Neutrophils are the first immune cells to arrive at
the site of inflammation, where they contribute to the destruction
and containment of pathogenic microorganisms through phagocytosis,
the release of bactericidal substances and the production of
pro-inflammatory cytokines (1).
Neutrophil activation is initiated by both microbial and endogenous
stimuli, resulting in the release of chromatin and granule
proteins, including neutrophil elastase (NE), myeloperoxidase (MPO)
and other components such as histone G and defensins. These
elements collectively form extracellular structures termed
neutrophil extracellular traps (NETs) (2). The process, termed NETosis, is
integral to the pathophysiology of myocardial ischemia-reperfusion
injury (MIRI) by modulating mechanisms such as reactive oxygen
species (ROS) production, inflammation and energy metabolism,
thereby contributing to the advancement of myocardial injury
(3,4). NETosis plays an important role in the
physiopathology of aseptic inflammatory conditions, such as MIRI,
by affecting ROS production, inflammation, energy metabolism and
other such mechanisms, but the specific mechanisms and directions
of its action are not yet fully understood.
Despite the development of more efficient and
effective reperfusion techniques, including percutaneous coronary
intervention and novel antiplatelet and antithrombotic therapies,
effective treatments to prevent MIRI remain limited (5,6).
Recent data show that cardiovascular disease is responsible for
~20% of all deaths worldwide each year (7). MIRI refers to myocardial injury that
occurs after rapid restoration of blood flow in the ischemic area
and accounts for 50% of the final infarct size (8). These injuries can be devastating,
leading to fatal myocardial damage, myocardial dysfunction,
vascular injury and tissue edema (9). The pathological mechanisms underlying
these injuries include apoptosis, autophagy, endoplasmic reticulum
stress, ferroptosis, pyroptosis, and ultimately, exacerbated
oxidative stress, calcium imbalance, activation of inflammatory
cascades, endothelial dysfunction and microvascular injury.
Recently, it has been shown that specific biomarkers (such as DNA,
histones and NE) that are released during NETosis may provide novel
biomarkers for the early diagnosis of MIRI (10). By regulating the NETosis process,
the development of novel therapeutic strategies, such as inhibiting
the formation of NETosis or promoting its clearance, may help to
mitigate MIRI and improve the prognosis of patients.
The present review systematically compiles and
analyzes the research progress on the role of NETosis in MIRI in
recent years, focusing on the formation mechanism of NETosis, its
dynamic changes during myocardial ischemia-reperfusion and the
potential therapeutic mechanism of NETosis-targeted therapy for
MIRI.
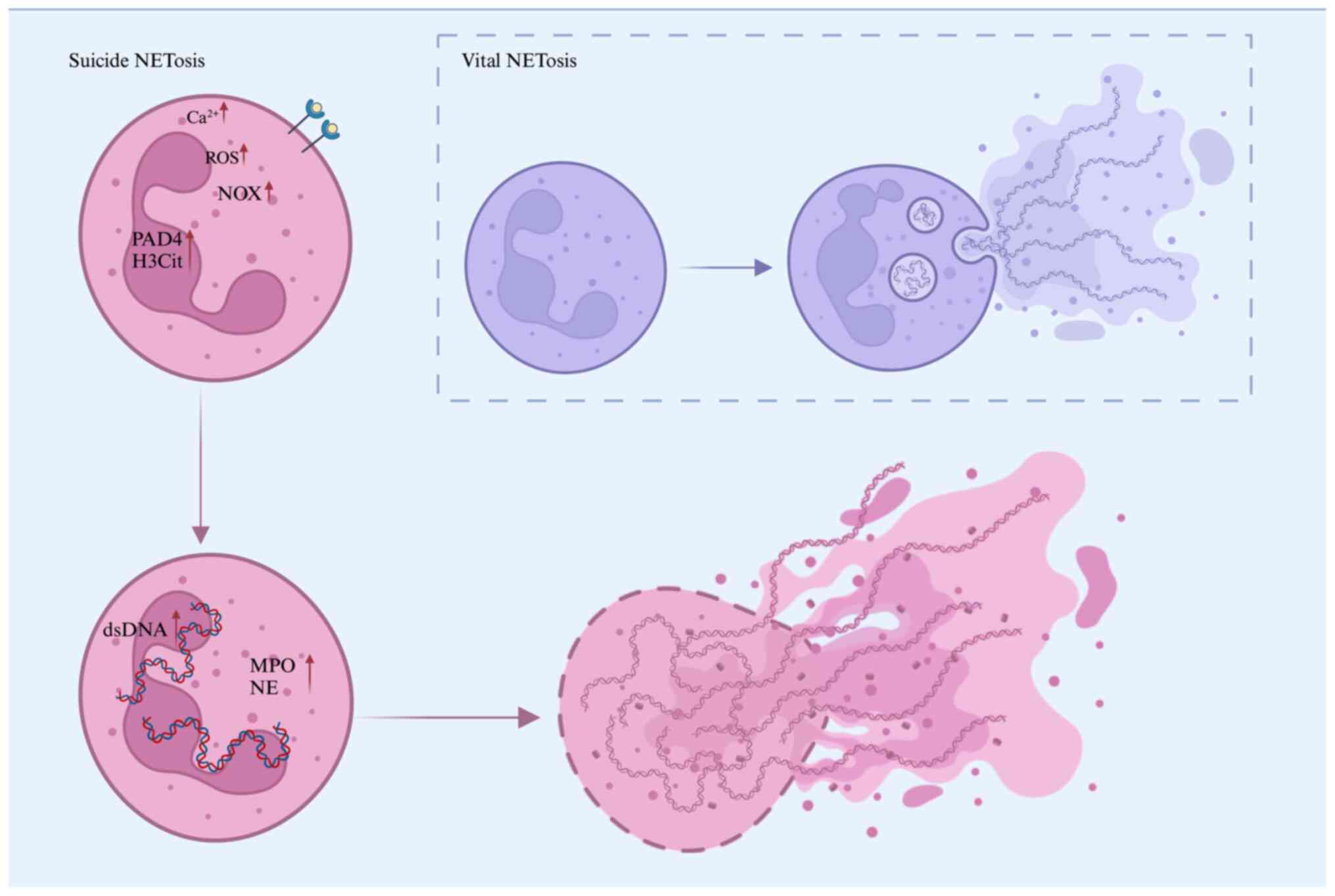
To date, the formation of NETosis has been
classified into two forms: i) Suicidal NETosis; and ii) vital
NETosis (11). Regardless of the
stimulus, NETosis formation involves the cellular process of
chromatin decondensation, which is essential for the release of
NETs (12).
