Introduction
Current radiotherapy for tumors can achieve
noninvasive local control, and plays an important role in cancer
therapy (1). However, despite
advances in irradiation technology, there are cases in which local
control of the tumor fails and recurrence or metastasis occurs.
From the viewpoint of radiation biology, cancer cells that exhibit
intrinsic or extrinsic radioresistance during fractionated
radiation therapy survive after treatment causing recurrence and
metastasis (2). Elucidation of the
mechanisms of radioresistance is an urgent issue for improving the
outcomes of radiation therapy.
The major cell-killing effect of ionizing radiation
is DNA damage [i.e., DNA double-strand breaks (DSBs)] caused by
reactive oxygen species (ROS) (3,4). This
lethal effect of ionizing radiation is modified by linear energy
transfer, dose rate, oxygen concentration, cell cycle distribution,
and the DNA damage response (DDR) of cells (5-9).
In addition, the authors previously reported that an increase in
the number of cancer stem cells (CSCs) among radioresistant cell
lines is involved in the increase in cell survival following
fractionated irradiation (i.e., sub-lethal damage repair) (10-12).
DSB repair is completed mainly by non-homologous end joining (NHEJ)
and homologous recombination (HR) (13). NHEJ is the dominant repair pathway
throughout the cell cycle, and HR functions from the S phase to the
M phase because it requires sister chromosomes (14). HR is frequently reported to be
associated with radioresistance owing to error-free repair
(15), whereas error-prone NHEJ has
been reported to induce genome instability and allow radioresistant
clones to expand (16). However,
the detailed DNA repair pathway in radioresistant cancer cells
remains unclear.
The radioresistant cell lines established after
long-term exposure to fractionated irradiation exhibit clinically
relevant radioresistance (17,18).
It has been suggested that cancer cells acquire radioresistance
during fractionated radiotherapy, resulting in poor prognosis.
However, the mechanisms underlying radioresistance in cancer cells
have not been fully clarified. Herein, to provide means of
overcoming radioresistance, the differences in DDR between
non-radioresistant and radioresistant cell lines using DSB repair
protein-specific inhibitors were investigated, and may serve as a
therapeutic target for eliminating radioresistant cancer cells in
radiotherapy.
Materials and methods
Reagents
The DNA-dependent protein kinase (DNA-PK) inhibitor,
NU7441, and the Rad51 inhibitor, IBR2, were purchased from Selleck
Chemicals and MedChemExpress, respectively. These inhibitors were
dissolved in 1 mM dimethyl sulfoxide (DMSO; i.e., 1.2092 ml for 5
mg NU7441 and 1.2485 ml for 5 mg IBR2; Sigma-Aldrich; Merck KGaA),
administered at the indicated concentrations (i.e., 0.5, 1, 5 and
10 µM for NU7441; and 1, 5, 10 and 15 µM for IBR2) 1 h before
irradiation, and then washed out using phosphate-buffered saline
(PBS) without magnesium and calcium (Takara Bio, Inc.) 24 h after
administration. Based on the pharmacokinetics that 80-90% of most
anticancer drugs are excreted from the body within 24 h after
administration (19), these were
washed out after 24 h.
Cell culture
Human oral squamous carcinoma cell lines, HSC2 and
HSC2-R, were used as models for non-radioresistant cells (HSC2) and
radioresistant cells (HSC2-R). The HSC2 cell line (ID: TKG 0487)
was provided by Cell Resource Center for Biomedical Research,
Institute of Development, Aging and Cancer, Tohoku University
(Sendai, Japan). HSC2-R cells were established by long-term
exposure to fractionated irradiation (i.e., 2 Gy/day, over 60 Gy)
(17). To confirm the origin of the
HSC2 and HSC2-R cells, a short tandem repeat analysis was performed
using a contract research service (BEX Co., Ltd.). The analysis
revealed that both cell lines were the same as HSC2 cells
registered at the National Institutes of Biomedical Innovation,
Health and Nutrition (Osaka, Japan). These cell lines were
maintained at 37˚C and 5% CO2 in Roswell Park Memorial
Institute 1640 medium (Thermo Fisher Scientific, Inc.) supplemented
with 10% heat-inactivated fetal bovine serum (FBS; Japan Bio Serum)
and 1% penicillin/streptomycin (Thermo Fisher Scientific,
Inc.).
Irradiation
The cultured cells were irradiated using an X-ray
generator (MBR-1520R-3; Hitachi, Ltd.) with 0.5-mm aluminum +
0.3-mm copper filters at a distance of 45 cm between the focus and
target (150 kV, 20 mA, 1.0 Gy/min). During the X-ray exposure, the
total dose and dose rate were monitored using a thimble ionization
chamber placed next to the sample. The uncertainty in the absorbed
dose measured by the thimble ionization chamber is ±1%.
Flow cytometric analysis
Apoptotic cells were detected using fluorescein
isothiocyanate (FITC)-conjugated Annexin V (cat. no. 640906;
BioLegend, Inc.) and propidium iodide (PI) (cat. no. 421301;
BioLegend, Inc.). Trypsinized cells were adjusted to a density of
1x106 cells/ml and washed with PBS without magnesium and
calcium. Next, the cells were incubated for 20 min at 4˚C in the
dark after the addition of FITC-Annexin V (5 µl/106
cells) and PI (10 µl/106 cells) and, analyzed by direct
immunofluorescence flow cytometry using CytoFLEX (Beckman Coulter,
Inc.). The percentage of FITC-Annexin V-positive cells was defined
as the percentage of apoptotic cells i.e., FITC-Annexin V (+)/PI
(-) fraction and FITC-Annexin V (+)/PI (+) fraction. To compensate
for histone protein levels, the cell cycle distribution and
phosphorylated-H2A histone family member X (γH2AX)-positive cells
were measured using double staining (20). In particular, trypsinized cells were
fixed with chilled 70% ethanol at -20˚C for 30 min, and then
stained with PI (15 µl/106 cells) and FITC-γH2AX (5
µl/106 cells) in the presence of RNase (0.2 mg/ml,
Nippon Gene Co., Ltd.) at room temperature in the dark for 15 min.
