1. Introduction
Type 1 diabetes mellitus (T1DM) is a chronic
metabolic disorder of autoimmune nature, characterized by the
selective and progressive destruction of insulin-producing
pancreatic β-cells by autoreactive T cells, leading to loss of
secretory function and insufficient hormone production (1-3).
This immune system dysregulation is caused by the combined action
of genetic, epigenetic, and environmental factors, through a
complex and varied network of interactions (4,5).
In mammals, insulin is an essential anabolic hormone
with multiple effects on glucose, lipid, protein, and mineral
metabolism. It is required for the entry of glucose into muscle and
fat cells, in addition to stimulating the liver to store glucose in
the form of glycogen and to synthesize fatty acids. Insulin is also
required to stimulate amino acid uptake, inhibit the breakdown of
fat in adipose tissue, and stimulate the uptake of potassium by
cells (6).
Given its importance, glucose metabolism needs to be
strictly regulated to ensure a sufficient energy supply to vital
tissues and organs, but not supply an excess. In this context, the
liver plays a critical role in glucose metabolism homeostasis,
regulating multiple metabolic pathways, including glycogenesis,
glycogenolysis, glycolysis, and gluconeogenesis (7). Thus, the controlled expression of
genes encoding the liver enzymes involved in these pathways plays a
critical role in the complex network of interactions between
multiple organs. Therefore, the regulation of the expression of
these genes is essential for the maintenance of long-term glucose
metabolism homeostasis (8).
Control of blood glucose levels depends on a complex
and tightly controlled network of hormones and neuropeptides
released by the brain, pancreas, liver, intestine, adipose tissue,
and muscles (9,10). However, the pancreas plays a
critical role within this network since the pancreatic islets
contain insulin-producing β-cells and glucagon-producing α-cells.
Through these two hormones, the pancreas regulates glucose
metabolism, where insulin lowers blood glucose levels, while its
counterpart, glucagon, helps maintain necessary blood glucose
levels; thus, acting together antagonistically, they maintain
energy homeostasis (9). The loss of
this balance, due to the absence or dysfunction of β-cells,
generates insulinogenic and hyperglycemia, which are the classic
phenotypes of T1DM (11).
Evidence suggests that genetic characteristics,
notably the genetics of HLA, specifically of the molecules DQ2 and
DQ8, are a necessary but insufficient condition for the development
of T1DM. Thus, other additional factors, including those related to
environmental stress, the individual's microbiome, and the
epigenome, are also important in the pathogenesis of the disease
(1,12). The increase in the incidence of T1DM
in recent decades cannot be explained solely based on genetic
factors, although HLA-DR3-DQ2 or HLA-DR4-DQ8 haplotypes, or both,
are considered risk factors for the disease (13,14).
This suggests that the origin of the disease depends
on the interaction between multiple factors, involving predisposing
genes and exposure to environmental factors that act as triggers.
Evidence of this is that these processes are heterogeneous even
among genetically related individuals, suggesting an interaction
between a genetic predisposition and other non-genetic factors,
including viral infections (15).
Three phenomena acting in parallel contribute to the onset of T1DM:
Early childhood lifestyle and factors, the dynamics of immune
system development, and the maturation of the gut microbiome
(16).
The potential of viral infections to trigger
pancreatic islet autoimmunity in patients with T1DM has been a
longstanding hypothesis. There is an increasing body of evidence
that implicates persistent infections by certain viruses,
particularly human enteroviruses, as more likely environmental
triggers that may contribute to different stages of disease
development (17).
Studies show that infections with certain viruses
play a crucial role in the pathogenesis of T1DM and can determine
whether a genetically susceptible individual will develop this
metabolic disease (3,18). Growing evidence points to a causal
relationship between infections by certain viruses, especially
human enteroviruses, and disease onset. Although the participation
of viruses in the etiology of T1DM has been extensively explored,
definitive proof of this causality is still lacking (18,19).
However, there are reports of viral infections in several
individuals with autoimmunity and damage to insulin-producing
pancreatic islet β-cells, in which enteroviruses are the prime
suspects (20).
Studies performed in Finland show that viral effects
can encompass all stages of the β-cell damage process, including
those that precede the onset of the disease (18). Among the strongest candidate
viruses, enteroviruses detected in the pancreas of patients with
T1DM stand out. In addition, epidemiological research, including a
meta-analysis of 56 studies, has shown more infections by these
enteroviruses in diabetic patients than in healthy subjects
(18,21,22).
Enteroviruses cause T1DM during experimental
infection of animals. Epidemiological studies in humans indicate
that they are associated with an increased risk of disease and have
been detected in the pancreas of diabetic patients (23,24).
These results provide consistent evidence that enteroviruses
trigger the autoimmune response against pancreatic islets resulting
in inflammation, destruction of β-cells, and initiation of T1DM
(20,25). Among the enteroviruses, Coxsackie B
appears to be the primary candidate for the trigger or acceleration
of islet β-cell autoimmunity in genetically susceptible
individuals, resulting in T1DM (26).
The present review presents and discusses the most
recent advances regarding the role of viruses as an environmental
trigger, contributing to the loss of immune self-tolerance and
triggering autoimmunity. A literature review was conducted using
the PubMed, Embase, and Scopus databases, employing the
descriptors: Type 1 diabetes, pathogenesis, virus, and associated
terms with the appropriate Boolean modifiers. Studies involving
humans or animals, original articles, and review articles regarding
meta-analysis published in recent years were included, while
abstracts, conference papers, editorials, and unrelated studies
were excluded.
2. Pathogenesis of T1DM
T1DM is an organ-specific autoimmune disease that
leads to the destruction of insulin-producing pancreatic β-cells,
where the balance between regulatory and effector T cells
determines the risk of developing the disease, activation time, and
duration (27). Its origin is
multifactorial, and the pathogenesis is complex, involving humoral
and cellular immune response abnormalities (28). The mechanisms that trigger the
disease involve the production of autoantibodies against
autoantigens produced by insulin-producing pancreatic islet
β-cells. Although these autoantibodies do not play a significant
role in the destruction of these cells, they serve as indicators of
an ongoing destructive process and as a strong predictive marker of
the future development of T1DM (1,29,30).
Pancreatic islet β cells produce self-antigens among them: Islet
cell antigens (ICA), glutamic acid decarboxylase 65-kilodalton
(GAD65), insulinoma antigen-2 (IA-2), insulin antigen (IA) and zinc
transporter 8 antigen (ZnT8A), which induce the production of their
respective autoantibodies (31,32).
A long-term follow-up longitudinal study showed that
pancreatic islet autoimmunity precedes the onset of T1DM in
prediabetic children and the presence of autoantibodies does not
necessarily lead to overt disease with clinical symptoms (33). The disease progresses in three
predictable and identifiable sequential steps before the onset of
symptoms: i) Humoral autoimmunity with the presence of
autoantibodies against antigens produced by pancreatic islet
β-cells but without dysglycemia; ii) cellular autoimmunity with the
presence of autoreactive cells against β-cells with dysglycemia but
no symptoms; iii) and the final stage is characterized by
hyperglycemia and the presence of symptoms such as polyuria,
thirst, hunger, and weight loss (1,34)
(Fig. 1). Of note, children with
autoantibodies to IA, GAD, and IA-2 showed different progression
paths, and insulin autoantibodies were most correlated with the
development of T1DM (33).
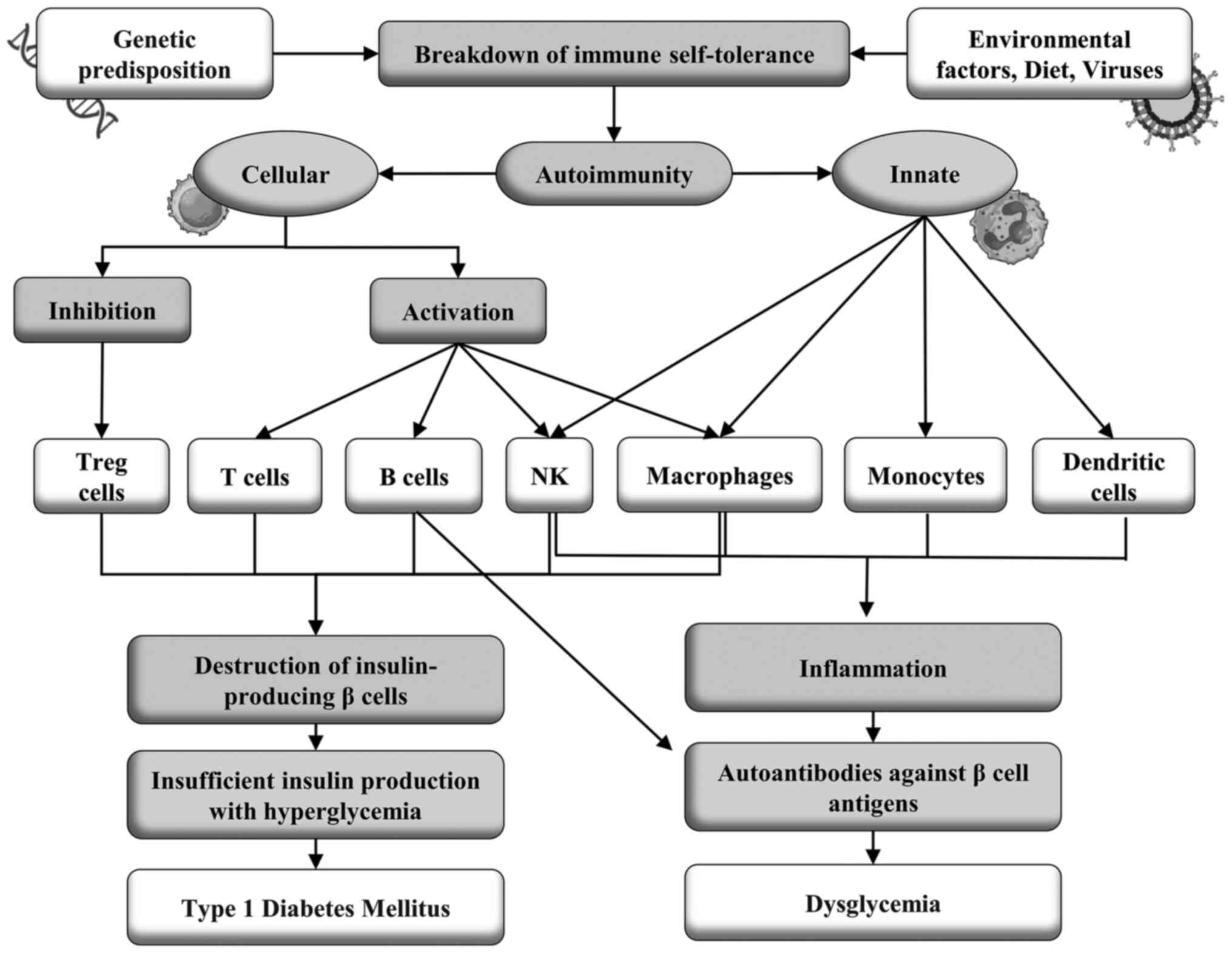 | Figure 1The onset of the disease occurs when
an individual with a genetic predisposition, particularly linked to
HLA Class I, specifically genotypes such as DR3-DQ2/DR3-DQ2 and
DR3-DQ2/DR4-DQ8, as well as haplotypes including DQA10501 and
DQB10302, is exposed to various environmental triggers. These
triggers encompass factors such as immune system maturation
conditions, infections by specific viruses, and dietary elements
rich in AGEs. This exposure can precipitate the breakdown of immune
self-tolerance, subsequently leading to the initiation of
autoimmunity, primarily of an innate immune response. This early
phase of autoimmunity is characterized by the infiltration of
monocytes, macrophages, dendritic cells, NK cells, and autoreactive
B cells into the pancreatic islets, inciting an inflammatory
cascade. Following this, a phase involving acquired humoral
immunity is activated. This results in autoantibodies targeting
antigens produced by β-cells, thereby inducing dysglycemia. After
the humoral response, cellular immunity is triggered, leading to
the proliferation of self-reactive TCD4+ and TCD8+ cells directed
against β-cells. This immune reaction is accompanied by Treg
suppression, ultimately culminating in the selective destruction of
insulin-producing β-cells within the pancreatic islets.