Suicidal NETosis denotes a mechanism of NET
formation that is concomitant with neutrophil apoptosis. Upon
activation by stimuli including phorbol ester, specific
autoantibodies, immune complexes or conditions characterized by
elevated calcium ion (Ca²+) concentrations, neutrophils
initiate the NADPH oxidase complex (NOX), leading to the production
of ROS such as superoxide anion, hydroxyl radical and hydrogen
peroxide (13,14). These ROS are highly reactive and
play a crucial role in facilitating NOX-dependent NET formation
(15). The release of ROS activates
peptidylarginine deaminase 4 (PAD4), the only nuclear-localized
isoenzyme in the PAD family, which is expressed in both the nucleus
and cytoplasmic granules of immune cells (16). PAD4 facilitates the citrullination
of core and linker histones, which diminishes their affinity for
DNA, thereby promoting chromatin decondensation and the unfolding
of densely packed chromatin structures (17,18).
Activation of PAD4 also triggers the translocation
of NE and MPO from azurophilic granules to the nucleus (19), which further enhances histone H3
citrullination (H3Cit) and chromatin decondensation, a pivotal
process in the formation of NETs (20,21).
Consequently, H3Cit is considered a specific and well-established
marker for NETs (22). With
chromatin decondensation and nuclear membrane rupture, the
decondensed chromatin mixes with granule proteins and is released
into the cytoplasm (23).
Eventually, the cytoplasmic membrane leaks, and the modified
chromatin is expelled from the neutrophil, forming extracellular
NETs in a meshwork structure. NETs are released into the
extracellular space 3-8 h after neutrophil activation, leading to
cell death due to membrane disintegration (24). Plasma levels of MPO have been shown
to be positively correlated with the risk of coronary artery
disease (25), and MPO-DNA
complexes are associated with severe adverse cardiovascular events
(26).
Moreover, the serum concentrations of critical
markers of NETs, such as double-stranded DNA (dsDNA), MPO and NE,
are markedly elevated in patients with coronary atherosclerotic
disease (27). Additionally,
extracellular H3Cit exhibits significant cytotoxicity, contributing
to tissue damage and thereby elevating the risk of mortality among
patients with cardiovascular disease (28). These observations indicate that MPO,
NE and H3Cit are integral to NET formation, further implicating
NETosis in the pathophysiological processes underlying MIRI.
Vital NET formation represents a type of NETosis
that transpires shortly after neutrophil activation, distinguished
by the generation of NETs without compromising the structural
integrity of the neutrophil (29).
In contrast to suicidal NETosis, this process involves the
encapsulation of modified chromatin within vesicles that are
secreted by the nucleus and subsequently expelled from the cell,
thereby preserving neutrophil integrity (30). Consequently, neutrophils remain
intact and retain the ability to perform additional functions
(Fig. 1).
Myocardial ischemia is primarily attributed to
atherosclerosis, thrombosis, insufficient coronary blood supply and
elevated myocardial oxygen demand, with atherosclerosis and luminal
thrombosis identified as the predominant mechanisms.
NETosis is a critical factor in the pathophysiology
of myocardial ischemia, particularly within pro-inflammatory and
pro-thrombotic contexts. NETs are observed to form in regions of
elevated cholesterol within atherosclerotic lesions. Elevated blood
cholesterol levels result in the deposition of cholesterol crystals
within the vessel walls, which are subsequently recognized and
phagocytosed by macrophages (31).
This process induces lysosomal damage and activates the NLR family
pyrin domain containing 3 (NLRP3) inflammasome within macrophages
(32). PAD4 plays a critical role
in the pathogenesis of atherosclerosis by regulating
apoptosis-associated speck-like proteins containing a caspase
recruitment domain (ASC proteins) and NLRP3 protein levels, thereby
facilitating the assembly of NLRP3 inflammasomes (33). Upon activation, these multiprotein
complexes lead to the production of pro-inflammatory cytokines such
as interleukin (IL)-1β and IL-18 (34,35).
These factors recruit more immune cells to the
vascular endothelium, which exacerbates the inflammatory response
and further activates neutrophils leading to NETosis (36). The release of NETs can exacerbate
tissue damage and contribute to a pro-inflammatory milieu.
Activated neutrophils secrete cytokines and chemokines, which
further recruit additional immune cells and amplify the
inflammatory response. This phenomenon is particularly evident in
conditions such as acute myocardial infarction, where enhanced
neutrophil-platelet interactions lead to increased thrombus
formation (37). Thrombus formation
plays a critical role in the pathogenesis of myocardial ischemia.
For example, Zhou et al (38) identified elevated levels of NETs in
coronary thrombosis among patients with ST-segment elevated
myocardial infarction. Neutrophils retained within thrombi play a
crucial role in initiating the endogenous coagulation cascade
through the release of NETs, which facilitate the activation of
coagulation factor XII. This process can subsequently lead to
thrombin generation and platelet activation (39). The structural network of NETs
further supports the deposition of platelet adhesion molecules and
promotes thrombin-mediated fibrin formation (40).