The fluorescence data were analyzed using the CytExpert software
ver. 2.4 (Beckman Coulter, Inc.). The concentrations of NU7441 and
IBR2 used were 5 and 10 µM, respectively. The assessment time
points were 1, 6, 24, and 48 h after 6 Gy irradiation in
combination with NU7441 or IBR2 administration.
Colony formation assay
Clonogenic potency following treatment with 6 Gy
irradiation and/or NU7441 or IBR2 administration was evaluated
using a colony formation assay. For the non-irradiated and
irradiated group, 400 and 10,000 cells were seeded on φ60 cell
culture dishes, respectively. After 6 h of incubation at 37˚C, to
allow the cells to adhere to the bottom of the dish, NU7441 or IBR2
was administered. Subsequently, 1 h after administration, the cells
were irradiated. After 24 h of administration, NU7441 or IBR2 were
washed out with PBS and cells were cultured for an 7-10 additional
days. The cells were then fixed with methanol (Wako Pure Chemical
Industries, Ltd.) for 1 min and stained with Giemsa staining
solution (Wako Pure Chemical Industries) for 2 h. These
aforementioned procedures were performed at room temperature.
Colonies with >50 cells were counted manually. The surviving
fraction for each cell line was calculated from the ratio of the
plating efficiency of the irradiated cells to that of the untreated
group.
Statistical analysis
The significance of the differences between the
control and experimental cultures was determined using the
Tukey-Kramer post hoc test after one-way analysis of variance.
Statistical analyses were performed using Microsoft Excel 2010
(Microsoft Corporation) with the add-on software Statcel v3 (OMS
Publishing). The data shown in this manuscript were obtained by
repeating the experiments thrice, and are presented as the mean ±
SD. P<0.05 was considered to indicate a statistically
significant difference.
Results
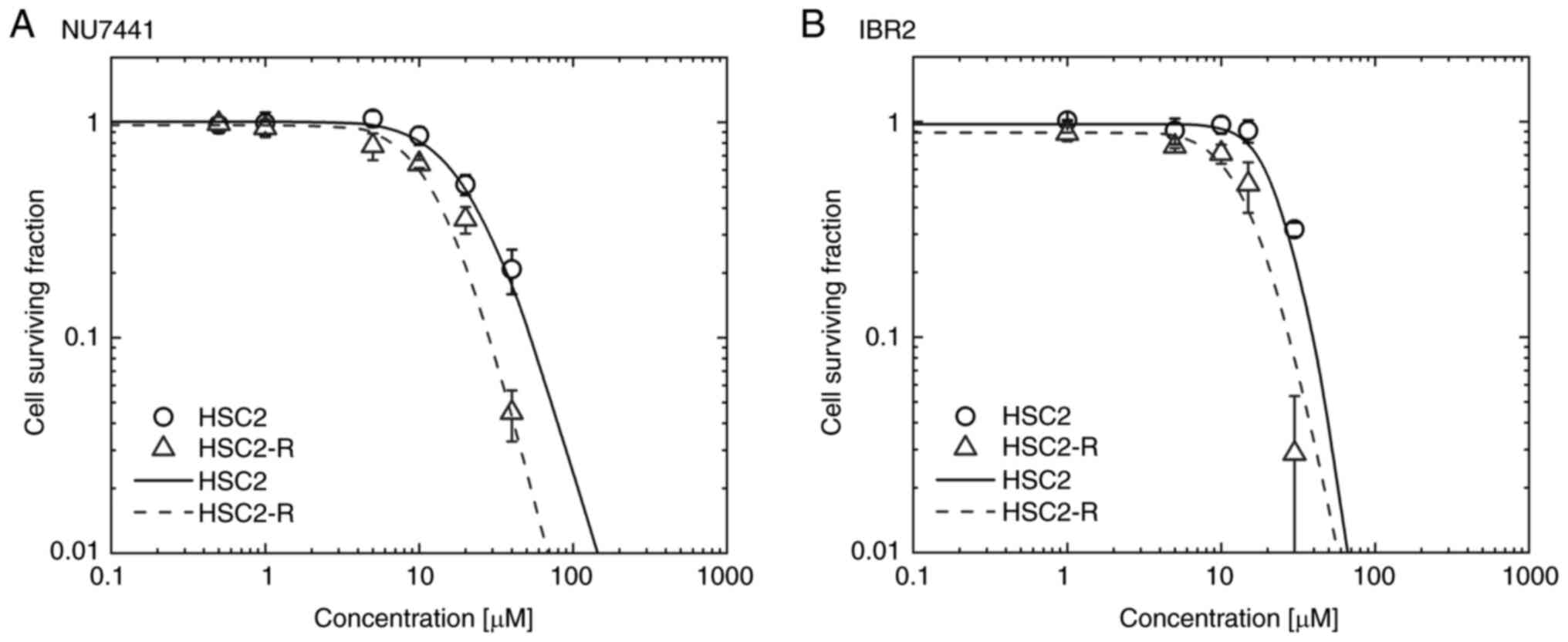
Cytotoxicity of NU7441 and IBR2
To investigate NU7441 and IBR2 toxicity in HSC2 and
HSC2-R cells, the surviving cell fraction at 0.5, 1, 5, and 10 µM
for NU7441 and 1, 5, 10, and 15 µM for IBR2 was measured using a
colony formation assay. The surviving cell fraction was fitted
using log logistic regression, and the 50% inhibitory dose
(IC50) was calculated. Following NU7441 administration,
the cell surviving fraction of HSC2 and HSC2-R cells was decreased
at 10 µM (Fig. 1A), and the
IC50 was 21.21 µM for HSC2 and 13.44 µM for HSC2-R cells
indicating HSC2-R cells were more sensitive to NU7441 than HSC2
cells (Fig. 1A). Following IBR2
administration, the surviving fraction decreased at 20 µM for HSC2
cells and 10 µM for HSC2-R cells (Fig.