Consequently, there is inadequate insulin production, leading to
hyperglycemia and the clinical manifestation of T1DM. HLA, human
leukocyte antigen; AGE, advanced glycation end product; NK, natural
killer; Tregs, regulatory T cells; T1DM, type 1 diabetes
mellitus. |
The consumption of advanced glycation end products
(AGEs) may be an environmental component that together with genetic
factors may play a role in the initiation of T1MD. AGEs bind to
specific cell surface receptors (RAGE) and have a pro-inflammatory
role (35). In the NOD mouse model,
the first cells to infiltrate the pancreas are those of innate
immunity, such as monocytes, natural killer cells, CD11c +
dendritic cells, and ER-MP23 macrophages, with the occurrence of
insulitis, self-reactive B cells and later, self-reactive T cells
(27).
Humoral autoimmunity. In an observational
cohort study, the majority of the patients tested positive for ≥1
type of autoantibodies against the autoantigens GAD, IA-2, and
ZnT8. The presented symptoms were similar among participants with
or without autoantibodies, as was the frequency of ketoacidosis.
The positivity for autoantibodies decreased with age, particularly
in men compared to women and non-white individuals compared to
white individuals. The body mass index (BMI) was higher in adults
without autoantibodies than in adults with autoantibodies.
Individuals negative for autoantibodies were found to more likely
to have a parent with diabetes and less likely to have another
autoimmune disease (36).
The development of T1DM is preceded by an initial
phase characterized by the presence of insulitis, a type of
inflammatory response mediated by T lymphocytes, followed by the
detection of one or more types of autoantibodies against the
autoantigens produced by the β-cells of the pancreatic islets, such
as IA, GAD, protein tyrosine phosphatase, IA-2 or IA-2β, and ZnT8,
which are indicative of the immunological onset of T1DM (37-39).
The presence of >1 type of autoantibody marks the first stage of
the disease; the second stage is marked by dysglycemia or glucose
intolerance, both asymptomatic. The third stage, in turn, is
defined by the clinical manifestation of T1DM with symptoms of
hyperglycemia, such as polyuria, polydipsia, enuresis, weight loss,
blurred vision, and sometimes ketoacidosis or diabetic hyperosmolar
syndrome (37,39).
A longitudinal follow-up study of a patient who
developed T1DM 23 years after the idiopathic CD4 lymphocytopenia
diagnosis found that the anti-GAD antibody had already been present
in low titers for at least 16 years and began to increase 6 months
before the onset of illness. Seroconversion to IA and IA-2
autoantigens was detected during the onset of disease symptoms. The
proportion of CD8/CD4 lymphocytes increased gradually for 8 years
before the disease began, in particular CD8+ T cells autoreactive
against proteins related to the catalytic subunit of
glucose-6-phosphatase specific to pancreatic islets at the
beginning of T1DM. The patient in this study had idiopathic CD4
lymphocytopenia and a high number of islet antigen-specific CD8+ T
cells that may contribute to the autoimmune destruction of
insulin-producing β-cells (40).
Seroconversion to pancreatic islet autoantibodies
precedes disease onset by many years, but the role of humoral
autoimmunity in the initiation and progression of T1DM is still
unclear. A new autoantibody directed to extracellular epitopes of
zinc transporter 8 antigen (ZnT8ec), expressed in pancreatic islet
cells, was identified in newly diagnosed patients with the disease,
detected by immunofluorescence. When T1DM patients were compared
with healthy controls the positivity rate for the antibody
(ZnT8ecA) was 23.6% (41). The
sequential expression of the autoantibodies: IAA, GADA, IA-2A, and
ZnT8ecA was evaluated in a group of 30 children in a longitudinal
follow-up study from the evolution to the clinical manifestation of
T1DM, and 10 of them were positive for ZnT8ecA. Notably, ZnT8ecA
was the first antibody to appear in all 10 children (41).
Glutamate is the primary excitatory neurotransmitter
and is inactivated by cellular uptake via subtypes of glutamate
transporters GLT-1 (EAAT2) and GLAST (EAAT1) (42). It has been shown that GLT1/EAAT2 is
expressed in the membrane of pancreatic islet β-cells and regulates
extracellular glutamate concentrations, preventing induced β-cell
death. Patients with T1DM had autoantibodies against β-cells, but
the target antigen and pathogenic mechanisms are unknown (43,44).
It was hypothesized that GLT1 could be the target of autoantibodies
and may be associated with the pathogenesis of T1DM. ELISA showed
that sera from individuals with T1DM recognized GLT1 expressed in
the brain and pancreatic islets of mice and COS7 cells transfected
with the GLT1 gene. These findings were validated in two cohorts of
patients with T1DM by immunofluorescence assays detecting
autoantibodies against GLT1 in 37% of subjects with the disease and
none of the healthy controls (45).
Furthermore, in the absence of complement, autoantibodies against
GLT1 markedly reduced the activity of this transporter in βTC3
cells, inducing the internalization of GLT1, leading to the death
of β-cells. Thus, GLT1 is a novel T1DM autoantigen and anti-GLT1
autoantibodies cause β-cell death in the presence and absence of
complement (45).
The concentrations of autoantibodies GADA and IA2A
showed no statistical difference between the two groups, but the
positivity rate for ZnT8A was higher in the group with ketoacidosis
(46,47). ZnT8A-positive patients had higher
IA2A titers and a higher prevalence of GADA and IA2A compared to
ZnT8A-negative patients. Multivariate logistic regression analysis
showed that positivity for ZnT8A was associated with an increased
risk of microalbuminuria independent of age, sex, and BMI. This
study concluded that the autoantibody, ZnT8A, had diagnostic value
for ketoacidosis in children with T1DM, showing greater specificity
than the other two autoantibody types. In addition, positivity for
ZnT8A was related to a higher titer of IA2A and a higher frequency
of multiple diabetes-related autoantibodies, a risk factor for
T1DM, independent of microalbuminuria. Thus, positivity for ZnT8A
may be related to ketoacidosis and microalbuminuria, accelerating
the progression of T1DM (48).
A case-controlled study involving 20 patients with
recent-onset T1DM, with six months or less of diagnosis and testing
positive for ≥1 islet autoantibodies and 20 healthy controls
negative for islet autoantibodies found that 474 genes were
differentially expressed in individuals with T1DM, most of which
were related to host defense, inflammatory, antibacterial, and
antiviral effects, and cell cycle progression. Conversely,
downregulated genes were involved in T1DM target cell repair,
inflammation control, and immune tolerance. Among the genes were
AREG, which is related to the expression of FOXP3, and the SMAD6
gene which is associated with immunological tolerance. SMAD6
expression was found to be negatively correlated with islet ZnT8
autoantibody. PDE12 gene expression, which resists viral pathogens,
was reduced in T1DM and negatively related to the ZnT8A and GADA
autoantibodies levels. Conversely, diabetic patients showed
increased expression of genes encoding long non-coding RNAs, MALAT1
and NEAT1, which are related to inflammatory mediators, autoimmune
diseases, and innate immune response against viral infections
(49).
Cellular autoimmunity. Cellular immunity
plays a critical role in the selective and progressive destruction
of insulin-producing pancreatic islets β-cells, with a drastic
reduction in the production of this hormone and, consequently, in
the pathology of T1DM. Evidence of this is the presence of
infiltration of TCD4+ and TCD8+ lymphocytes, B lymphocytes, natural
killer (NK) cells, dendritic cells (DCs), macrophages, and other
autoreactive immune cells that recognize pancreatic islets as
foreign, resulting in their destruction, and establishment of the
clinical disease (1,29,50).
CD8+ T cells are responsible for the autoimmune
destruction of insulin-producing β-cells. Rapid disease progression
is associated with an increased number of islet-specific CD8+ T
cells with a transitional memory phenotype (51). They analyzed the phenotype and
function of these cells in the progression of T1DM and identified
islet-specific CD8+ T cells by high-content single-cell mass
cytometry in combination with peptide-loaded Human Leukocyte
Antigen (HLA) tetramer staining. A novel analytical method,
DISCOV-R, was used to characterize rare subsets of CD8+ T cells.
Autoreactive T cells are phenotypically heterogeneous and differ as
a function of the rate of disease progression. Activated
islet-specific memory CD8+ T cells were prevalent in T1DM patients
with a rapid loss of C-peptide. In contrast, the slow progression
of the disease is correlated with a profile of exhaustion and
expression of multiple inhibitory receptors, reduced production of
cytokines, and low proliferative capacity. Thus, the correlation
between these properties of autoreactive CD8+ T cells and the rate
of progression of T1DM after its onset can be considered an
attractive phenotypic biomarker of the disease trajectory (52).
The class I HLA-B*3906 and HLA-A*2402 genes are
associated with a higher risk of T1DM and promote the early onset
of the disease, suggesting that CD8+ T cells that recognize the
peptides presented in the context of these class I molecules on
cells β play a major role in the autoimmune response that results
in disease (53). A study analyzed
the frequency and phenotype of CD8+ T cells specific for the β-cell
antigens: Pre-proinsulin (PPI) and circulating B-specific insulin
(InsB) in HLA-B*3906+ children newly diagnosed T1DM and in
high-risk HLA-A*2402+ children prior to the appearance of
disease-associated autoantibodies and before the diagnosis as T1DM.
Antigen-specific CD8+ T cells were detected using these HLA class I
tetramers, and memory status was assessed by flow cytometry.
HLA-B*3906+ children with T1DM had an increase in memory CD8+ T
cells specific for the preproinsulin epitope PPI5-12
compared to healthy subjects, matched by age and HLA genotype. In
high-risk HLA-A*2402+ children, the percentage of terminal effector
cells within InsB15-24-specific CD8+ T cells increased before
diagnosis compared to samples collected before the appearance of
autoantibodies. These results indicate that CD8+ T cells
autoreactive to β-cells restricted by disease-associated HLA class
I molecules exhibit an experienced phenotype to antigens and show
enhanced effector function during the period leading up to clinical
diagnosis (54).