Calcium ions are crucial for the maintenance of
normal cardiomyocyte function, with mitochondrial Ca2+
levels modulating the rate of oxidative metabolism to align with
ATP depletion in the cytoplasm. This regulation ensures that
cardiomyocytes receive adequate energy to sustain their normal
systolic and diastolic functions (52). During reperfusion injury,
cardiomyocytes exacerbate injury by reversing the
Na+/Ca²+ exchanger, leading to sodium efflux
and subsequent intracellular calcium overload. Excessive calcium
efflux contributes to mitochondrial permeability transition pore
opening and mitochondrial ROS production (53,54).
In a high Ca²+ concentration environment, calcium ions
can directly or indirectly activate NOX by altering its
conformation or active state, triggering NOX activation and
promoting NETosis (55). This
pathway is referred to as NOX-independent NETosis. Research has
shown that direct activation of small-conductance calcium-activated
potassium (SK) channels can induce NETosis, with the involvement of
SK3 (56,57).
Extracellular histones, as critical DAMPs,
contribute to myocardial injury during MIRI (63). Extracellular histones exacerbate the
inflammatory response by activating TLR2 and TLR4, leading to the
production of pro-inflammatory cytokines. Histone interactions with
T cells promote Th17 cytokine production, further amplifying the
inflammatory state (64-67).
High mobility group protein B1, a critical DAMP released from
necrotic cardiac tissue following prolonged ischemic injury,
interacts with the receptor for advanced glycation end-products.
This interaction facilitates the recruitment of a substantial
number of neutrophils to the injured myocardium, thereby promoting
the production of IL-6 through signaling pathways such as MAPK and
nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB),
along with the release of other inflammatory mediators, ultimately
triggering NET formation (68). The
inflammatory pathway factors involved in NETosis during reperfusion
injury and potential treatments are described in Table I.
Damaged endothelial cells release a variety of
pro-inflammatory factors, such as IL-1β and IL-6, which further
amplify the inflammatory response (73). NETs can induce NF-κB-dependent
endothelial angiogenesis, altering the plaque microenvironment and
promoting the development of unstable plaques (74,75).
Angiotensin II has been demonstrated to induce NETosis through both
the ROS/PAD4 pathway and autophagy-dependent mechanisms (76).
Subsequent to cardiomyocyte injury and necrosis,
there is a pronounced accumulation of extracellular matrix
components, accompanied by the differentiation of cardiac
fibroblasts into myofibroblasts under stress conditions. These
myofibroblasts exhibit enhanced proliferation and secretion
activities, culminating in the development of cardiac fibrosis
(77). NETs are intricately linked
to inflammatory processes and may significantly influence the
fibroblastic response during myocardial injury. Additionally,
non-classical monocytes, integral components of the immune system,
play a crucial role in myocardial healing. It has been observed
that overproduction of NETs can reduce the expression of CX3C
chemokine receptor 1 on non-classical monocytes at the site of
injury. This reduction may impair the migration of non-classical
monocytes to ischemic tissues, resulting in delayed or compromised
myocardial healing (78).
The release of NETs also stimulates the
proliferation and activation of cardiac fibroblasts, which secrete
collagen and other fibrosis-associated proteins, thus contributing
to the progression of myocardial fibrosis (79). NETs recognize and bind endocytosed
dsDNA via TLR9, which activates the myeloid differentiation factor
88 and, in turn, triggers the NF-κB signaling pathway (80,81).
Upon activation of the TLR-NF-κB signaling axis, myofibroblast
differentiation is promoted by regulating the expression of genes
such as transforming growth factor-β, collagen I (a major component
of the cardiac extracellular matrix) and α-smooth muscle actin, a
signature protein of myofibroblasts. These changes are accompanied
by stress fiber formation and contribute to the progression of
myocardial fibrosis (82).
During myocardial ischemia-reperfusion, excessive
production of NETs can facilitate thrombosis and worsen
microvascular obstruction, thereby elevating the risk of myocardial
infarction. Chang et al (83) concluded that the combined use of
circulating cell-free DNA (cfDNA) and creatine kinase-MB may
enhance the diagnostic accuracy and sensitivity for acute
myocardial ischemia, enabling a more comprehensive evaluation of
the condition of patients. Recent research has demonstrated that in
patients with acute myocardial ischemia, plasma cfDNA is
predominantly released through NETosis, further substantiating its
potential as a diagnostic marker for acute myocardial ischemia
(84).
Additionally, markers such as platelet count,
MPO-DNA complexes, NE-DNA complexes, H3Cit and soluble platelet
selectin have been identified as independent predictors of major
adverse cardiovascular events 1 year after myocardial infarction
(85). A prospective clinical study
demonstrated that plasma levels of
bactericidal/permeability-increasing protein (BPI) in patients
experiencing acute myocardial ischemia exhibited a positive
correlation with IL-1β, high-sensitivity C-reactive protein,
MPO-DNA and S100A8/A9. Consequently, BPI may serve as a novel
biomarker for myocardial ischemia, with its primary mechanism
potentially involving the mediation of NET formation through
interactions between platelets and neutrophils (86,87).
Animal studies have found that colchicine alleviates
inflammation and cardiac remodeling after acute myocardial
infarction by inhibiting the formation of NETs, thereby improving
cardiac function (88).
Additionally, colchicine reduces microvascular obstruction by
suppressing the proliferation of neutrophils in the bone marrow and
their migration to the ischemia-reperfusion injury area (89). Building on these findings from
animal studies, clinical research has further validated the
potential application of colchicine in cardiovascular diseases.
