS, Pili R, Kim
SK, et al: Phase I trials of anti-ENPP3 antibody-drug conjugates in
advanced refractory renal cell carcinomas. Clin Cancer Res.
24:4399–4406. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Doñate F, Raitano A, Morrison K, An Z,
Capo L, Aviña H, Karki S, Morrison K, Yang P, Ou J, et al: AGS16F
is a novel antibody drug conjugate directed against ENPP3 for the
treatment of renal cell carcinoma. Clin Cancer Res. 22:1989–1999.
2016.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Trapero C, Jover L, Fernández-Montolí ME,
García-Tejedor A, Vidal A, Gómez de Aranda I, Ponce J, Matias-Guiu
X and Martín-Satué M: Analysis of the ectoenzymes ADA, ALP, ENPP1,
and ENPP3, in the contents of ovarian endometriomas as candidate
biomarkers of endometriosis. Am J Reprod Immunol.
79:2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Mille F, Tamayo-Orrego L, Lévesque M,
Remke M, Korshunov A, Cardin J, Bouchard N, Izzi L, Kool M,
Northcott PA, et al: The Shh receptor Boc promotes progression of
early medulloblastoma to advanced tumors. Dev Cell. 31:34–47.
2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Landskron J, Kraggerud SM, Wik E, Dørum A,
Bjørnslett M, Melum E, Helland Ø, Bjørge L, Lothe RA, Salvesen HB
and Taskén K: C77G in PTPRC (CD45) is no risk allele for ovarian
cancer, but associated with less aggressive disease. PLoS One.
12(e0182030)2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Dargiene G, Streleckiene G, Skieceviciene
J, Leja M, Link A, Wex T, Kupcinskas L, Malfertheiner P and
Kupcinskas J: TLR1 and PRKAA1 gene polymorphisms in the development
of atrophic gastritis and gastric cancer. J Gastrointestin Liver
Dis. 27:363–369. 2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Pradere JP, Dapito DH and Schwabe RF: The
Yin and Yang of Toll-like receptors in cancer. Oncogene.
33:3485–3495. 2014.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Demicco EG, Boland GM, Brewer Savannah KJ,
Lusby K, Young ED, Ingram D, Watson KL, Bailey M, Guo X, Hornick
JL, et al: Progressive loss of myogenic differentiation in
leiomyosarcoma has prognostic value. Histopathology. 66:627–638.
2015.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Skonier J, Neubauer M, Madisen L, Bennett
K, Plowman GD and Purchio AF: cDNA cloning and sequence analysis of
beta ig-h3, a novel gene induced in a human adenocarcinoma cell
line after treatment with transforming growth factor-beta. DNA Cell
Biol. 11:511–522. 1992.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Wen G, Partridge MA, Li B, Hong M, Liao W,
Cheng SK, Zhao Y, Calaf GM, Liu T, Zhou J, et al: TGFBI expression
reduces in vitro and in vivo metastatic potential of lung and
breast tumor cells. Cancer Lett. 308:23–32. 2011.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Calaf GM, Echiburú-Chau C, Zhao YL and Hei
TK: BigH3 protein expression as a marker for breast cancer. Int J
Mol Med. 21:561–568. 2008.PubMed/NCBI
|
|
67
|
Ahmed AA, Mills AD, Ibrahim AE, Temple J,
Blenkiron C, Vias M, Massie CE, Iyer NG, McGeoch A, Crawford R, et
al: The extracellular matrix protein TGFBI induces microtubule
stabilization and sensitizes ovarian cancers to paclitaxel. Cancer
Cell. 12:514–527. 2007.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Brazão-Silva MT, Rodrigues MF, Eisenberg
AL, Dias FL, de Castro LM, Nunes FD, Faria PR, Cardoso SV, Loyola
AM and de Sousa SC: Metallothionein gene expression is altered in
oral cancer and may predict metastasis and patient outcomes.
Histopathology. 67:358–367. 2015.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Blaveri E, Simko JP, Korkola JE, Brewer
JL, Baehner F, Mehta K, Devries S, Koppie T, Pejavar S, Carroll P
and Waldman FM: Bladder cancer outcome and subtype classification
by gene expression. Clin Cancer Res. 11:4044–4055. 2005.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Sanchez-Carbayo M, Socci ND, Lozano J,
Saint F and Cordon-Cardo C: Defining molecular profiles of poor
outcome in patients with invasive bladder cancer using
oligonucleotide microarrays. J Clin Oncol. 24:778–789.
2006.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Stransky N, Vallot C, Reyal F,
Bernard-Pierrot I, de Medina SG, Segraves R, de Rycke Y, Elvin P,
Cassidy A, Spraggon C, et al: Regional copy number-independent
deregulation of transcription in cancer. Nat Genet. 38:1386–1396.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
72
|
Dyrskjøt L, Thykjaer T, Kruhøffer M,
Jensen JL, Marcussen N, Hamilton-Dutoit S, Wolf H and Orntoft TF: .
Identifying distinct classes of bladder carcinoma using
microarrays. Nat Genet. 33:90–96. 2003.PubMed/NCBI View
Article : Google Scholar
|