The population of CD8+ T cells specific for β-cell
antigens in T1DM maintains its self-reactive potential despite
having access to a permanent source of these antigens. The
longevity of these cells was evaluated, and the T-cell multipotency
index was established based on DNA methylation levels.
β-cell-specific CD8+ T cells maintained a stem-like epigenetic
multipotency score. Single-cell assay for transposase-accessible
chromatin using sequencing revealed the coexistence of epigenetic
programs of naive T cells associated with those of individual
β-cell specific effector CD8+ T cells. Analysis of the anatomical
distribution of β-cell-specific CD8+ T cells and the setting of
stem-associated epigenetic programs showed that self-reactive CD8+
T cells isolated from mouse lymph nodes maintained developmentally
plastic phenotypic and epigenetic profiles relative to the same
cells isolated from the pancreas. These data point to a new vision
into the lifespan of β-cell-specific CD8+ T cell responses
(55).
Analysis of gene expression by RNA sequencing in
CD4+ and CD8+ T cells, natural killer (NK) and B cells, and
chromatin accessibility by transposase accessible chromatin
sequencing assay (ATAC-seq) occurred. Gene expression was evaluated
in five genetically at-risk children with islet autoantibodies,
with a median age of 3 years, who progressed to T1DM and compared
with five sex, age, and HLA-DR-matched children who did not develop
the disease. In children with the disease, the differentially
expressed genes (DEGs) were mostly confined to CD4+ T cells and
enriched for genes and pathways linked to cytotoxicity (56). Several highly expressed DEGs were
validated in a semi-independent cohort of 13 children who
progressed to the disease and 11 who did not. Flow cytometry
revealed that disease progression was associated with the expansion
of CD4+ cells with a cytotoxic phenotype. The ATAC-seq method
showed that progression to T1DM was associated with the
reconfiguration of chromatin regulatory regions in CD4+ cells, some
of them linked to differentially expressed genes associated with
cytotoxicity. These findings suggest that cytotoxic CD4 T cells
play a role in the progression of T1DM (56,57).
Autoimmune diseases mediated by CD8 T cells result
from the breakdown of self-tolerance mechanisms in self-reactive
CD8 T cells (58). In T1DM, there
is a loss of tolerance to pancreatic islet autoantigens due to
defects in both central tolerance, which does not eliminate
potentially self-reactive lymphocytes, and peripheral tolerance,
which fails to control self-reactive T cells that have escaped the
thymus. This breakdown of the self-tolerance mechanisms of
β-cell-specific self-reactive CD8 T cells results in the
destruction of these insulin-producing cells (27,59).
To understand the mechanism by which this occurs,
the fate of β-cell-specific CD8 T cells was analyzed in non-obese
diabetic (NOD) mice. A stem-like autoimmune progenitor population
was identified in the pancreatic draining lymph node that
self-renewed giving rise to autoimmune mediators that migrate to
the pancreas, where they further differentiate and destroy β-cells.
The transfer of only 20 cell autoimmune progenitors induced T1DM,
while up to 100,000 autoimmune pancreatic mediators could not.
Pancreatic autoimmune mediators are short-lived, stem-like
autoimmune progenitors that must continually seed the pancreas to
sustain β-cell destruction. Single-cell RNA sequencing and clonal
analysis revealed that autoimmune CD8 T cells are unique T cell
differentiation states with identified characteristics that suggest
the transition from autoimmune progenitor to autoimmune mediator
(60).
The molecule CD137 is a member of the tumor necrosis
factor receptor family and modulator of T1DM progression in NOD
mice. CD137 expression on CD4 T cells inhibits the development of
the disease, but on CD8 T cells it promotes T1DM, increasing CD8 T
cells self-reactivity to β-cells (61). CD137 is expressed in a subpopulation
of FOXP3+ CD4 regulatory T cells (Tregs), which constitutes the
primary source of soluble CD137 that suppresses T cells, binding to
its ligand, CD137L, whose expression is increased on activated T
cells. NOD mice transfected with the CD137L gene (NOD.Tnfsf9-/-)
significantly delayed the development of T1DM, had less insulitis,
and had a reduced quantity of CD8 T cells autoreactive to β-cells
when compared to wild-type NOD mice. Adoptive T cell transfer
showed that CD137L deficiency on myeloid APCs was associated with
T1DM suppression. Conversely, the lack of CD137L in T cells
increased their diabetogenic activity. Neither CD137 nor CD137L is
required for the development and homeostasis of FOXP3+ Treg cells.
However, CD137 was critical for T1DM suppressive activity in
vivo by FOXP3+ Treg cells, suggesting that the interaction
between CD137 and CD137L regulates this function (62).
Regulatory CD4+ T cells (Tregs) act as protectors
against T1DM, recognizing antigens such as insulin or pancreatic
islet peptides, presented in the context of HLA-DQ6 or DR15, which
are protective haplotypes against diabetes (63). This interaction generates the
production of anti-inflammatory cytokines, such as IL-10 and TGF-β
and this activation of antigen-specific Tregs protects β-cells from
destruction through four mechanisms: i) Damage to cells occurs when
CD4+ T cell effectors recognize antigens of islet cells presented
by APCs, in the HLA-DQ8 or DR4 context, which is a risk for T1DM,
and are activated and migrate to the pancreas, where they induce
β-cell destruction (64).
Antigen-specific Tregs act by suppressing effector CD4+ T cells and
preventing their migration to the pancreas through the release of
anti-inflammatory cytokines. ii) Effector CD8+ T cells in
pancreatic islets recognize self-antigens presented in the HLA-A2
context by β-cells and destroy these cells. Tregs can suppress
effector CD8+ T cells and prevent β-cell destruction by releasing
anti-inflammatory cytokines. iii) Tregs can reduce
antigen-independent granzyme-mediated APC death. iv) Alternatively,
Tregs can induce antigen-dependent APC death when the antigen is
presented to the T cell receptor (TCR) of Tregs (63).
3. Role of viruses in the development of
T1DM
The rapid growth in the incidence of T1DM suggests
that environmental factors, including viruses, play a significant
role in the pathogenesis of the disease. Some microbial agents may
act as risk or protective factors for the disease. To date, two
hypotheses have attempted to explain these effects: The hygiene
hypothesis suggests that exposure to these agents in early
childhood stimulates immunoregulatory mechanisms that protect
against autoimmunity. The triggering hypothesis suggests that
specific agents can damage insulin-producing β-cells and contribute
to autoimmunity (23,65,66).
Certain viruses, particularly enteroviruses, are the primary
suspects and candidates for risk factors of T1DM. According to
epidemiological studies, they can cause disease in animals and are
associated with an increased risk of diabetes in humans, in
addition to being detected in the pancreas of patients with T1DM.
The possible protective effect of certain microbes has been
investigated in animal models and epidemiological studies, in which
certain enteric agents and gut microbiome patterns were associated
with a reduced risk of T1DM (20,23).
Growing evidence continues to implicate
enteroviruses as the most likely triggering agents of autoimmunity
in T1DM patients, possibly through persistent infections,
contributing to different stages of the disease (17,67). A
meta-analysis included 38 studies covering 5,921 subjects from all
continents, including 2,841 T1DM patients and 3,080 healthy
controls. Pooled analysis showed that enterovirus infection was
positively associated with disease in European, African, Asian,
Australian, and Latin American populations, but inconclusive for
North America. This association between enterovirus infection and
T1DM was found in blood and tissue samples, but no association was
observed in stool samples (68).
Among the viruses that may be associated with an increased risk for
T1DM, enteroviruses, primarily Coxsackie, echoviruses, in addition
to rotavirus and, to a lesser extent, mumps, parainfluenza,
rubella, and cytomegaloviruses stand out (20,23,68,69).
The long-term interaction between genetic,
epigenetic, and environmental factors, including viral infection,
can increase or decrease an individual's likelihood of developing
an autoimmune disease, depending on the imbalance between risk
factors and protective effects; three main mechanisms have been
proposed to explain the development of autoimmunity in T1DM:
molecular mimicry, epitope spread, and bystander activation
(70).
Studies report the association between viral
infections and the development of T1DM, but the immunological
mechanisms involved and the link between viral infections and
disease onset or progression are still unclear (71). One of the most commonly discussed
possibilities is molecular mimicry which involves cross-reactive
immunity against epitopes shared between viruses and human
pancreatic β-cells. Thus, molecular mimicry may cause the
activation of self-reactive T cells in T1DM, activated by a virus
that carries an epitope bearing a strong similarity to certain
structures present in human β-cells (72). This triggers a cross-reactive
autoimmune response that eliminates the viral infection and the
β-cells of the pancreatic islets. This mechanism may explain how
certain viruses may play a role in triggering T1DM (71,73).
Certain viruses infect and damage pancreatic β
cells, whose innate antiviral immune response can be modulated by
specific viral RNA receptors and sensors, including the melanoma
differentiation-associated gene 5 (MDA5), encoded by the IFIH1 gene
(74). MDA5 has been specifically
linked to inflammation and cell death of pancreatic cells from
rotavirus-infected mice models (75). Activation of the MDA5 receptor
limits rotavirus infection through the induction of the interferons
and pro-inflammatory cytokine production but may also facilitate
progression to an autoimmune response with the destruction of
pancreatic β-cells. Polymorphisms in the IFIH1 gene, which encodes
MDA5, are associated with an increased risk of T1DM (76).
MDA5 receptor activation is associated with
inflammatory and immunoregulatory responses from pancreatic islets
to viral infections and is expressed in pancreatic endocrine cells
with preferential localization to α-cells compared to β-cells,
suggesting that α-cells are better equipped than β-cells to respond
to viral infections and initiate viral clearance mechanisms. This
difference appears to make β-cells more susceptible to the
establishment of persistent low-grade viral infections. MDA5 is
increased in patients with new-onset T1DM, possibly because of
elevated inflammatory conditions (77).
Growing evidence shows that the post-translational
processing of peptides may play a role in the immune response under
pathological conditions by molecular mimicry. This mechanism
appears particularly relevant in T1DM, as epitopes resulting from
post-translational splicing derived from disease-related antigens
linked to HLA class I and II complexes. CD4+ and CD8+ T
cell-mediated immune responses in T1DM patients confirmed its
immunogenicity (78). Peptide
processing theoretically generates large sequencing variability,
increasing the frequency of human-viral zwitter peptides, which
share complete sequence homology, regardless of whether they
originate from human or viral antigens, impairing discrimination
between self and non-self-antigens by T cells, increasing the risk
of virus-triggered autoimmune responses (79).
Linear sequence similarities in amino acid motifs
are not the only criterion for developing molecular mimicry
autoimmunity. Autoreactive immune cells are prepared by molecular
mimicry, becoming sensitized but not causing apparent disease.
However, subsequent environmental insults can induce these
previously sensitized autoreactive cells to induce autoimmune
disease (80). The association
between viral infections and autoimmune diseases has long been
reported as events that precede target organ inflammation (81). Virally-infected mice that were later
challenged following viral shedding, with a nonspecific immune
insult developed autoimmune disease (82). Conventional inflammatory responses
to specific pathogens induced autoimmune disease in animals primed
with a molecular mimic for a CNS antigen (83). Therefore, activation of the immune
system alone is not a necessary condition for autoimmune disease,
but the environment in which sensitized immune cells are exposed to
antigens is an important factor in the onset of the disease
(80).