Clinical studies have shown that low-dose colchicine provides
significant clinical benefits in post-myocardial infarction
patients, reducing the incidence of cardiovascular events with a
favorable safety profile, suggesting its potential applicability in
MIRI (90,91).
NETs may exert a dual role in the management of
MIRI. Modulating NETs represents a potentially significant
therapeutic strategy for the treatment of MIRI. By inhibiting the
formation of NETs or facilitating their degradation, it may be
possible to mitigate myocardial tissue damage and enhance tissue
repair. There are no clinical or randomized controlled trials on
the mechanism of NETosis in MIRI, and more basic research is needed
to support this area of study. However, clinical studies on NETosis
in other diseases, such as pneumonia and cancer, offer valuale
insights (92-95).
Considering that NETosis is primarily reliant on
PAD4, targeting PAD4 represents a promising therapeutic strategy.
The inhibitor GSK484 has demonstrated significant attenuation of
MIRI in an animal model (96).
Additionally, BB-CLA, a potent PAD4 inhibitor, effectively
prevented the formation and release of NETs induced by polyinosinic
acid-polycytidylic acid, a synthetic analog of viral
double-stranded RNA (97).
Additionally, inhibition of von Willebrand factor (VWF) activity,
which mediates leukocyte recruitment, also affects PAD4 activity
and promotes NET formation, suggesting that drugs targeting both
VWF and NETs may offer a novel therapeutic approach to reduce
ischemia-related myocardial injury (98). Currently, studies on the combination
therapy of NETosis in MIRI are limited and mainly focused on the
animal experimental stage. Combination treatment of DNase I with
rhADAMTS13 or rt-PA effectively improved myocardial infarct size
and microcirculatory impairment in rats with MIRI (99). Combination of NETosis inhibitors
with other anti-inflammatory or antithrombotic drugs may have a
synergistic effect and significantly attenuate the pathological
damage in MIRI (100). Based on
the pathological mechanism of NETosis and related therapeutic
targets, it is theorized that combination therapies, such as
combining NETosis inhibitors with anti-inflammatory, antioxidant,
or antithrombotic drugs may have greater therapeutic potential. The
use of combination therapy in MIRI will be further explored in
future studies.
The activity of DNase, an enzyme that degrades NETs,
plays a crucial role in the clearance of NETs and subsequent
modulation of inflammation and thrombosis. Alterations in DNase
activity have been linked to ST-segment regression and the onset of
myocardial infarction (101).
DNase treatment can reduce the cytotoxicity of NETs by degrading
their DNA backbone, thus offering potential for reducing thrombotic
inflammation (102). However,
DNase primarily targets DNA and has limited effects on other NET
components, such as histones, which remain capable of causing
vascular damage (103). Despite
these limitations, dual-active DNase formulations, capable of
targeting both DNA and histones, may offer further promise in
modulating thrombotic inflammation (104). Recent studies have also suggested
that DNase could serve as a prognostic biomarker in patients with
atherosclerosis, offering new insights into recurrent ischemic
events (105).
The glutathione metabolic pathway plays a crucial
antioxidant role in NETosis. Isocrystalline xanthophylls have been
demonstrated to mitigate reperfusion-induced oxidative stress
injury and ferroptosis in cardiomyocytes by upregulating
glutathione peroxidase 4(106).
Studies indicate that the application of leukotriene C4
(LTC4) receptor antagonists can obstruct the LTC4
signaling pathway, thereby inhibiting NETosis and reducing MIRI
(107,108). The expression of the transcription
factor HIF-1α is upregulated under hypoxic conditions; notably, in
myocardial infarction, the expression level of HIF-1α is
significantly increased in neutrophils (109). HIF-1α induction may help promote
neutrophil survival, mitigate oxidative stress and inhibit NET
formation.
Krüppel-like factor 2 (KLF2) is a prominent member
of the zinc finger protein transcription factor family. Under
homeostatic conditions, KLF2 acts to inhibit HIF-1 signaling. The
regulation of the neutrophil KLF2-NETosis-thrombosis pathway may
represent a novel therapeutic target for the treatment of
reperfusion injury (110). SkQ1, a
mitochondria-targeted antioxidant, effectively scavenges ROS within
mitochondria during NETosis (111). Furthermore, colchicine treatment
has been shown to inhibit the formation of NETs and reduce
inflammation by decreasing NOX2/ROS production, which significantly
enhances survival rates and improves left ventricular ejection
fraction in murine models (88).
Chemokines such as chemokine (C-X-C motif) ligand 4
(CXCL4) and chemokine (C-C motif) ligand 5 (CCL5) play a pivotal
role in the inflammatory response. A synthetic peptide, MKEY, has
been shown to block the CCL5-CXCL4 interaction, preventing NET
formation by neutrophils in vivo (112). Adenosine, an immunosuppressive
molecule, inhibits neutrophil activation and NET release (113). Inhibition of ectonucleotidases
CD39 and CD73, which play a role in purinergic signaling, has
garnered attention as a potential immunotherapeutic strategy in
cancer treatment (114). Previous
studies indicate that hydrogel-delivered CD39 and CD73 can reduce
NETosis and immune infiltration, improving MIRI outcomes (115,116).