1B). The IC50 value indicating sensitivity to IBR2
was 25.75 µM for HSC2 cells and 15.59 µM for HSC2-R cells.
Radioresistant HSC2-R cells were more sensitive to both the
DNA-PKcs inhibitor, NU7441, and the Rad51 inhibitor, IBR2, than
non-radioresistant HSC2 cells.
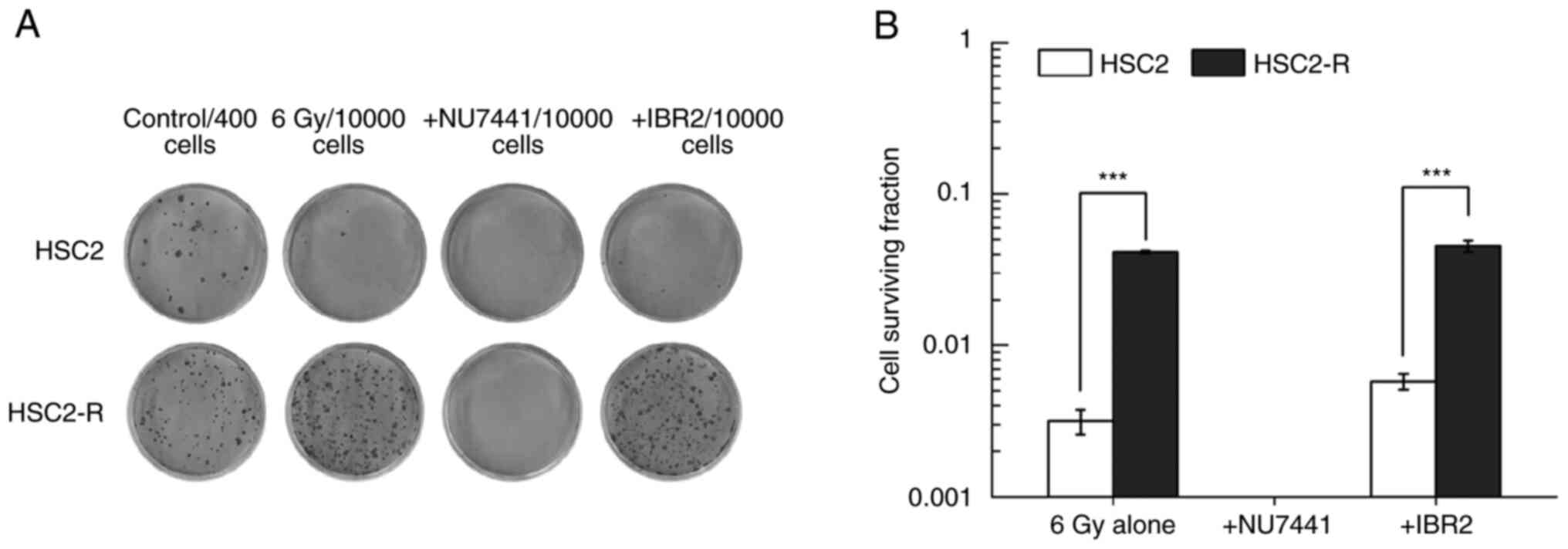
The radiosensitization effects of
NU7441 and IBR2
The surviving cell fraction following treatment with
a combination of 6 Gy irradiation and NU7441 or IBR2 was measured
using a colony formation assay. Both inhibitors, NU7441 and IBR2,
were used at concentrations that did not reduce or slightly affect
cell survival (i.e., 5 µM for NU7441 and 10 µM for IBR2). The
surviving fraction of radioresistant HSC2-R cells was significantly
higher than that of non-radioresistant HSC2 cells following
treatment with 6 Gy irradiation alone (4.14±0.08% vs. 0.32±0.06%)
(Fig. 2A and B). Although HSC2 and HSC2-R cells did not
exhibit decreased cell survival after administration of 5 µM NU7441
alone (Fig. 1A), no countable
colonies were observed following combination treatment with 6 Gy
irradiation and NU7441 (Fig. 2A).
There was no significant difference between 6 Gy irradiation alone
and the combination of 6 Gy irradiation and IBR2 administration in
either cell line.