During viral infections, a significant quantity of T
cells are activated in a T cell receptor (TCR)-independent and
cytokine-dependent manner, by a mechanism known as ‘bystander
activation’. Type I interferons, IL-18, and IL-15, are the most
important factors that induce bystander activation of T cells, each
of which plays a somewhat different role (84). Bystander-activated T cells do not
have specificity for the pathogen but can affect the course of the
immune response to an infection. For example, Bystander-activated
CD8+ T cells participate in protective immunity by secreting
cytokines, such as interferon-γ. Conversely, they also cause damage
to the host by exerting cytotoxic effects facilitated by NK cell
activation receptors, such as NKG2D, and cytolytic molecules, such
as granzyme B. Interestingly, there is a strong association between
the cytolytic function of bystander-activated CD8+ T cells and
liver injury in patients with acute infection with the hepatitis A
virus (84,85).
Bystander activation occurs when CD8+ and CD4+ T
cells are activated in an antigen-independent manner in the absence
of TCRs. This alternative mechanism encompasses activation through
soluble factors or membrane-bound molecules that bind to receptors
other than the TCR (84). In
certain viral infections, this activation is beneficial for
clearing the virus but also triggers the activation of autoreactive
T cells in individuals genetically predisposed to autoimmunity
(86). Memory T cells are the most
vulnerable to bystander activation as they express cytokines and
co-signaling receptors. This activation occurs due to an
inflammatory environment, co-signaling ligands, and interactions
with neighboring cells (70).
Several types of T cell populations, such as NKT,
and Tγδ, in addition to conventional CD4+ and CD8+ T cells, can be
induced to exhibit innate-type effector function through bystander
activation (86,87). Antigen-specific T cells are the
hallmark of the adaptive immune response, while non-antigen-related
T cells in an inflammatory environment proliferate and synthesize
effector cytokines. In a viral infection, antigen-specific T cells
are activated by cytokines to ensure protective immunity. Bystander
activation of various types of T cells in the absence of antigen
recognition with the production of inflammatory cytokines may also
contribute to the elimination of a pathogen. Conversely, the
function of bystander-activated T cells may also be a mechanism in
the pathogenesis of autoimmune disease. Studies have reported that
non-antigen-related T cell infiltration into inflammatory tissues
participates in an autoimmune response (87).
Role of enteroviruses. Human enteroviruses
are small, non-enveloped, positive-sense, single-stranded RNA
viruses belonging to the Picornaviridae family and the
Enterovirus genus, which includes 7 species involved in human
diseases, including Poliovirus and Coxsackie, which
constitute five groups: The A group with 24 serotypes, the B group
with 6 serotypes, the C group with 23 serotypes, the D group with 5
serotypes and the Echoviruses group (88,89).
They are responsible for several human infections, ranging from
asymptomatic to mild and severe diseases, as well as certain
chronic autoimmune diseases, including T1DM (25).
Although they are highly cytolytic, enteroviruses
can persist in various tissues, including pancreatic islets, and
this persistence is hypothesized to play a role in the pathogenesis
of T1DM. Viral persistence is the result of virus-host coevolution
leading to cellular resistance to lysis due to mutations or
downregulation of the viral receptor and reduced replication
(90). Group B coxsackieviruses are
more commonly implicated in the development of T1DM. Its
persistence in pancreatic cells can trigger autoimmunity in
genetically predisposed individuals, destroying insulin-producing
β-cells through the activation of inflammation. The persistence of
this virus in the intestine, blood cells, and thymus has been
reported and possibly contributes to a certain extent to the
pathogenesis of T1DM (26,90).
Environmental factors can initiate and possibly
sustain, accelerate, or delay pancreatic β-cell damage. The role of
these factors has been extensively studied, and viruses, especially
enteroviruses, stand out as the most likely candidates. The
detection of enteroviruses in T1DM patients in randomized trials
confirmed the importance of human enteroviruses in the pathogenesis
of the disease (17). Genetic
susceptibility and innate and acquired immune responses against
enteroviruses may somehow contribute to the breakdown of tolerance
to self-antigens. The frequency with which this occurs, the
mechanisms and pathways of activation of virus-induced
autoimmunity, and the destruction of β-cells in T1DM have yet to be
determined (91).
The detection of enteroviruses in the serum, blood,
feces, nasal swabs, and pancreatic tissue of patients with T1DM,
before or close to the clinical onset of the disease, suggests an
association between infections with these viruses and the disease
(24). However, definitive evidence
of the role of these pathogens in the onset and progression of the
disease is still lacking. Emerging evidence suggests that chronic
human enterovirus infections occur in the pancreas of patients with
T1DM. However, the lack of sensitive molecular techniques capable
of detecting low quantities of viral proteins and RNA remains an
obstacle. Enterovirus RNA has been frequently detected in various
tissues of patients with T1DM and the presence of the virus in
pancreatic islets suggests persistent slow-replicating infection
that may have a role in the autoimmune response, which results in
T1DM (24,92).
Functionally, the thymus can be compared to a
computer highly specialized in orchestrating central immune
self-tolerance, a necessary condition for the survival of the
species, from the evolutionary pressure imposed by the hostile
environment and the autotoxicity inherent to the stochastic
generation of the diversity of immune cell receptors that
characterize the adaptive immune response. The presentation of
self-antigens in the thymus is responsible for the clonal deletion
of autoreactive T cells, which arise during the random
recombination of gene segments that encode variable parts of the
TCR. At the same time, the presentation of autoantigens in the
thymus generates Tregs that can inhibit, in the periphery, the
autoreactive T cells that have escaped negative selection in the
thymus. Thus, the autoimmunity against neuroendocrine glands is due
to a defect in intrathymic programming breaking the immune
self-tolerance that can be genetic or acquired during, for example,
an enteroviral infection (93).
Whether or not viruses can disrupt thymus function
and play a role in the pathogenesis of autoimmune diseases is an
open question (94). It is known
that through the synthesis and presentation of HLA-linked
neuroendocrine autoantigens, thymic epithelial cells (TECs) play a
crucial role in programming central immune self-tolerance to
neuroendocrine functions (93).
Insulin-like growth factor-2 (IGF-2) is the dominant polypeptide of
the insulin family expressed in these cells in different animal
species and humans. Thymic infection by coxsackievirus B4 (CV-B4)
may compromise the thymus function and interfere with the
intrathymic programming of tolerance to insulin antigen family and
secondarily to insulin-secreting pancreatic islet β-cells (95). Productive thymus infection by this
virus occurs after oral inoculation in mice. Infection of a murine
medullary TEC line induces a significant decrease in Igf2 gene
expression and IGF-2 production. It is hypothesized that inhibition
of Igf2 expression in infected TECs can lead to a breakdown of
central immune self-tolerance. This leads to the triggering of
autoimmune responses against self-antigens produced by
insulin-secreting pancreatic islet β-cells (96).
Thymus gland dysfunction can be a crucial step in
organ-specific autoimmune diseases such as T1DM. Viruses can
disrupt thymic functions by inducing thymic atrophy, dysfunction,
apoptosis, and impaired lymphocyte maturation (97). Enteroviruses have been widely
associated with the pathogenesis of T1DM due to enterovirus-induced
thymic dysfunction in natal or perinatal life. Infection of the
thymus by enteroviruses can interfere with T cell maturation or
lead to the production of autoreactive T cells, both being
potentially involved in the development of T1DM. It has been shown
that CV-B4 infection can have multiple effects on the thymus
resulting in the dysregulation of tolerance involved in the
pathogenesis of T1DM, but the specifics require further study
(95). The possible mechanisms by
which CV-B4 can trigger the autoimmune response that leads to T1DM
are shown in Fig. 2.
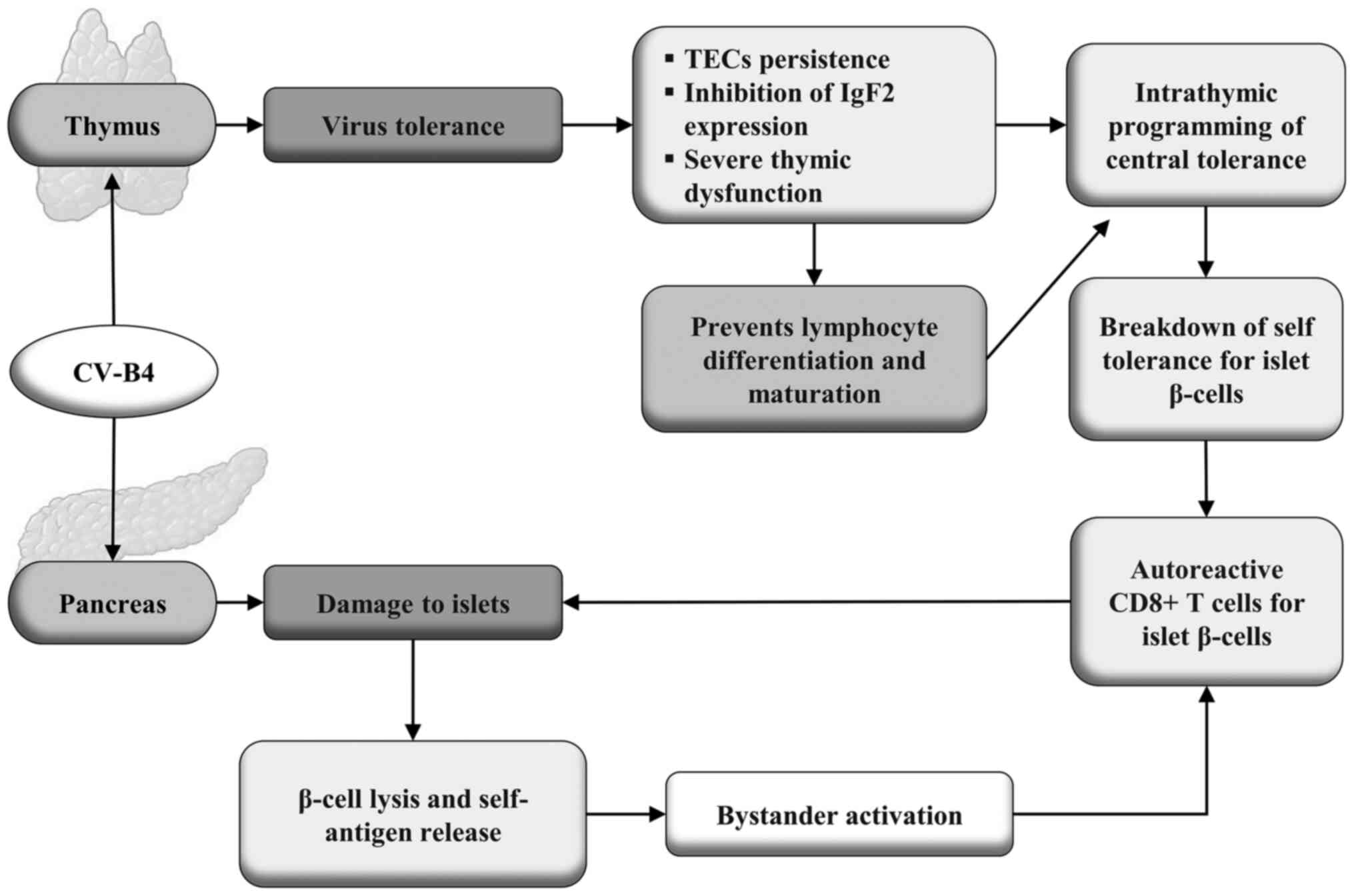 | Figure 2Infection of the thymus by CV-B4 can
induce tolerance to the virus, leading to persistent infection of
TECs, which play a crucial role in programming central immune
self-tolerance. This infection causes severe thymic dysfunction and
inhibits the expression of the gene encoding Igf2. Furthermore, it
prevents the differentiation and maturation of lymphocytes,
interfering with the intrathymic programming of central
immunological self-tolerance, resulting in the breakdown of
self-tolerance for the β-cells of the pancreatic islets. This leads
to the production of self-reactive cytotoxic CD8+ T cells against
insulin-producing β-cells, promoting their destruction. This same
virus can also infect the spleen, causing the lysis of β-cells
resulting in the release of autoantigens, leading to increased
bystander activation of cytotoxic CD8+ T cells auto-reactive
against β-cells, creating a positive feedback of an autoimmune
process that can lead to T1DM. CV-B4, Coxsackievirus B4; TEC,
thymic epithelial cell; Igf2, Insulin-like growth factor-2; T1DM,
type 1 diabetes mellitus. |
It has been suggested that only a minority of
pancreatic β-cells appear to be infected at any one time.