Recent research has demonstrated that in the context
of coronary thrombosis, NETs primarily activate neutrophils and
erythrocytes as the predominant constituents of most coronary
thrombi, as opposed to the traditional emphasis on platelet
aggregation (117). The inhibition
of NET formation through VWF-mediated leukocyte recruitment
presents a potentially novel therapeutic approach to mitigating
ischemia-related myocardial injury. Research utilizing Lnk gene
deletion mice (Lnk-/-) and LNK(TT) [neutrophils derived from human
induced pluripotent stem cells with LNK(TT) mutations] indicates
that the associated pathologies primarily manifest as abnormalities
in platelets and neutrophils. These abnormalities include
conditions such as coronary artery disease, thrombocytosis and
neutropenia. The Lnk-/- and LNK(TT) models may exhibit heightened
sensitivity of neutrophils to external stimuli, such as oxidized
phospholipids (OxPL), which subsequently increases their
susceptibility to NETosis (118).
Consequently, therapeutic interventions targeting OxPL could be
advantageous for genetically predisposed human populations.
Recombinant tissue plasminogen activator (rt-PA) is a recombinant
tissue-type plasminogen activator that restores blood flow in large
vessels by dissolving thrombi. rt-PA combined with DNase I can
simultaneously solve the problems of microcirculation and
obstruction of large vessels, improve reperfusion and alleviate the
phenomenon of ‘no reflow’.
In the complex pathological process of MIRI,
neutrophils exert multifaceted effects. They contribute to
cardiomyocyte death, thereby exacerbating myocardial injury, and
significantly impact normal vascular function. Additionally,
neutrophils promote inflammation by releasing various cytokines and
inflammatory mediators, which hinder the post-injury repair
process, and their functions and phenotypes can vary depending on
the inflammatory environment and stimuli present.
In detailed investigations of molecular mechanisms,
PAD4 facilitates the formation of NETs through its interaction with
granule proteins, thereby contributing to MIRI. Nonetheless, the
excessive production and release of NETs in the context of MIRI
present a dualistic effect. While they aid in the clearance of
pathogens from the injury site, they also intensify cardiomyocyte
apoptosis and the inflammatory response, potentially impairing
cardiac function.
Recent therapeutic approaches for MIRI focus on
targeting the NETosis process to attenuate or prevent the injury,
including the use of PAD4 inhibitors, antioxidants,
anti-inflammatory drugs and other related therapies. As research
advances, the identification of different neutrophil subtypes with
distinct functional properties in the inflammatory response has
broadened the scope for potential treatments. Although these
NETosis-based treatments show promise in laboratory and animal
models, their clinical application requires further validation.
Therapeutic strategies targeting NETosis represent a
promising novel approach for the treatment of MIRI. Although
significant progress has been made in animal models, the
translation of these strategies into clinical practice continues to
face substantial challenges. Clinical trial outcomes have often
been inconsistent, potentially due to individual variations in
susceptibility to reperfusion injury and the limited therapeutic
time window for intervention (119). Furthermore, there is a notable
lack of high-quality clinical trials to rigorously validate the
safety and efficacy of targeted therapies or to elucidate their
precise mechanisms of action. Consequently, future research must
focus on deepening the understanding of the specific mechanisms by
which NETosis contributes to MIRI, as well as on developing more
effective and clinically applicable interventions.
In conclusion, targeting NETosis in MIRI represents
a promising therapeutic avenue that may significantly improve
patient prognosis. By reducing myocardial damage and enhancing
repair mechanisms, these therapies are expected to change the way
MIRI is managed, providing patients with a better prognosis and
quality of life. Future clinical trials should prioritize patient
stratification, biomarker development, and evaluation of
combination therapies to realise the full clinical potential of
NETosis-targeted therapies.
Not applicable.
Funding: The present study was supported by the National Key
Research and Development Program of China (grant no.
2022YFC3500705).
Not applicable.
ZZ and YW conceived the review topic, conducted the
literature search, and wrote the original draft. TL and HW reviewed
and edited the manuscript. All authors have read and approved the
final manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Brinkmann V and Zychlinsky A: Neutrophil
extracellular traps: Is immunity the second function of chromatin?
J Cell Biol. 198:773–783. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Metzler KD, Fuchs TA, Nauseef WM, Reumaux
D, Roesler J, Schulze I, Wahn V, Papayannopoulos V and Zychlinsky
A: Myeloperoxidase is required for neutrophil extracellular trap
formation: Implications for innate immunity. Blood. 117:953–959.
2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Remijsen Q, Kuijpers TW, Wirawan E,
Lippens S, Vandenabeele P and Vanden Berghe T: Dying for a cause:
NETosis, mechanisms behind an antimicrobial cell death modality.
Cell Death Differ. 18:581–588. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the Nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Welt FGP, Batchelor W, Spears JR, Penna C,
Pagliaro P, Ibanez B, Drakos SG, Dangas G and Kapur NK: Reperfusion
injury in patients with acute myocardial infarction: JACC
scientific statement. J Am Coll Cardiol. 83:2196–2213.
2024.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Global Cardiovascular Risk Consortium.
Magnussen C, Ojeda FM, Leong DP, Alegre-Diaz J, Amouyel P,
Aviles-Santa L, De Bacquer D, Ballantyne CM, Bernabé-Ortiz A, et
al: Global effect of modifiable risk factors on cardiovascular
disease and mortality. N Engl J Med. 389:1273–1285. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tsao CW, Aday AW, Almarzooq ZI, Alonso A,
Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP,
Commodore-Mensah Y, et al: Heart disease and stroke statistics-2022
update: A report from the American Heart Association. Circulation.
145:e153–e639. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Han T, Tang H, Lin C, Shen Y, Yan D, Tang
X and Guo D: Extracellular traps and the role in thrombosis. Front
Cardiovasc Med. 9(951670)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yipp BG and Kubes P: NETosis: how vital is
it? Blood. 122:2784–2794. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Thiam HR, Wong SL, Qiu R, Kittisopikul M,
Vahabikashi A, Goldman AE, Goldman RD, Wagner DD and Waterman CM:
NETosis proceeds by cytoskeleton and endomembrane disassembly and
PAD4-mediated chromatin decondensation and nuclear envelope
rupture. Proc Natl Acad Sci USA. 117:7326–7337. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hakkim A, Fuchs TA, Martinez NE, Hess S,
Prinz H, Zychlinsky A and Waldmann H: Activation of the Raf-MEK-ERK
pathway is required for neutrophil extracellular trap formation.