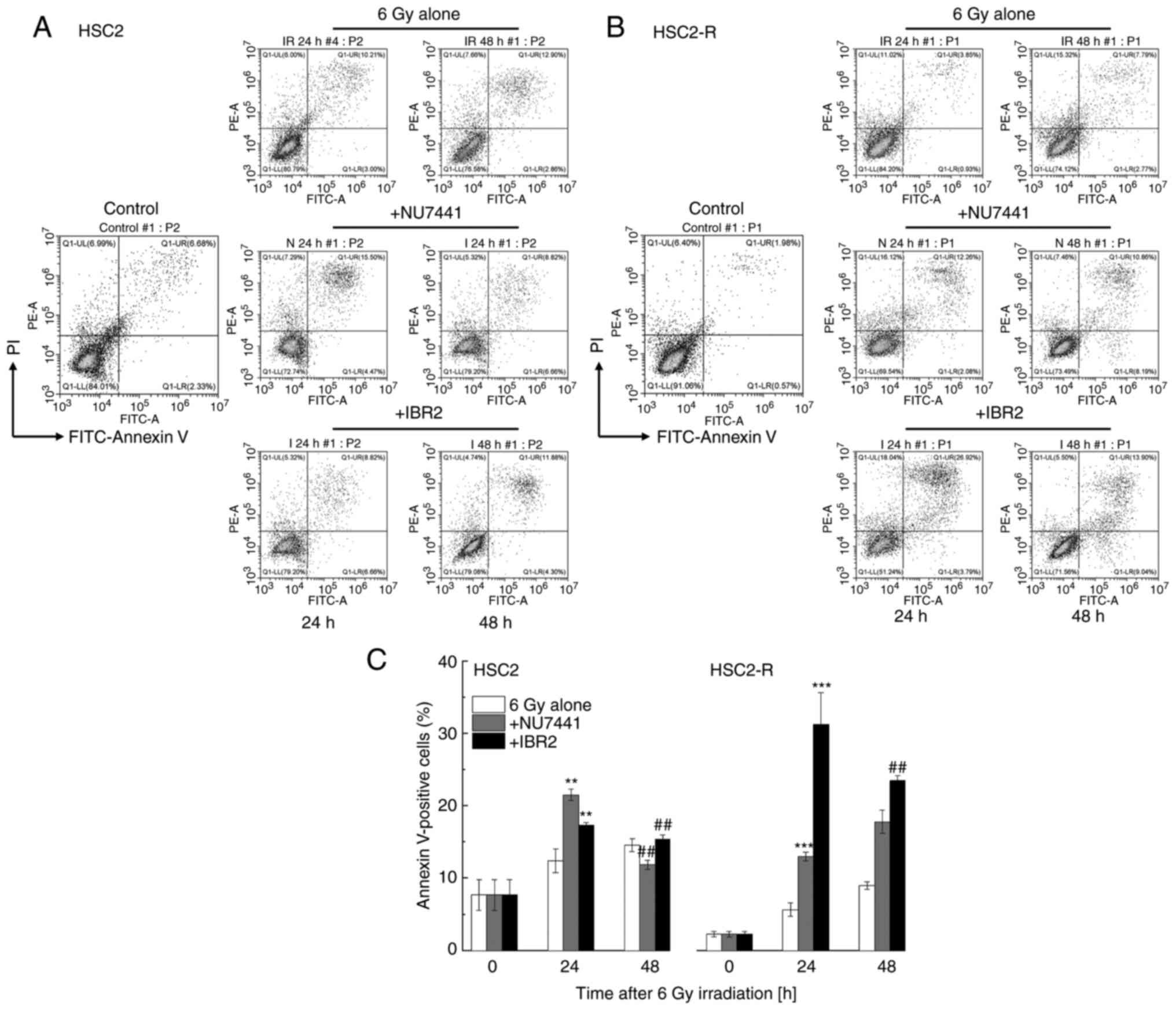
Apoptosis induction under radiation
combined with NU7441 or IBR2
The percentage of apoptotic cells was assessed after
24 and 48 h of treatment using Annexin V and PI staining. Apoptotic
cells were defined as Annexin V-positive cells [i.e., Annexin V
(+)/PI (-) and Annexin V (+)/PI (+) fractions] (Fig. 3A and B). The percentage of apoptotic HSC2-R
cells 24 h after treatment with 6 Gy irradiation alone was
significantly lower than that of apoptotic HSC2 cells (5.66±0.93%
vs. 12.38±1.64%, respectively) (Fig.
3C). At 48 h after 6 Gy irradiation, the percentage of
apoptotic HSC2-R cells significantly increased compared to that at
24 h after treatment with irradiation (8.98±0.50%) but was lower
than that of apoptotic HSC2 cells (14.55±0.88%). Following
treatment with the combination of radiation and NU7441, the
percentage of apoptotic HSC2 cells was significantly increased
(21.52±0.76%) compared to that of HSC2-R cells (13.02±0.61%) at 24
h. At 48 h after treatment, the percentage of apoptotic HSC2 cells
was then decreased (11.86±0.64%), and that of HSC2-R cells was
further increased (17.77±1.61%). Meanwhile, IBR2 treatment for 24 h
induced more apoptosis in HSC2-R cells (31.29±4.36%) than in HSC2
cells (17.32±0.37%), and then decreased at 48 h (15.35±0.59% for
HSC2 cells; 23.48±0.67% for HSC2-R cells) (Fig. 3C). Although the radioresistant
HSC2-R cells were apoptosis-resistant compared with HSC2 cells,
both inhibitors significantly induced apoptosis at least at 24
h.
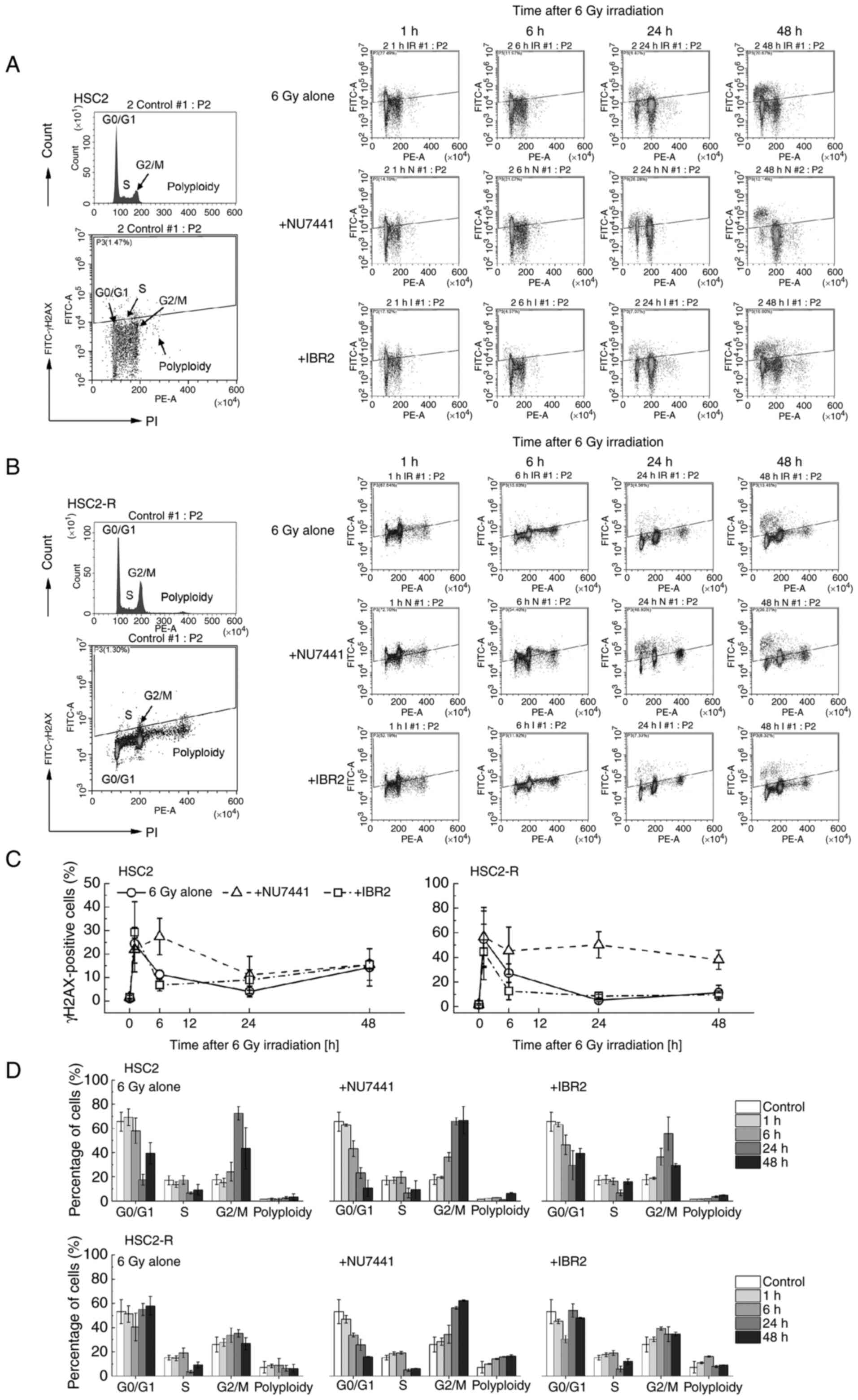
DNA damage response following
administration of NU7441 or IBR2 in combination with 6 Gy
irradiation
To compensate for the amount of histone protein that
depends on DNA content, the cell cycle distribution of cells and
γH2AX dynamics were assessed concurrently using flow cytometry. The
measurement time points were 1, 6, 24, and 48 h after 6 Gy
irradiation in combination with NU7441 or IBR2 administration
(Fig. 4). The cell cycle
distribution-dependent γH2AX-positive fraction was observed 48 h
after treatment, and the G2/M phase fraction was found to be
decreased, especially in the 6 Gy + NU7441 group (Fig. 4A and B). The number of γH2AX-positive HSC2 and
HSC2-R cells increased at 1 h after treatment and then, it was
rapidly decreased after 6 h of treatment in the 6 Gy alone group
and the 6 Gy + IBR2 group, but not in the 6 Gy + NU7441 group
(Fig. 4C). At 24-48 h of treatment
with 6 Gy irradiation alone or in combination with IBR2, the number
of γH2AX-positive HSC2 cells gradually increased, whereas that of
HSC2-R cells did not change. Upon NU7441 administration, the number
of γH2AX-positive HSC2 cells began to decrease between 6 and 24 h,