Furthermore, enteroviruses can become defective and stop
replicating, which may explain why they are rarely isolated from
pancreatic tissues. It appears that enteroviral infection of
β-cells largely depends on the innate and adaptive immunity of the
host. Thus, viruses may play a role in T1DM at multiple levels,
including stimulating chronic autoimmunity and generating
inflammation that leads to β-cell dysfunction or destruction. This
stressful situation contributes to autoimmunity, creating a vicious
circle (98).
The Environmental Determinants of Diabetes in the
Young (TEDDY) study is the largest multicenter prospective study of
young children with genetic susceptibility to T1DM that aimed to
identify the environmental causes of the disease. Metagenomic
sequencing of monthly stool samples paired with healthy controls
collected from newborns until the detection of islet autoimmunity
or T1DM was performed (99).
Enterovirus B was the only virus identified in the human virome
with a significant association with islet autoimmunity. Although
there was no difference between the frequencies between cases and
controls, children with prolonged excretion of enterovirus B were
more likely to develop T1DM. Interestingly, human mastadenovirus C
infection early in life was less frequent in children who developed
islet autoimmunity than in those who did not, suggesting a
protective effect. An association between a polymorphism in the
coxsackie virus and the adenovirus receptor human gene with
susceptibility to T1DM led the authors to propose that competition
for receptor binding between the adenovirus and the coxsackie virus
conferred the protective effect of the adenovirus (69).
The coincidence of periodic epidemiological patterns
of incidence of infections by enteroviruses and T1DM stimulated the
first suspicions of the association of these viruses with the onset
of the disease (100).
Furthermore, there is a strong correlation between genetic
susceptibility to islet autoimmunity and enterovirus infection.
Clinical and epidemiological findings point to the coxsackie B
virus as a potential triggering mechanism of an autoimmune reaction
against β-cells, leading to an increase in inflammation,
destruction of these cells, and T1DM development (25). Conversely, NOD mice immunized with a
vaccine against coxsackievirus Bs were protected from the
accelerated onset of the disease without causing inflammation of
the pancreatic islets and preventing the progression of the disease
induced by this virus in the animal model that develops the disease
spontaneously (101).
Several serotypes of group B coxsackieviruses result
in chronic infections in human cells both in vivo and in
vitro, but the mechanisms leading to the persistence of these
enteroviruses and autoimmunity against pancreatic β-cells are still
not fully understood (102). A
carrier-state-type persistent infection model using a PANC-1 human
pancreatic cell line and two different coxsackie B strains was used
to assess virus-induced changes in the cell transcriptome. Clear
changes were observed in the gene expression of factors associated
with the pancreatic microenvironment, such as the secretory pathway
and lysosomal biogenesis during persistent infection. In addition,
the antiviral response pathways were activated differently by the
two viral strains. It was revealed that there were extensive
transcriptional responses in persistently infected pancreatic cells
resulting in notable alterations, with some opposing and some
similar changes between the two strains (103).
A study in Taiwan showed that the incidence of T1DM
was significantly higher in children aged 0-6 years old, decreasing
in adolescents aged 13-19 years old. The risk of developing the
disease in children aged 0-6 years infected with an enterovirus was
significantly higher than in uninfected individuals. Furthermore,
the incidence of T1DM in children aged 0-6 years old was positively
correlated with isolation rates of the Coxsackie virus A species,
but no association after 7 years of age (104). The complex interactions of
epigenetic and environmental genetic factors, especially the
viruses, can trigger autoimmune reactions against autoantigens
produced by β cells responsible for destroying these cells, and
thus, the subsequent development of T1DM. Growing evidence
continues to implicate persistent enteroviral infections as the
most likely potential environmental trigger for T1DM. Consistent
evidence also suggests that enteroviral infections may contribute
by acting at different stages of T1DM development (17).
The NOD female mouse model is suitable for studying
T1DM since these mice spontaneously develop the disease, and this
can be accelerated by certain viruses. Toll-like 3 (Tlr3-/-) and
wild-type (Tlr3+/+) knockout female NOD mice were used to assess
the role of this receptor in triggering T1DM. Islet samples from
Tlr3+/+ and Tlr3-/-animals infected and uninfected with CV-B4 were
analyzed by immunostaining, laser capture microdissection, and
real-time reverse transcription-quantitative PCR. Tlr3+/+ mice
showed a higher incidence of insulitis and expression of CXCL10,
IL1β, TNFα, and TGFβ1 compared with Tlr3-/-. After CV-B4 infection,
NOD Tlr3+/+ mice had a higher incidence of insulitis and T cell
infiltration 3 days post-infection compared with Tlr3-/-mice also
infected with CV-B4. The results indicated that the Toll-like
receptor 3 was required to create an inflammatory microenvironment
of pancreatic islets, increasing insulitis and the expression of
cytokines that favor the development of CV-B4-induced T1DM in
female NOD mice (105).
Role of rotaviruses. Rotaviruses have long
been implicated as a potential environmental agent that triggers
T1DM. The proposed mechanism involves molecular mimicry between the
external capsid antigen of human rotavirus, VP7 protein, and the
autoantigen tyrosine phosphatase IA-2 of the pancreatic islets, the
molecular target of autoimmunity in T1DM. The major epitope of the
intracytoplasmic domain of the IA-2 antigen has been shown to share
56% homology with a sequence of the rotavirus VP7 protein (106). The role of rotavirus VP7 protein
in accelerating diabetes has also been demonstrated experimentally
in NOD mice infected with rhesus monkey rotavirus (107,108).
Rotavirus and other viruses have historically been
considered one of the environmental factors that trigger T1DM
(108). A positive association was
found between seroconversion to rotavirus VP7 protein and the
increase in autoantibodies to GAD, IA-2, and insulin antigens
associated with T1DM; thus, rotavirus infection is a trigger for
the development of pancreatic islet autoimmunity in genetically
predisposed children. A later study reinforced the theory of
molecular mimicry between the rotavirus protein VP7 and the
pancreatic islet peptides, IA2 and GAD5, demonstrating how the same
epitope sequences in these peptides bind to T1DM-associated
HLA-DRB1*04 molecules and can induce T cell proliferative responses
(109,110).
Rotavirus infection generates activated immune cells
with a Th1 phenotype that migrates to the pancreas where it can
trigger autoimmunity through two mechanisms: i) molecular mimicry
in which cross-reactive Th1 cells that recognize both the rotavirus
and host peptide results in the release of cytokines and
chemokines, which recruit and activate cytotoxic T cells that
damage the islets resulting in the subsequent release of islet
antigens that activate APCs, which then feedback in to the
autoimmune process. ii) Bystander activation of nonspecific
autoreactive Th1 cells and rotavirus-specific Th1 cells leads to
inflammation, increasing infiltration of cytotoxic T cells and
destruction of islets promoting the release of autoantigens, which
activates autoreactive Th1 cells by a TCR-dependent mechanism. The
self-reactive T cells activated in this manner mediate islet damage
by releasing more antigens that feedback on the autoimmune response
(108,111). In Fig.
3, a proposal showing the possible mechanisms by which
rotaviruses trigger the autoimmune response that leads to T1DM is
shown.