Nat Chem Biol. 7:75–77. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Steinberg BE and Grinstein S:
Unconventional roles of the NADPH oxidase: Signaling, ion
homeostasis, and cell death. Sci STKE. 2007(pe11)2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nishinaka Y, Arai T, Adachi S,
Takaori-Kondo A and Yamashita K: Singlet oxygen is essential for
neutrophil extracellular trap formation. Biochem Biophys Res
Commun. 413:75–79. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Christophorou MA, Castelo-Branco G,
Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA,
Bertone P, Silva JC, Zernicka-Goetz M, et al: Citrullination
regulates pluripotency and histone H1 binding to chromatin. Nature.
507:104–108. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ho JW, Quan C, Gauger MA, Alam HB and Li
Y: Role of peptidylarginine deiminase and neutrophil extracellular
traps in injuries: Future novel diagnostics and therapeutic
targets. Shock. 59:247–255. 2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang Y, Li M, Stadler S, Correll S, Li P,
Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, et al: Histone
hypercitrullination mediates chromatin decondensation and
neutrophil extracellular trap formation. J Cell Biol. 184:205–213.
2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Papayannopoulos V, Metzler KD, Hakkim A
and Zychlinsky A: Neutrophil elastase and myeloperoxidase regulate
the formation of neutrophil extracellular traps. J Cell Biol.
191:677–691. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zollet V, Arenas Hoyos I, Hirsiger S,
Brahim BB, Petrucci MF, Casoni D, Wang J, Spirig R, Nettelbeck K,
Garcia L, et al: Neutrophil extracellular traps and citrullinated
fibrinogen contribute to injury in a porcine model of limb ischemia
and reperfusion. Front Immunol. 15(1436926)2024.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen Z, Zhang H, Qu M, Nan K, Cao H, Cata
JP, Chen W and Miao C: Review: The emerging role of neutrophil
extracellular traps in sepsis and sepsis-associated thrombosis.
Front Cell Infect Microbiol. 11(653228)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Thålin C, Hisada Y, Lundström S, Mackman N
and Wallén H: Neutrophil extracellular traps: Villains and targets
in arterial, venous, and cancer-associated thrombosis. Arterioscler
Thromb Vasc Biol. 39:1724–1738. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Leshner M, Wang S, Lewis C, Zheng H, Chen
XA, Santy L and Wang Y: PAD4 mediated histone hypercitrullination
induces heterochromatin decondensation and chromatin unfolding to
form neutrophil extracellular trap-like structures. Front Immunol.
3(307)2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Papayannopoulos V: Neutrophil
extracellular traps in immunity and disease. Nat Rev Immunol.
18:134–147. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Friedman GD, Klatsky AL and Siegelaub AB:
The leukocyte count as a predictor of myocardial infarction. N Engl
J Med. 290:1275–1278. 1974.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Borissoff JI, Joosen IA, Versteylen MO,
Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Ten Cate H,
Hofstra L, et al: Elevated levels of circulating DNA and chromatin
are independently associated with severe coronary atherosclerosis
and a prothrombotic state. Arterioscler Thromb Vasc Biol.
33:2032–2040. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang Y, Yang M, Xu Y, Yan S, Jin E and Li
X: Neutrophil extracellular trap burden correlates with the
stenosis of coronary atherosclerosis. PeerJ.
11(e15471)2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kawasaki H and Iwamuro S: Potential roles
of histones in host defense as antimicrobial agents. Infect Disord
Drug Targets. 8:195–205. 2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yipp BG, Petri B, Salina D, Jenne CN,
Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert
HC, et al: Infection-induced NETosis is a dynamic process involving
neutrophil multitasking in vivo. Nat Med. 18:1386–1393.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pilsczek FH, Salina D, Poon KK, Fahey C,
Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, et
al: A novel mechanism of rapid nuclear neutrophil extracellular
trap formation in response to Staphylococcus aureus. J Immunol.
185:7413–7425. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Schmitt MMN, Megens RTA, Zernecke A,
Bidzhekov K, van den Akker NM, Rademakers T, van Zandvoort MA,
Hackeng TM, Koenen RR and Weber C: Endothelial junctional adhesion
molecule-a guides monocytes into flow-dependent predilection sites
of atherosclerosis. Circulation. 129:66–76. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yalcinkaya M, Liu W, Xiao T, Abramowicz S,
Wang R, Wang N, Westerterp M and Tall AR: Cholesterol trafficking
to the ER leads to the activation of CaMKII/JNK/NLRP3 and promotes
atherosclerosis. J Lipid Res. 65(100534)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Münzer P, Negro R, Fukui S, di Meglio L,
Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, et
al: NLRP3 Inflammasome assembly in neutrophils is supported by PAD4
and promotes NETosis under sterile conditions. Front Immunol.
12(683803)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Warnatsch A, Ioannou M, Wang Q and
Papayannopoulos V: Inflammation. Neutrophil extracellular traps
license macrophages for cytokine production in atherosclerosis.
Science. 349:316–320. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yalcinkaya M, Liu W, Thomas LA, Olszewska
M, Xiao T, Abramowicz S, Papapetrou EP, Westerterp M, Wang N, Tabas
I and Tall AR: BRCC3-Mediated NLRP3 deubiquitylation promotes
inflammasome activation and atherosclerosis in Tet2 clonal
hematopoiesis. Circulation. 148:1764–1777. 2023.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Westerterp M, Fotakis P, Ouimet M, Bochem
AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van
Gemert S, Wang N, et al: Cholesterol efflux pathways suppress
inflammasome activation, NETosis, and atherogenesis. Circulation.