and then gradually increased until 48 h. Meanwhile, the number of
γH2AX-positive HSC2-R cells slightly decreased at 6 h compared with
that at 1 h, but remained high compared to the 6 Gy alone and 6 Gy
+ IBR2 groups at 24 to 48 h.
The cell cycle distribution of HSC2 cells after 6 Gy
irradiation did not change until 6 h. At 24 h, the percentage of
cells in the G2/M phase was markedly higher than that at 0, 1, and
6 h, while the percentage of cells in the G0/G1 and S phases
decreased at this time point, suggesting that cells were arrested
at the G2/M checkpoint (Fig. 4D).
At 48 h, this arrest tended to be released. The kinetics of the
cell cycle distribution were the same as those of the 6 Gy + IBR2
group. Meanwhile, in the 6 Gy + IBR2 treatment group of HSC2-R
cells, the cells in the G0/G1 phase were decreased and the cells in
the G2/M phase were slightly increased at the 6-h time point, and
then arrest was released after 24 h (Fig. 4D). In the 6 Gy + NU7441 group, the
number of cells in the G0/G1 phase decreased continuously 1 h after
treatment in both cell lines, while the G2/M phase cell population
increased. NU7441 combined with 6 Gy irradiation extended G2/M
arrest. Notably, in HSC2-R cells, polyploid cell populations were
more abundant than in HSC2 cells and increased over time following
NU7441 administration.
Discussion
In the present study, the DDR of radioresistant
HSC2-R cells and the non-radioresistant parental HSC2 cells was
investigated using the DNA-PKcs inhibitor, NU7441, and Rad51
inhibitor, IBR2. DNA-PKcs is a major NHEJ protein, whereas Rad51 is
a major HR protein (21,22). Both DNA repair mechanisms are
important in radiation-induced lethal DSBs. In the present study,
it was determined that radioresistant HSC2-R cells were more
sensitive to both DNA repair inhibitors than non-radioresistant
HSC2 cells, as indicated by the corresponding IC50
values (Fig. 1). In addition, the
combination of NU7441 administration and 6 Gy irradiation
eliminated colony formation in HSC2 and HSC2-R cells, whereas IBR2
administration with 6 Gy irradiation did not affect colony
formation after 24 h of exposure. Because exposure to both
inhibitors for >24 h completely eliminated colony formation,
cell survival at other experimental time points was not
feasible.
It has been reported that oral squamous cell
carcinoma cell lines such as SAS and HSC4 are HR deficient
(23). There are no reports on the
HR capacity of HSC2 and HSC2-R cells, but the lack of colony
formation inhibitory effects of IBR2 administration with 6 Gy
irradiation suggests that it may not be very proficient. In
addition, long-term exposure of HepG2 and HeLa cells to
fractionated radiation was revealed to increase the protein levels
of phosphorylated DNA-PKcs (24).