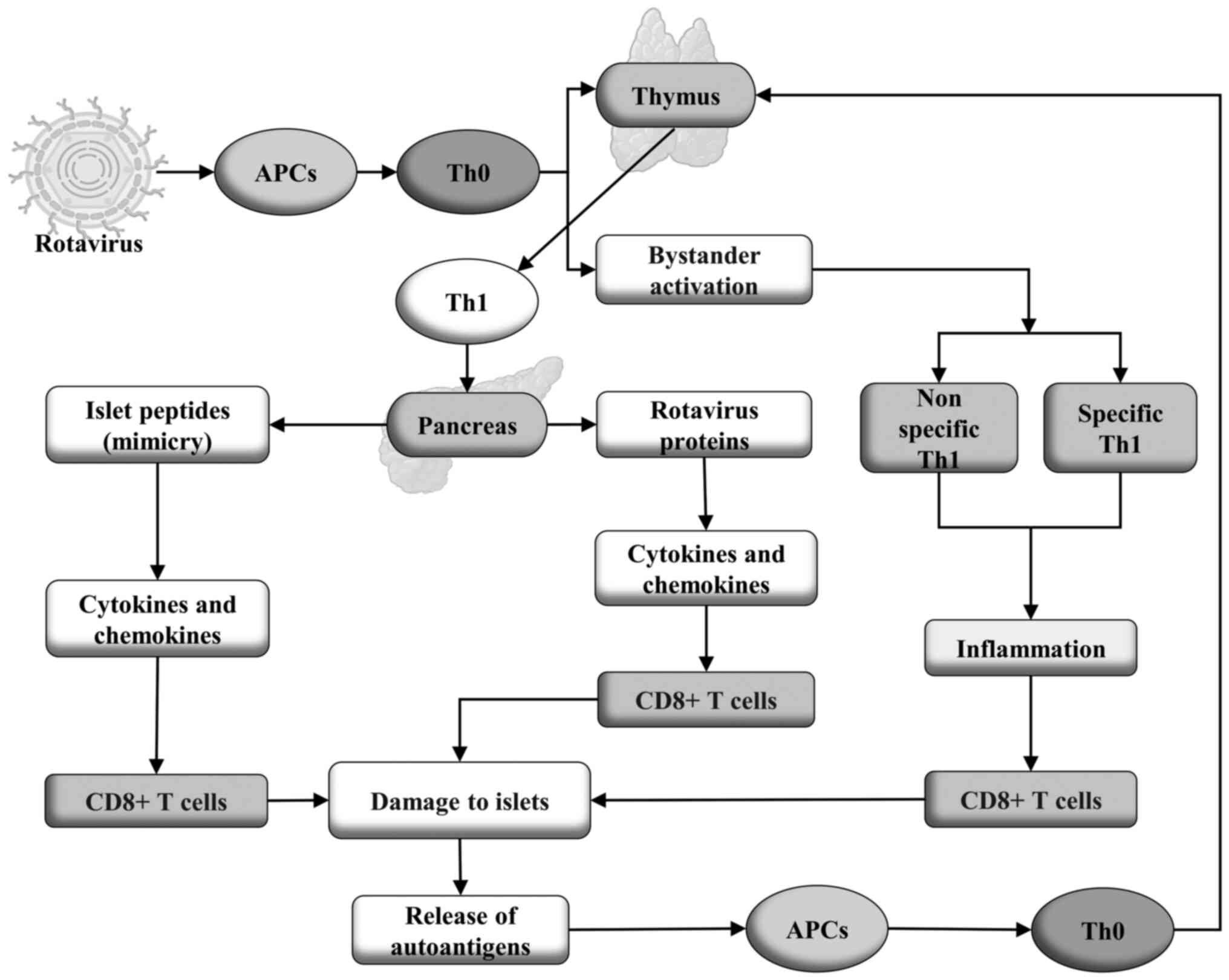 | Figure 3In persistent infection of thymic
epithelial cells by rotavirus, the viral proteins are presented by
APCs to Th0 cells that subsequently differentiate into Th1 cells,
which recognize not only viral proteins but also peptides produced
by β-cells of pancreatic islets, through the mechanism of molecular
mimicry. These cells migrate to the pancreas and interact with the
cells that exhibit class I MHC presentation of both antigens,
resulting in the production of cytokines and chemokines that
activate cytotoxic CD8+ T cells auto-reactive against β-cells, and
thus causing damage to the pancreatic islets. This results in the
release of self-antigens that are again presented by APCs to Th0
cells which migrate to the thymus, where they differentiate into
Th1 cells, thus generating a positive feedback loop. Rotavirus also
activates Th1 cells via a bystander pathway, independent of TCR,
generating non-specific and rotavirus-specific auto-reactive Th1
cells, which produce cytokines and chemokines, resulting in
inflammation and activation of self-reactive cytotoxic CD8+ T cells
that cause damage to the pancreatic islets. This generates an
autoimmune process that can lead to T1DM. T1DM, type 1 diabetes
mellitus; APC, antigen-presenting cell; MHC, major
histocompatibility complex; TCR, T cell receptor. |
Rotavirus infections were initially identified as
possible triggers for the initiation of T1DM due to the
similarities between viral peptide sequences and human pancreatic
islet peptide sequences in carriers of the disease (111). Furthermore, rotavirus infection
increases the risk of T1DM in NOD mice. Research on the association
between rotavirus infections with the risk of the disease in humans
has produced mixed results and suggested the involvement of other
factors, such as age and diet. With the global availability of
rotavirus vaccines, studies have evaluated whether rotavirus
vaccination alters the incidence of T1DM and have found no
associations, protective or otherwise. These studies suggest a
possible etiologic relationship between certain wild-type rotavirus
infections and T1DM (110).
Epidemiological and immunological data suggest a
strong link between rotavirus infection and two high-incidence
autoimmune pathologies: Celiac disease (CD) and T1DM. The role of
current oral rotavirus vaccines is being elucidated, with a
positive protective association against these autoimmune diseases
so far demonstrated (108),
highlighting the potential role of rotaviruses as triggers of T1DM.
Two recent studies suggest that the complete routine vaccination
schedule against rotavirus decreased the incidence of T1DM in
children. The first was performed in Australia (112). The second was a longitudinal
cohort study involving 1,474,535 children in the United States,
conducted by Rogers et al (113), from 2001 to 2017, using data from
a health insurer in the country. Participants were classified as
vaccinated when they received the series of one of the rotavirus
vaccines (three doses of RotaTeq or two doses of Rotarix), or at
least one dose without completing the series; as unvaccinated if
they had not received any of the vaccines, and those born before
its existence. Children who completed the rotavirus vaccine series
had a 33% reduced risk of T1DM compared to unvaccinated children.
Those who completed the RotaTeq pentavalent vaccine series had a
37% lower risk of developing the disease. Partial vaccination was
not associated with the incidence of T1DM. The authors concluded
that rotavirus vaccination is associated with a reduction in T1DM
incidence, and that rotavirus immunization may be the first
practical measure for prevention of T1DM (113). However, a less comprehensive
cohort study involving 880,629 children found no evidence that
rotavirus vaccination prevented T1DM (114).
In addition to viruses from the Picornaviridae
family, which includes enteroviruses, such as Coxsackievirus B, and
from the Reoviridae family, including Rotavirus, other viruses
belonging to different families, such as the Togaviridae family
(for example Rubella virus), the Paramyxoviridae family (for
example Mumps virus), the Herpesviridae family (for example
Cytomegalovirus and Epstein Barr virus), and the Coronaviridae
family (for example SARS-CoV-2), have been identified as potential
environmental factors associated with the risk of developing T1DM
(3).
4. Conclusions
Viral infections are potential candidates as
environmental triggers of T1DM. However, certain aspects require
further clarification, such as the absence of generalized
cytopathic effects on the pancreas of patients with the disease.
Additionally, there is no closely related temporal association
between such infections and disease onset, although there is strong
evidence that at least some viruses act as environmental triggers
in T1DM. The exact mechanisms by which this occurs remains to be
definitively proven. Furthermore, viruses have rarely been isolated
from the pancreas of individuals with T1DM, possibly due to the
inaccessibility of the organ. Data available in the literature
point to the role of viruses, notably enteroviruses, in the
etiopathogenesis of the disease, especially during possible
persistent or chronic recurrent infections of pancreatic β-cells.
The detection, with a certain frequency, of RNA and viral proteins
in the pancreatic tissues of individuals with T1DM represents
strong evidence of latent infection or slow replication of these
viruses. However, the mechanism by which they act to trigger the
autoimmune process that leads to selective and progressive
destruction of insulin-producing pancreatic β-cells, with disease
initiation and progression, is still unclear.
Acknowledgements
Not applicable
Funding
Funding: This study was financed in part by the Coordenação de
Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES)
(Finance Code, 001).
Availability of data and materials
Not applicable.
Authors' contributions
JJPA, JVF, and JVA conceived the study, performed
the literature review, and drafted the manuscript. MTFO and VDA
drafted the manuscript. FLF, FLB, and TAAMF critically revised the
manuscript for important intellectual content. DCFL, JMGA, and VSA
were involved in the formal analysis of the article included in the
review. Data authentication is not applicable. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Katsarou A, Gudbjörnsdottir S, Rawshani A,
Dabelea D, Bonifacio E, Anderson BJ, Jacobsen LM, Schatz DA and
Lernmark Å: Type 1 diabetes mellitus. Nat Rev Dis Primers.
3(17016)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Roep BO, Thomaidou S, van Tienhoven R and
Zaldumbide A: Type 1 diabetes mellitus as a disease of the β-cell
(do not blame the immune system?). Nat Rev Endocrinol. 17:150–161.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zorena K, Michalska M, Kurpas M, Jaskulak
M, Murawska A and Rostami S: Environmental factors and the risk of
developing type 1 diabetes-old disease and new data. Biology
(Basel). 11(608)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
de Azevedo JCV, de Medeiros Fernandes TAA,
Cavalcante GA, de Medeiros IACM, Lanza DCF, de Araújo JMG, Bezerra
FL and Fernandes JV: Biology and natural history of type 1 diabetes
mellitus. Curr Pediatr Rev. 19:253–275. 2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cerna M: Epigenetic regulation in etiology
of type 1 diabetes mellitus. Int J Mol Sci. 21(36)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lucier J, Weinstock RS and Doerr C:
Diabetes mellitus type 1 (Nursing). StatPearls Publishing, Treasure
Island, FL, 2023.
|
|
7
|
Alamri ZZ: The role of liver in
metabolism: An updated review with physiological emphasis. Int J
Basic Clin Pharmacol. 7:2271–2276. 2018.
|
|
8
|
Han HS, Kang G, Kim JS, Choi BH and Koo
SH: Regulation of glucose metabolism from a liver-centric
perspective. Exp Mol Med. 48(e218)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Röder PV, Wu B, Liu Y and Han W:
Pancreatic regulation of glucose homeostasis. Exp Mol Med.
48(e219)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chadt A and Al-Hasani H: Glucose
transporters in adipose tissue, liver, and skeletal muscle in
metabolic health and disease. Pflugers Arch. 472:1273–1298.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zheng P, Li Z and Zhou Z: Gut microbiome
in type 1 diabetes: A comprehensive review. Diabetes Metab Res Rev.
34(e3043)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cohn A, Sofia AM and Kupfer SS: Type 1
diabetes and celiac disease: Clinical overlap and new insights into
disease pathogenesis. Curr Diab Rep. 14(517)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pociot F and Lernmark Å: Genetic risk
factors for type 1 diabetes. Lancet. 387:2331–2339. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Abela AG and Fava S: Why is the incidence
of type 1 diabetes increasing? Curr Diabetes Rev.
17(e030521193110)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zajec A, Trebušak Podkrajšek K, Tesovnik
T, Šket R, Čugalj Kern B, Jenko Bizjan B, Šmigoc Schweiger D,
Battelino T and Kovač J: Pathogenesis of type 1 diabetes:
Established facts and new insights. Genes (Basel).
13(706)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Siljander H, Honkanen J and Knip M:
Microbiome and type 1 diabetes. EBioMedicine. 46:512–521.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lloyd RE, Tamhankar M and Lernmark Å:
Enteroviruses and type 1 diabetes: Multiple mechanisms and factors?
Annu Rev Med. 73:483–499. 2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hyöty H: Viruses in type 1 diabetes.
Pediatr Diabetes. 17 (Suppl 22):S56–S64. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Alhazmi A, Sane F, Lazrek M, Nekoua MP,
Badia-Boungou F, Engelmann I, Alidjinou EK and Hober D:
Enteroviruses and type 1 diabetes mellitus: An overlooked
relationship in some regions. Microorganisms.
8(1458)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Isaacs SR, Foskett DB, Maxwell AJ, Ward
EJ, Faulkner CL, Luo JYX, Rawlinson WD, Craig ME and Kim KW:
Viruses and type 1 diabetes: From enteroviruses to the Virome.
Microorganisms. 9(1519)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Geravandi S, Richardson S, Pugliese A and
Maedler K: Localization of enteroviral RNA within the pancreas in
donors with T1D and T1D-associated autoantibodies. Cell Rep Med.