138:898–912. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li H, Tang C, Zhu X, Zhang W, Abudupataer
M, Ding S, Duan C, Yang X and Ge J: Histamine deficiency
facilitates coronary microthrombosis after myocardial infarction by
increasing neutrophil-platelet interactions. J Cell Mol Med.
24:3504–3520. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhou J, Chen R, Liu C, Zhou P, Li J, Wang
Y, Zhao X, Zhao H, Song L and Yan H: Associations of NETs with
inflammatory risk and atherosclerotic severity in ST-segment
elevation myocardial infarction. Thromb Res. 203:5–11.
2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Stakos DA, Kambas K, Konstantinidis T,
Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki
A, Skendros P, Konstantinides S and Ritis K: Expression of
functional tissue factor by neutrophil extracellular traps in
culprit artery of acute myocardial infarction. Eur Heart J.
36:1405–1414. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fuchs TA, Brill A, Duerschmied D,
Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW,
Hartwig JH and Wagner DD: Extracellular DNA traps promote
thrombosis. Proc Natl Acad Sci USA. 107:15880–15885.
2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Liang GY, Cai QY, Niu YM, Zheng H, Gao ZY,
Liu DX and Xu G: Cardiac glucose uptake and suppressed
expression/translocation of myocardium glucose transport-4 in dogs
undergoing ischemia-reperfusion. Exp Biol Med (Maywood).
233:1142–1148. 2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Remijsen Q, Vanden Berghe T, Wirawan E,
Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M,
Willems J and Vandenabeele P: Neutrophil extracellular trap cell
death requires both autophagy and superoxide generation. Cell Res.
21:290–304. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tian H, Zhao X, Zhang Y and Xia Z:
Abnormalities of glucose and lipid metabolism in myocardial
ischemia-reperfusion injury. Biomed Pharmacother.
163(114827)2023.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zamanian M, Hajizadeh M, Shamsizadeh A,
Moemenzadeh M, Amirteimouri M, Elshiekh M and Allahtavakoli M:
Effects of naringin on physical fatigue and serum MMP-9
concentration in female rats. Pharm Biol. 55:423–427.
2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zamanian M, Shamsizadeh A, Esmaeili Nadimi
A, Hajizadeh M, Allahtavakoli F, Rahmani M, Kaeidi A, Safari
Khalegh H and Allahtavakoli M: Short-term effects of troxerutin
(vitamin P4) on muscle fatigue and gene expression of Bcl-2 and Bax
in the hepatic tissue of rats. Can J Physiol Pharmacol. 95:708–713.
2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chouchani ET, Pell VR, Gaude E,
Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord
ENJ, Smith AC, et al: Ischaemic accumulation of succinate controls
reperfusion injury through mitochondrial ROS. Nature. 515:431–435.
2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Awasthi D, Nagarkoti S, Sadaf S, Chandra
T, Kumar S and Dikshit M: Glycolysis dependent lactate formation in
neutrophils: A metabolic link between NOX-dependent and independent
NETosis. Biochim Biophys Acta Mol Basis Dis.
1865(165542)2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Sun L, Wu Q, Nie Y, Cheng N, Wang R, Wang
G, Zhang D, He H, Ye RD and Qian F: A role for MK2 in enhancing
neutrophil-derived ROS production and aggravating liver
ischemia/reperfusion injury. Front Immunol. 9(2610)2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yu J, Fu Y, Gao J, Zhang Q, Zhang N, Zhang
Z, Jiang X, Chen C and Wen Z: Cathepsin C from extracellular
histone-induced M1 alveolar macrophages promotes NETosis during
lung ischemia-reperfusion injury. Redox Biol.
74(103231)2024.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Stojkov D, Gigon L, Peng S, Lukowski R,
Ruth P, Karaulov A, Rizvanov A, Barlev NA, Yousefi S and Simon HU:
Physiological and Pathophysiological Roles of Metabolic Pathways
for NET Formation and Other Neutrophil Functions. Front Immunol.
13(826515)2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Kurian GA, Rajagopal R, Vedantham S and
Rajesh M: The role of oxidative stress in myocardial ischemia and
reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev.
2016(1656450)2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bolli R and Marbán E: Molecular and
cellular mechanisms of myocardial stunning. Physiol Rev.
79:609–634. 1999.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Meng M, Jia R, Wei M, Meng X, Zhang X, Du
R, Sun W, Wang L and Song L: Oxidative stress activates Ryr2-Ca2+
and apoptosis to promote PM2.5-induced heart injury of
hyperlipidemia mice. Ecotoxicol Environ Saf.
232(113228)2022.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Gorski PA, Jang SP, Jeong D, Lee A, Lee P,
Oh JG, Chepurko V, Yang DK, Kwak TH, Eom SH, et al: Role of SIRT1
in modulating acetylation of the sarco-endoplasmic reticulum
Ca2+-ATPase in heart failure. Circ Res. 124:e63–e80.
2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Fuchs TA, Abed U, Goosmann C, Hurwitz R,
Schulze I, Wahn V, Weinrauch Y, Brinkmann V and Zychlinsky A: Novel
cell death program leads to neutrophil extracellular traps. J Cell
Biol. 176:231–241. 2007.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Ellson CD, Davidson K, Ferguson GJ,
O'Connor R, Stephens LR and Hawkins PT: Neutrophils from p40phox-/-
mice exhibit severe defects in NADPH oxidase regulation and
oxidant-dependent bacterial killing. J Exp Med. 203:1927–1937.