Based on these findings, the main DNA repair pathway in
radioresistant HSC2-R cells is suggested to be NHEJ. By contrast, a
nasopharyngeal carcinoma cell line exhibited increased levels of
HR-associated repair proteins RPA1, BRCA2, BRCA1, and Rad51 after
long-term exposure to fractionated irradiation (25). These differences are thought to
depend on the phenotype of the parental cell line; however,
alternative epigenetic repair mechanisms may be induced by genomic
instability (26). Indeed, HepG2
cells that acquired radioresistance had a high mutation frequency
at the hypoxanthine phosphoribosyltransferase locus compared
to parental cells (27). It should
be noted that genome instability is also induced by error-prone
backup DSB repair mechanisms, that is, alternative-end joining and
single-strand annealing (28), but
its association with radioresistance of cells has not yet been
reported.
The apoptotic fraction of HSC2-R cells following 6
Gy irradiation was lower than that of HSC2 cells, and both NU7441
and IBR2 significantly promoted apoptosis in both cell lines 24 h
after administration (Fig. 3).
Several studies have reported the induction of apoptosis by the
combined use of Rad51 inhibitor and other cytotoxic agents. B02, a
RAD51-targeting agent, synergistically increased cytotoxicity
stimulated by doxorubicin (29) or
AZD1775(30). In addition, Rad51
inhibition enhanced radiation-induced cell death (31,32).
In HSC2 cells, the apoptotic fraction decreased at 48 h after
administration of both inhibitors compared with that at 24 h, and
this phenomenon was observed only after IBR2 administration in
HSC2-R cells. According to recent views, apoptosis is reversible, a
process referred to as anastasis, and caspases outside of apoptosis
promote tumor repopulation (33).
Therefore, the decrease in apoptotic cell fraction may be due to
anastasis. Most anticancer agents are excreted from the human body
within 24 h. Based on the pharmacokinetics, the inhibitors were
washed out after 24 h. Such experimental manipulations may also
affect the fraction of apoptotic cells. In addition, exposure to
inhibitors was only performed for 24 h in the experiment of the
present study. Both inhibitors should be investigated at further
experimental time points in the future. A further limitation of the
present study, was that the apoptotic cell fraction under the
treatment of NU7441 or IBR2 without 6 Gy irradiation was not
confirmed. The apoptotic effects of NU7441 and IBR2 could modify
the cell surviving fraction; however, the colony formation was not
significantly decreased at the concentration used in the present
study (Fig. 1). Regarding the
concentration i.e., 5 µM NU7441 and 10 µM IBR2 used, previous
studies have reported that these concentrations do not
significantly induce apoptosis (34,35).
The cell cycle distribution and γH2AX dynamics of
HSC2 and HSC2-R cells was assessed by flow cytometric analysis. The
γH2AX-positive fraction of HSC2 and HSC2-R cells was increased 1 h
after treatment, and then immediately reduced (i.e., 6 h after
treatment) (Fig. 4). Only NU7441
administration maintained the size of the fractions, suggesting
that NHEJ was predominant in the early phase of the repair process.
Strong G2/M phase arrest was observed following NU7441
administration in HSC2 and HSC2-R cells. Although HSC2 cells
exhibited reduced γH2AX-positive fractions, those of γH2AX-positive
HSC2-R cells remained high from 24 to 48 h. Following IBR2
administration in HSC2-R cells, cell cycle distribution and γH2AX
expression was not significantly altered compared with the
treatment with 6 Gy irradiation alone (Fig. 4C and D). The percentage of γH2AX-positive HSC2
cells increased again at 48 h, but not that of HSC2-R cells. HR is
the predominant repair pathway at the later phase of the repair
process that causes secondary replication-induced DSBs (36). It has been reported that DNA-PKcs
can regulate the selection of the DSB repair pathway according to
the cell cycle phase. In the S and G2 phases of the cell cycle,
DNA-PKcs dissociation from DSBs by autophosphorylation results in
the selection of the HR repair pathway to promote the end resection
of DSB sites (37,38). In addition, inducing HR deletions
results in greater radioresistance compared to the parental cell
line (39), and restoring the HR
mechanism in HR-deficient cancers renders them radiosensitive
(40). These observations and
studies suggest that the repair mechanism of HSC2-R cells strongly
depends on DNA-PKcs phosphorylation, and a weak repair mechanism
during the S and G2/M phases. In the present study, the protein
expression of DDR signaling pathways including targets of NU7441 or
IBR2, was not investigated. The protein expression related to cell
cycle regulation and DNA repair should be investigated in the
future. Furthermore, the potential radioresistance mechanisms in
HSC2-R cells may be associated with an increase in the polyploidy
fraction. It has been reported that the majority of polyploid giant
cancer cells (PGCCs), induced by irradiation, undergo cell death,
but some PGCCs exhibit proliferative capacity and undergo neosis,
which may result in tumor repopulation (41). NU7441 and IBR2 increased the
polyploid fraction in HSC2-R cells. However, the fate of the
polyploid fraction of cells requires long-term observation;
therefore, this population needs to be tracked further in the
future.
In the present study, using inhibitors targeting two
major DSB repair proteins, the mechanism of radioresistance
acquisition and targets for overcoming radioresistance of cells
were revealed. The DNA-PKcs specific inhibitor, NU7441, markedly
decreased colony formation in radioresistant HSC2-R cells by
suppressing DDR in the S and G2/M phases. Meanwhile, the Rad51
specific inhibitor, IBR2, promoted apoptosis in radioresistant
HSC2-R cells, but colony formation and DDR were not altered
compared to 6 Gy irradiation alone, suggesting that HR may not be
the primary DSB repair pathway in HSC2-R cells. Based on the
findings of the present study, the DSB repair pathway of
radioresistant cells depends on NHEJ. Therefore, it is suggested
that targeting DNA-PKcs aids in eliminating radioresistant cancer
cells, and thus overcoming treatment resistance and preventing
recurrence. However, the detailed molecular pathways underlying
this mechanism should be studied in the future.