2(100371)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Isaacs SR, Roy A, Dance B, Ward EJ,
Foskett DB, Maxwell AJ, Rawlinson WD, Kim KW and Craig ME:
Enteroviruses and risk of islet autoimmunity or type 1 diabetes:
Systematic review and meta-analysis of controlled observational
studies detecting viral nucleic acids and proteins. Lancet Diabetes
Endocrinol. 11:578–592. 2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kondrashova A and Hyöty H: Role of viruses
and other microbes in the pathogenesis of type 1 diabetes. Int Rev
Immunol. 33:284–295. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Oikarinen S, Krogvold L, Edwin B, Buanes
T, Korsgren O, Laiho JE, Oikarinen M, Ludvigsson J, Skog O,
Anagandula M, et al: Characterisation of enterovirus RNA detected
in the pancreas and other specimens of live patients with newly
diagnosed type 1 diabetes in the DiViD study. Diabetologia.
64:2491–2501. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Geravandi S, Liu H and Maedler K:
Enteroviruses and T1D: Is it the virus, the genes or both which
cause T1D. Microorganisms. 8(1017)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nekoua MP, Alidjinou EK and Hober D:
Persistent coxsackievirus B infection and pathogenesis of type 1
diabetes mellitus. Nat Rev Endocrinol. 18:503–516. 2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bluestone JA, Herold K and Eisenbarth G:
Genetics, pathogenesis and clinical interventions in type 1
diabetes. Nature. 464:1293–1300. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kahaly GJ and Hansen MP: Type 1 diabetes
associated autoimmunity. Autoimmun Rev. 15:644–648. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li M, Song LJ and Qin XY: Advances in the
cellular immunological pathogenesis of type 1 diabetes. J Cell Mol
Med. 18:749–758. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Knip M, Siljander H, Ilonen J, Simell O
and Veijola R: Role of humoral beta-cell autoimmunity in type 1
diabetes. Pediatr Diabetes. 17 (Suppl 22):S17–S24. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Winter WE, Harris N and Schatz D: Type 1
diabetes islet autoantibody markers. Diabetes Technol Ther.
4:817–839. 2002.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Winter WE and Schatz DA: Autoimmune
markers in diabetes. Clin Chem. 57:168–175. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kwon BC, Anand V, Achenbach P, Dunne JL,
Hagopian W, Hu J, Koski E, Lernmark Å, Lundgren M, Ng K, et al:
Progression of type 1 diabetes from latency to symptomatic disease
is predicted by distinct autoimmune trajectories. Nat Commun.
13(1514)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Insel RA, Dunne JL, Atkinson MA, Chiang
JL, Dabelea D, Gottlieb PA, Greenbaum CJ, Herold KC, Krischer JP,
Lernmark Å, et al: Staging presymptomatic type 1 diabetes: A
scientific statement of JDRF, the Endocrine Society, and the
American Diabetes Association. Diabetes Care. 38:1964–1974.
2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Du C, Whiddett RO, Buckle I, Chen C,
Forbes JM and Fotheringham AK: Advanced glycation end products and
inflammation in the development of type 1 diabetes. Cells.
11(3503)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Bravis V, Kaur A, Walkey HC, Godsland IF,
Misra S, Bingley PJ, Williams AJK, Dunger DB, Dayan CM, Peakman M,
et al: Relationship between islet autoantibody status and the
clinical characteristics of children and adults with incident type
1 diabetes in a UK cohort. BMJ Open. 8(e020904)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dayan CM, Korah M, Tatovic D, Bundy BN and
Herold KC: Changing the landscape for type 1 diabetes: The first
step to prevention. Lancet. 394:1286–1296. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Beik P, Ciesielska M, Kucza M, Kurczewska
A, Kuźmińska J, Maćkowiak B and Niechciał E: Prevention of type 1
diabetes: Past experiences and future opportunities. J Clin Med.
9(2805)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
American Diabetes Association Professional
Practice Committee. 2. Classification and diagnosis of diabetes:
Standards of medical care in diabetes-2022. Diabetes Care. 45
(Suppl 1):S17–S38. 2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Maruyama K, Chujo D, Watanabe K, Kawabe A,
Sugiyama T, Ohsugi M, Tanabe A, Ueki K and Kajio H: Evaluation of
cellular and humoral autoimmunity before the development of type 1
diabetes in a patient with idiopathic CD4 lymphocytopenia. J
Diabetes Investig. 10:1108–1111. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Gu Y, Merriman C, Guo Z, Jia X, Wenzlau J,
Li H, Li H, Rewers M, Yu L and Fu D: Novel autoantibodies to the
β-cell surface epitopes of ZnT8 in patients progressing to type-1
diabetes. J Autoimmun. 122(102677)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bjørnsen LP, Hadera MG, Zhou Y, Danbolt NC
and Sonnewald U: The GLT-1 (EAAT2; slc1a2) glutamate transporter is
essential for glutamate homeostasis in the neocortex of the mouse.
J Neurochem. 128:641–649. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Di Cairano ES, Davalli AM, Perego L, Sala
S, Sacchi VF, La Rosa S, Finzi G, Placidi C, Capella C, Conti P, et
al: The glial glutamate transporter 1 (GLT1) is expressed by
pancreatic beta-cells and prevents glutamate-induced beta-cell
death. J Biol Chem. 286:14007–14018. 2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhou Y, Waanders LF, Holmseth S, Guo C,
Berger UV, Li Y, Lehre AC, Lehre KP and Danbolt NC: Proteome
analysis and conditional deletion of the EAAT2 glutamate
transporter provide evidence against a role of EAAT2 in pancreatic
insulin secretion in mice. J Biol Chem. 289:1329–1344.
2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Perego C, Di Cairano ES, Galli A, Moretti
S, Bazzigaluppi E, Centonze VF, Gastaldelli A, Assi E, Fiorina P,
Federici M, et al: Autoantibodies against the glial glutamate
transporter GLT1/EAAT2 in Type 1 diabetes mellitus-Clues to novel
immunological and non-immunological therapies. Pharmacol Res.
177(106130)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Juusola M, Parkkola A, Härkönen T,
Siljander H, Ilonen J, Åkerblom HK and Knip M: Childhood Diabetes
in Finland Study Group. Positivity for Zinc Transporter 8
Autoantibodies at diagnosis is subsequently associated with reduced
β-cell function and higher exogenous insulin requirement in
children and adolescents with type 1 diabetes. Diabetes Care.
39:118–121. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yohena S, Penas-Steinhardt A, Muller C,
Faccinetti NI, Cerrone GE, Lovecchio S, Ridner E, Valdez S and
Frechtel G: Immunological and clinical characteristics of latent
autoimmune diabetes in the elderly. Diabetes Metab Res Rev.
35(e3137)2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang M, Wang X, Wang R, Shu J, Zhi X, Gu
C, Pu L, Cai C, Yang W and Lv L: Clinical study of autoantibodies
in type 1 diabetes mellitus children with ketoacidosis or
microalbuminuria. J Clin Lab Anal. 36(e24164)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Santos AS, Cunha-Neto E, Gonfinetti NV,
Bertonha FB, Brochet P, Bergon A, Moreira-Filho CA, Chevillard C
and da Silva MER: Prevalence of inflammatory pathways over
immuno-tolerance in peripheral blood mononuclear cells of
recent-onset type 1 diabetes. Front Immunol.
12(765264)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zirpel H and Roep BO: Islet-resident
dendritic cells and macrophages in type 1 diabetes: In search of
Bigfoot's print. Front Endocrinol (Lausanne).
12(666795)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wong FS and Wen L: A predictive
CD8+ T cell phenotype for T1DM progression. Nat Rev
Endocrinol. 16:198–199. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Wiedeman AE, Muir VS, Rosasco MG, DeBerg
HA, Presnell S, Haas B, Dufort MJ, Speake C, Greenbaum CJ, Serti E,
et al: Autoreactive CD8+ T cell exhaustion distinguishes subjects
with slow type 1 diabetes progression. J Clin Invest. 130:480–490.
2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Schloss J, Ali R, Racine JJ, Chapman HD,
Serreze DV and DiLorenzo TP: HLA-B*39:06 efficiently mediates type
1 diabetes in a mouse model incorporating reduced thymic insulin
expression. J Immunol. 200:3353–3363. 2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Yeo L, Pujol-Autonell I, Baptista R,
Eichmann M, Kronenberg-Versteeg D, Heck S, Dolton G, Sewell AK,
Härkönen T, Mikk ML, et al: Circulating β cell-specific
CD8+ T cells restricted by high-risk HLA class I
molecules show antigen experience in children with and at risk of
type 1 diabetes. Clin Exp Immunol. 199:263–277. 2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Abdelsamed HA, Zebley CC, Nguyen H,
Rutishauser RL, Fan Y, Ghoneim HE, Crawford JC, Alfei F, Alli S,
Ribeiro SP, et al: Beta cell-specific CD8+ T cells
maintain stem cell memory-associated epigenetic programs during
type 1 diabetes. Nat Immunol. 21:578–587. 2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Bediaga NG, Garnham AL, Naselli G,
Bandala-Sanchez E, Stone NL, Cobb J, Harbison JE, Wentworth JM,
Ziegler AG, Couper JJ, et al: Cytotoxicity-related gene expression
and chromatin accessibility define a subset of CD4+ T cells that
mark progression to type 1 diabetes. Diabetes. 71:566–577.
2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ramos-Rodríguez M, Raurell-Vila H, Colli
ML, Alvelos MI, Subirana-Granés M, Juan-Mateu J, Norris R,
Turatsinze JV, Nakayasu ES, Webb-Robertson BM, et al: The impact of
proinflammatory cytokines on the β-cell regulatory landscape
provides insights into the genetics of type 1 diabetes. Nat Genet.
51:1588–1595. 2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Burrack AL, Martinov T and Fife BTT: T
Cell-mediated beta cell destruction: Autoimmunity and Alloimmunity
in the context of type 1 diabetes. Front Endocrinol (Lausanne).
8(343)2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Rathod S: Novel Insights into the
immunotherapy-based treatment strategy for autoimmune type 1
diabetes. Diabetology. 3:79–96. 2022.
|
|
60
|
Gearty SV, Dündar F, Zumbo P,
Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, Socci ND,
Trivedi P, Lowe SW, Lauer P, et al: An autoimmune stem-like CD8 T
cell population drives type 1 diabetes. Nature. 602:156–161.
2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Forsberg LA, Gisselsson D and Dumanski JP:
Mosaicism in health and disease-clones picking up speed. Nat Rev
Genet. 18:128–142. 2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Foda BM, Ciecko AE, Serreze DV, Ridgway
WM, Geurts AM and Chen YG: The CD137 ligand is important for type 1
diabetes development but dispensable for the homeostasis of
disease-suppressive CD137+ FOXP3+ regulatory
CD4 T cells. J Immunol. 204:2887–2899. 2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Mitchell AM and Michels AW: Self-Antigens
targeted by regulatory T cells in type 1 diabetes. Int J Mol Sci.
23(3155)2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Jacobsen LM, Newby BN, Perry DJ, Posgai
AL, Haller MJ and Brusko TM: Immune mechanisms and pathways
targeted in type 1 diabetes. Curr Diab Rep. 18(90)2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Bach JF: Revisiting the hygiene hypothesis
in the context of autoimmunity. Front Immunol.