Acknowledgements
We would like to thank Dr Tomita (Kagoshima
University, Kagoshima, Japan) and Dr Kuwahara (Tohoku Medical and
Pharmaceutical University, Sendai, Japan) for donating the
radioresistant cells.
Funding
Funding: Funding was provided by Japan Society for the Promotion
of Science (grant nos. 20K16814 and 19K08141). The funders had no
role in the study design, data collection and analysis, decision to
publish, or preparation of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KOh and RS conceived the study, participated in its
design and coordination, and drafted the manuscript. KOh, RS and KH
participated in the experiments, and performed the analysis and
interpretation of the data. ET, YH, MF and KOk critically reviewed
the article for important intellectual content. KH, ET, YH, MF and
KOk confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Atun R, Jaffray DA, Barton MB, Bray F,
Baumann M, Vikram B, Hanna TP, Knaul FM, Lievens Y, Lui TY, et al:
Expanding global access to radiotherapy. Lancet Oncol.
16:1153–1186. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Olivares-Urbano MA, Griñán-Lisón C,
Marchal JA and Núñez MI: CSC Radioresistance: A therapeutic
challenge to improve radiotherapy effectiveness in cancer. Cells.
9(1651)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bajinskis A, Natarajan AT, Erixon K and
Harms-Ringdahl M: DNA double strand breaks induced by the indirect
effect of radiation are more efficiently repaired by non-homologous
end joining compared to homologous recombination repair. Mutat Res.
756:21–29. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Vignard J, Mirey G and Salles B:
Ionizing-radiation induced DNA double strand breaks: A direct and
indirect lighting up. Radiother Oncol. 108:362–369. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hawkins RB and Inaniwa T: A
microdosimetric-kinetic model for cell killing by protracted
continuous irradiation including dependence on LET I: Repair in
cultured mammalian cells. Radiat Res. 180:584–594. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Matsuya Y, McMahon SJ, Tsutsumi K, Sasaki
K, Okuyama G, Yoshii Y, Mori R, Oikawa J, Prise KM and Date H:
Investigation of dose-rate effects and cell-cycle distribution
under protracted exposure to ionizing radiation for various
dose-rates. Sci Rep. 8(8287)2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Qi XS, Pajonk F, McCloskey S, Low DA,
Kupelian P, Steinberg M and Sheng K: Radioresistance of the breast
tumor is highly correlated to its level of cancer stem cell and its
clinical implication for breast irradiation. Radiother Oncol.
124:455–461. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Matsuya Y, McMahon SJ, Butterworth KT,
Naijo S, Nara I, Yachi Y, Saga R, Ishikawa M, Sato T, Date H and
Prise KM: Oxygen enhancement ratios of cancer cells after exposure
to intensity modulated X-ray fields: DNA damage and cell survival.
Phys Med Biol. 66:2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kim RK, Suh Y, Cui YH, Hwang E, Lim EJ,
Yoo KC, Lee GH, Yi JM, Kang SG and Lee SJ: Fractionated
radiation-induced nitric oxide promotes expansion of glioma
stem-like cells. Cancer Sci. 104:1172–1177. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fukui R, Saga R, Matsuya Y, Tomita K,
Kuwahara Y, Ohuchi K, Sato T, Okumura K, Date H, Fukumoto M and
Hosokawa Y: Tumor radioresistance caused by radiation-induced
changes of stem-like cell content and sub-lethal damage repair
capability. Sci Rep. 12(1056)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Saga R, Matsuya Y, Takahashi R, Hasegawa
K, Date H and Hosokawa Y: Analysis of the high-dose-range
radioresistance of prostate cancer cells, including cancer stem
calls, based on a stochastic model. J Radiat Res. 60:298–307.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Murata K, Saga R, Monzen S, Tsuruga E,
Hasegawa K and Hosokawa Y: Understanding the mechanism underlying
the acquisition of radioresistance in human prostate cancer cells.
Oncol Lett. 17:5830–5838. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shibata A: Regulation of repair pathway
choice at two-ended DNA double-strand breaks. Mutat Res.
803-805:51–55. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shibata A and Jeggo PA: DNA double-stand
break repair in a cellular context. Clin Oncol (R Coll Radiol).
26:243–249. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Karanam K, Kafri R, Loewer A and Lahav G:
Quantitative live cell imaging reveals a gradual shift between DNA
repair mechanisms and a maximal use of HR in mid S phase. Mol Cell.
47:320–329. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang Y, Xu H, Liu T, Huang M, Butter PP,
Li C, Zhang L, Kao GD, Gong Y, Maity A, et al: Temporal DNA-PK
activation drives genomic instability and therapy resistance in
glioma stem cells. JCI insight. 3(e98096)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kuwahara Y, Mori M, Oikawa T, Shimura T,
Ohtake Y, Mori S, Ohkubo Y and Fukumoto M: The modified
high-density survival assay is the useful tool to predict the
effectiveness of fractionated radiation exposure. J Radiat Res.
51:297–302. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kuwahara Y, Roudkenar MH, Urushihara Y,
Saito Y, Tomita K, Roushandeh AM, Sato T, Kurimasa A and Fukumoto
M: Clinically relevant radioresistant cell line: A simple model to
understand cancer radioresistance. Med Mol Morphol. 50:195–204.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liston DR and Davis M: Clinically relevant
concentrations of anticancer drugs: A guide for nonclinical
studies. Clin Cancer Res. 23:3489–3498. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Huang X, Okafuji M, Traganos F, Luther E,
Holden E and Darzynkiewicz Z: Assessment of histone H2AX
phosphorylation induced by DNA topoisomerase Ⅰ and Ⅱ inhibitors
topotecan and mitoxantrone and by the DNA corss-linking agent
cisplatin. Cytometry A. 58:99–110. 2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Difilippantonio MJ, Zhu J, Chen HT, Meffre
E, Nussenzweig MC, Max EE, Ried T and Nussenzweig A: DNA repair
protein Ku80 suppresses chromosomal aberrations and malignant
transformation. Nature. 404:510–514. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Tachon G, Cortes U, Guichet PO, Rivet P,
Balbous A, Masliantsev K, Berger A, Boissonnade O, Wager M and
Karayan-Tapon L: Cell cycle changes after glioblastoma stem cell
irradiation: The major role of RAD51. Int J Mol Sci.
19(3018)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wurster S, Hennes F, Parplys AC, Seelbach
JI, Mansour WY, Zielinski A, Petersen C, Clauditz TS, Münscher A,
Friedl AA and Borgmann K: PARP1 inhibition radiosensitizes HNSCC
cells deficient in homologous recombination by disabling the DNA
replication fork elongation response. Oncotarget. 7:9732–9741.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shimura T, Kakuda S, Ochiai Y, Nakagawa H,
Kuwahara Y, Takai Y, Kobayashi J, Komatsu K and Fukumoto M:
Acquired radioresistance of human tumor cells by
DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene.
29:4826–4837. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Z, Zuo W, Zeng Q, Li Y, Lu T, Bu Y
and Hu G: The homologous recombination repair pathway is associated
with resistance to radiotherapy in nasopharyngeal carcinoma. Int J
Biol Sci. 16:408–419. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bakhoum SF and Cantley LC: The
multifaceted role of chromosomal instability in cancer and its
microenvironment. Cell. 174:1347–1360. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kuwahara Y, Roudkenar MH, Urushihara Y,
Saito Y, Tomita K, Roushandeh AM, Sato T, Kurimasa A and Fukumoto
M: X-ray induced mutation frequency at the Hypoxanthine
Phosphoribosyltransferase locus in clinically relevant
radioresistant cells. Int J Med Phs Clin Eng Radiat Oncol.
6:377–391. 2017.
|
|
28
|
Ceccaldi R, Rondinelli B and D'Andrea AD:
Repair pathway choices and consequences at the double-strand break.
Trends Cell Biol. 26:52–64. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Schurmann L, Schumacher L, Roquette K,
Brozovic A and Fritz G: Inhibition of the DSB repair protein RAD51
potentiates the cytotoxic efficacy of doxorubicin via promoting
apoptosis-related death pathways. Cancer Lett. 520:361–373.
2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lindemann A, Patel AA, Tang L, Tanaka N,
Gleber-Netto FO, Bartels MD, Wang L, McGrail DJ, Lin SY, Frank SJ,
et al: Combined Inhibition of Rad51 and wee1 enhances cell killing
in HNSCC through induction of apoptosis associated with excessive
DNA damage and replication stress. Mol Cancer Ther. 20:1257–1269.
2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sak A, Stueben G, Groneberg M, Bocker W
and Struschke M: Targeting of Rad51-dependent homologous
recombination: Implications for the radiation sensitivity of human
lung cancer cell lines. Br J Cancer. 92:1089–1097. 2005.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wéra AC, Lobbens A, Stoyanov M, Lucas S
and Michiels C: Radiation-induced synthetic lethality: Combination
of poly(ADP-ribose) polymerase and RAD51 inhibitors to sensitize
cells to proton irradiation. Cell Cycle. 18:1770–1783.
2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mirzayans R and Murray D: Intratumor
heterogeneity and therapy resistance: Contributions of dormancy,
apoptosis reversal (anastasis) and cell fusion to disease
recurrence. Int J Mol Sci. 21(1308)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yanai M, Makino H, Ping B, Takeda K,
Tanaka N, Sakamoto T, Yamaguchi K, Kodani M, Yamasaki A, Igishi T
and Shimizu E: DNA-PK inhibition by NU7441 enhances
chemosensitivity to topoisomerase inhibitor in non-small cell lung
carcinoma cells by blocking DNA damage repair. Yonago Acta Med.
60:9–15. 2017.PubMed/NCBI
|
|
35
|
Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G,
Qiu XL, Chen CF, Hu CM, Goldblatt E, et al: A novel small molecule
RAD51 inactivator overcomes imatinib-resistance in chronic myeloid
leukaemia. EMBO Mol Med. 5:353–365. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Groth P, Orta ML, Elvers I, Majumder MM,
Lagerqvist A and Helleday T: Homologous recombination repairs
secondary replication induced DNA double-strand breaks after
ionizing radiation. Nucleic Acids Res. 40:6585–6594.
2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yue X, Bai C, Xie D, Ma T and Zhou PK:
DNA-PKcs: A multi-faceted player in DNA damage response. Front
Genet. 11(607428)2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shibata A, Conrad S, Birraux J, Geuting V,
Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G,
Löbrich M and Jeggo PA: Factors determining DNA double-strand break
repair pathway choice in G2 phase. EMBO J. 30:1079–1092.
2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Frankenberg-Schwager M, Gebauer A, Koppe
C, Wolf H, Pralle E and Frankenberg D: Single-strand annealing,
conservative homologous recombination, nonhomologous DNA end
joining, and the cell cycle-dependent repair of DNA double-strand
breaks induced by sparsely or densely ionizing radiation. Radiat
Res. 171:265–273. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
40
|
Barazas M, Gasparini A, Huang Y,
Küçükosmanoğlu A, Annunziato S, Bouwman P, Sol W, Kersbergen A,
Proost N, de Korte-Grimmerink R, et al: Radiosensitivity is an
acquired vulnerability of PARPi-resistant BRCA1-deficient tumors.
Cancer Res. 79:452–460. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang Z, Feng X, Deng Z, Chen J, Wang Y,
Zhao M, Zhao Y, He S and Huang Q: Irradiation-induced polyploid
giant cancer cells are involved in tumor cell repopulation via
neosis. Mol Oncol. 15:2219–2234. 2021.PubMed/NCBI View Article : Google Scholar
|