11(615192)2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Rewers M and Ludvigsson J: Environmental
risk factors for type 1 diabetes. Lancet. 387:2340–2348.
2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Richardson SJ and Morgan NG: Enteroviral
infections in the pathogenesis of type 1 diabetes: New insights for
therapeutic intervention. Curr Opin Pharmacol. 43:11–19.
2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Wang K, Ye F, Chen Y, Xu J, Zhao Y, Wang Y
and Lan T: Association between enterovirus infection and type 1
diabetes risk: A meta-analysis of 38 case-control studies. Front
Endocrinol (Lausanne). 12(706964)2021.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Vehik K, Lynch KF, Wong MC, Tian X, Ross
MC, Gibbs RA, Ajami NJ, Petrosino JF, Rewers M, Toppari J, et al:
Prospective virome analyses in young children at increased genetic
risk for type 1 diabetes. Nat Med. 25:1865–1872. 2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Pacheco Y, Acosta-Ampudia Y, Monsalve DM,
Chang C, Gershwin ME and Anaya JM: Bystander activation and
autoimmunity. J Autoimmun. 103(102301)2019.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Op de Beeck A and Eizirik DL: Viral
infections in type 1 diabetes mellitus-why the β cells? Nat Rev
Endocrinol. 12:263–273. 2016.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Begum S, Aiman S, Ahmad S, Samad A,
Almehmadi M, Allahyani M, Aljuaid A, Afridi SG and Khan A:
Molecular mimicry analyses unveiled the human herpes simplex and
poxvirus epitopes as possible candidates to incite autoimmunity.
Pathogens. 11(1362)2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Smatti MK, Cyprian FS, Nasrallah GK, Al
Thani AA, Almishal RO and Yassine HM: Viruses and autoimmunity: A
review on the potential interaction and molecular mechanisms.
Viruses. 11(762)2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Dias Junior AG, Sampaio NG and Rehwinkel
J: A Balancing Act: MDA5 in antiviral immunity and
autoinflammation. Trends Microbiol. 27:75–85. 2019.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Dou Y, Yim HC, Kirkwood CD, Williams BR
and Sadler AJ: The innate immune receptor MDA5 limits rotavirus
infection but promotes cell death and pancreatic inflammation. EMBO
J. 36:2742–2757. 2017.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Looney BM, Xia CQ, Concannon P, Ostrov DA
and Clare-Salzler MJ: Effects of type 1 diabetes-associated IFIH1
polymorphisms on MDA5 function and expression. Curr Diab Rep.
15(96)2015.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Nigi L, Brusco N, Grieco GE, Fignani D,
Licata G, Formichi C, Aiello E, Marselli L, Marchetti P, Krogvold
L, et al: Increased expression of viral sensor MDA5 in pancreatic
islets and in hormone-negative endocrine cells in recent onset type
1 diabetic donors. Front Immunol. 13(833141)2022.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Mishto M, Mansurkhodzhaev A,
Rodriguez-Calvo T and Liepe J: Potential mimicry of viral and
pancreatic β cell antigens through non-spliced and cis-Spliced
Zwitter Epitope candidates in type 1 diabetes. Front Immunol.
12(656451)2021.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Jadeja SD and Tobin DJ: Autoantigen
discovery in the hair loss disorder, alopecia Areata: Implication
of post-translational modifications. Front Immunol.
13(890027)2022.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Cusick MF, Libbey JE and Fujinami RS:
Molecular mimicry as a mechanism of autoimmune disease. Clin Rev
Allergy Immunol. 42:102–111. 2012.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Fujinami RS, von Herrath MG, Christen U
and Whitton JL: Molecular mimicry, bystander activation, or viral
persistence: Infections and autoimmune disease. Clin Microbiol Rev.
19:80–94. 2006.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Theil DJ, Tsunoda I, Rodriguez F, Whitton
JL and Fujinami RS: Viruses can silently prime for and trigger
central nervous system autoimmune disease. J Neurovirol. 7:220–227.
2001.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Tsunoda I, Terry EJ, Marble BJ, Lazarides
E, Woods C and Fujinami RS: Modulation of experimental autoimmune
encephalomyelitis by VLA-2 blockade. Brain Pathol. 17:45–55.
2007.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Kim TS and Shin EC: The activation of
bystander CD8+ T cells and their roles in viral
infection. Exp Mol Med. 51:1–9. 2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Björkström NK, Strunz B and Ljunggren HG:
Natural killer cells in antiviral immunity. Nat Rev Immunol.
22:112–123. 2022.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Lee HG, Cho MZ and Choi JM: Bystander
CD4+ T cells: Crossroads between innate and adaptive
immunity. Exp Mol Med. 52:1255–1263. 2020.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Shim CH, Cho S, Shin YM and Choi JM:
Emerging role of bystander T cell activation in autoimmune
diseases. BMB Rep. 55:57–64. 2022.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Tapparel C, Siegrist F, Petty TJ and
Kaiser L: Picornavirus and enterovirus diversity with associated
human diseases. Infect Genet Evol. 14:282–293. 2013.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Zell R: Picornaviridae-the ever-growing
virus family. Arch Virol. 163:299–317. 2018.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Alidjinou EK, Sané F, Engelmann I, Geenen
V and Hober D: Enterovirus persistence as a mechanism in the
pathogenesis of type 1 diabetes. Discov Med. 18:273–282.
2014.PubMed/NCBI
|
|
91
|
Christoffersson G and Flodström-Tullberg
M: Mouse models of virus-induced type 1 diabetes. Methods Mol Biol.
2128:93–105. 2020.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Rodriguez-Calvo T: Enterovirus infection
and type 1 diabetes: Unraveling the crime scene. Clin Exp Immunol.
195:15–24. 2019.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Geenen V, Bodart G, Henry S, Michaux H,
Dardenne O, Charlet-Renard C, Martens H and Hober D: Programming of
neuroendocrine self in the thymus and its defect in the development
of neuroendocrine autoimmunity. Front Neurosci.
7(187)2013.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Jaïdane H, Sané F, Hiar R, Goffard A,
Gharbi J, Geenen V and Hober D: Immunology in the clinic review
series; focus on type 1 diabetes and viruses: Enterovirus, thymus
and type 1 diabetes pathogenesis. Clin Exp Immunol. 168:39–46.
2012.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Alhazmi A, Nekoua MP, Michaux H, Sane F,
Halouani A, Engelmann I, Alidjinou EK, Martens H, Jaidane H, Geenen
V, et al: Effect of Coxsackievirus B4 infection on the thymus:
Elucidating its role in the pathogenesis of type 1 diabetes.
Microorganisms. 9(1177)2021.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Michaux H, Martens H, Jaïdane H, Halouani
A, Hober D and Geenen V: How does thymus infection by
coxsackievirus contribute to the pathogenesis of type 1 diabetes?
Front Immunol. 6(338)2015.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Luo M, Xu L, Qian Z and Sun X:
Infection-associated thymic atrophy. Front Immunol.
12(652538)2021.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Dunne JL, Richardson SJ, Atkinson MA,
Craig ME, Dahl-Jørgensen K, Flodström-Tullberg M, Hyöty H, Insel
RA, Lernmark Å, Lloyd RE, et al: Rationale for enteroviral
vaccination and antiviral therapies in human type 1 diabetes.
Diabetologia. 62:744–753. 2019.PubMed/NCBI View Article : Google Scholar
|
|
99
|
TEDDY Study Group. The environmental
determinants of diabetes in the Young (TEDDY) Study. Ann N Y Acad
Sci. 1150:1–13. 2008.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Karaoglan M and Eksi F: The coincidence of
newly diagnosed type 1 diabetes mellitus with IgM antibody
positivity to Enteroviruses and respiratory tract viruses. J
Diabetes Res. 2018(8475341)2018.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Stone VM, Butrym M, Hankaniemi MM,
Sioofy-Khojine AB, Hytönen VP, Hyöty H and Flodström-Tullberg M:
Coxsackievirus B vaccines prevent infection-accelerated diabetes in
NOD mice and have no disease-inducing effect. Diabetes.
70:2871–2878. 2021.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Alidjinou EK, Engelmann I, Bossu J,
Villenet C, Figeac M, Romond MB, Sané F and Hober D: Persistence of
Coxsackievirus B4 in pancreatic ductal-like cells results in
cellular and viral changes. Virulence. 8:1229–1244. 2017.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Buchacher T, Honkimaa A, Välikangas T,
Lietzén N, Hirvonen MK, Laiho JE, Sioofy-Khojine AB, Eskelinen EL,
Hyöty H, Elo LL, et al: Persistent coxsackievirus B1 infection
triggers extensive changes in the transcriptome of human pancreatic
ductal cells. iScience. 25(103653)2021.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Shih WL, Tung YC, Chang LY, Fang CT and
Tsai WY: Increased incidence of pediatric type 1 diabetes with
novel association with coxsackievirus a species in young children
but declined incidence in adolescents in Taiwan. Diabetes Care.
44:1579–1585. 2021.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Benner SE, Walter DL, Thuma JR, Courreges
M, James CBL, Schwartz FL and McCall KD: Toll-like receptor 3 is
critical to the pancreatic islet milieu that is required for
Coxsackievirus B4-induced type 1 diabetes in female nonobese
diabetic mice. Pancreas. 51:48–55. 2022.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Honeyman MC, Brusic V, Stone NL and
Harrison LC: Neural network-based prediction of candidate T-cell
epitopes. Nat Biotechnol. 16:966–969. 1998.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Pane JA, Fleming FE, Graham KL, Thomas HE,
Kay TW and Coulson BS: Rotavirus acceleration of type 1 diabetes in
non-obese diabetic mice depends on type I interferon signalling.
Sci Rep. 6(29697)2016.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Gómez-Rial J, Rivero-Calle I, Salas A and
Martinón-Torres F: Rotavirus and autoimmunity. J Infect.
81:183–189. 2020.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Honeyman MC, Stone NL, Falk BA, Nepom G
and Harrison LC: Evidence for molecular mimicry between human T
cell epitopes in rotavirus and pancreatic islet autoantigens. J
Immunol. 184:2204–2210. 2010.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Burke RM, Tate JE, Jiang B and Parashar
UD: Rotavirus and type 1 diabetes-is there a connection? A
synthesis of the evidence. J Infect Dis. 222:1076–1083.
2020.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Harrison LC, Perrett KP, Jachno K, Nolan
TM and Honeyman MC: Does rotavirus turn on type 1 diabetes? PLoS
Pathog. 15(e1007965)2019.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Perrett KP, Jachno K and Nolan TM: Role of
rotavirus vaccination in decline in incidence of type 1
diabetes-reply. JAMA Pediatr. 173(895)2019.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Rogers MAM, Basu T and Kim C: Lower
incidence rate of type 1 diabetes after receipt of the rotavirus
vaccine in the United States, 2001-2017. Sci Rep.
9(7727)2019.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Inns T, Fleming KM, Iturriza-Gomara M and
Hungerford D: Paediatric rotavirus vaccination, coeliac disease and
type 1 diabetes in children: A population-based cohort study. BMC
Med. 19(147)2021.PubMed/NCBI View Article : Google Scholar
|