2006.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Douda DN, Khan MA, Grasemann H and
Palaniyar N: SK3 channel and mitochondrial ROS mediate NADPH
oxidase-independent NETosis induced by calcium influx. Proc Natl
Acad Sci USA. 112:2817–2822. 2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Liao X, Song X, Li J, Li L, Fan X, Qin Q,
Zhong C, Yang P, Zhan J and Cai Y: An injectable co-assembled
hydrogel blocks reactive oxygen species and inflammation cycle
resisting myocardial ischemia-reperfusion injury. Acta Biomater.
149:82–95. 2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Andreadou I, Cabrera-Fuentes HA, Devaux Y,
Frangogiannis NG, Frantz S, Guzik T, Liehn EA, Gomes CPC, Schulz R
and Hausenloy DJ: Immune cells as targets for cardioprotection: New
players and novel therapeutic opportunities. Cardiovasc Res.
115:1117–1130. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhang L, Deng S, Zhao S, Ai Y, Zhang L,
Pan P, Su X, Tan H and Wu D: Intra-peritoneal administration of
mitochondrial DNA provokes acute lung injury and systemic
inflammation via toll-like receptor 9. Int J Mol Sci.
17(1425)2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Xie L, He S, Kong N, Zhu Y, Tang Y, Li J,
Liu Z, Liu J and Gong J: Cpg-ODN, a TLR9 agonist, aggravates
myocardial ischemia/reperfusion injury by activation of TLR9-P38
MAPK signaling. Cell Physiol Biochem. 47:1389–1398. 2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Hidalgo A, Libby P, Soehnlein O, Aramburu
IV, Papayannopoulos V and Silvestre-Roig C: Neutrophil
extracellular traps: From physiology to pathology. Cardiovasc Res.
118:2737–2753. 2022.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Shah M, He Z, Rauf A, Beikoghli Kalkhoran
S, Heiestad CM, Stensløkken KO, Parish CR, Soehnlein O, Arjun S,
Davidson SM and Yellon D: Extracellular histones are a target in
myocardial ischaemia-reperfusion injury. Cardiovasc Res.
118:1115–1125. 2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Wilson AS, Randall KL, Pettitt JA, Ellyard
JI, Blumenthal A, Enders A, Quah BJ, Bopp T, Parish CR and Brüstle
A: Neutrophil extracellular traps and their histones promote Th17
cell differentiation directly via TLR2. Nat Commun.
13(528)2022.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Tsourouktsoglou TD, Warnatsch A, Ioannou
M, Hoving D, Wang Q and Papayannopoulos V: Histones, DNA, and
citrullination promote neutrophil extracellular trap inflammation
by regulating the localization and activation of TLR4. Cell Rep.
31(107602)2020.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H,
Wu D, Liu B, Li H, Li H, et al: Neutrophil extracellular traps are
indirectly triggered by lipopolysaccharide and contribute to acute
lung injury. Sci Rep. 6(37252)2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Clark SR, Ma AC, Tavener SA, McDonald B,
Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair
GD, et al: Platelet TLR4 activates neutrophil extracellular traps
to ensnare bacteria in septic blood. Nat Med. 13:463–469.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
68
|
Maugeri N, Campana L, Gavina M, Covino C,
De Metrio M, Panciroli C, Maiuri L, Maseri A, D'Angelo A, Bianchi
ME, et al: Activated platelets present high mobility group box 1 to
neutrophils, inducing autophagy and promoting the extrusion of
neutrophil extracellular traps. J Thromb Haemost. 12:2074–2088.
2014.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Kawashima S and Yokoyama M: Dysfunction of
endothelial nitric oxide synthase and atherosclerosis. Arterioscler
Thromb Vasc Biol. 24:998–1005. 2004.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Carlstrom M, Weitzberg E and Lundberg JO:
Nitric oxide signaling and regulation in the cardiovascular system:
Recent advances. Pharmacol Rev. 76:1038–1062. 2024.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Godo S, Takahashi J, Shiroto T, Yasuda S
and Shimokawa H: Coronary microvascular spasm: Clinical
presentation and diagnosis. Eur Cardiol. 18(e07)2023.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Zhou Q, Cao J and Chen L: Apelin/APJ
system: A novel therapeutic target for oxidative stress-related
inflammatory diseases (Review). Int J Mol Med. 37:1159–1169.
2016.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wu X, Xu M, Liu Z, Zhang Z, Liu Y, Luo S,
Zheng X, Little PJ, Xu S and Weng J: Pharmacological inhibition of
IRAK1 and IRAK4 prevents endothelial inflammation and
atherosclerosis in ApoE-/- mice. Pharmacol Res.
175(106043)2022.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Aldabbous L, Abdul-Salam V, McKinnon T,
Duluc L, Pepke-Zaba J, Southwood M, Ainscough AJ, Hadinnapola C,
Wilkins MR, Toshner M and Wojciak-Stothard B: Neutrophil
extracellular traps promote angiogenesis: Evidence from vascular
pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol.
36:2078–2087. 2016.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Cao Y, Chen M, Jiao X, Li S, Wang D, Zhan
Y, Li J, Hao Z, Li Q, Liu Y, et al: Neutrophil extracellular traps
mediate the crosstalk between plaque microenvironment and unstable
carotid plaque formation. Exp Mol Med. 56:1717–1735.
2024.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Chrysanthopoulou A, Gkaliagkousi E,
Lazaridis A, Arelaki S, Pateinakis P, Ntinopoulou M, Mitsios A,
Antoniadou C, Argyriou C, Georgiadis GS, et al: Angiotensin II
triggers release of neutrophil extracellular traps, linking
thromboinflammation with essential hypertension. JCI Insight.
6(e148668)2021.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling-concepts and clinical implications: A consensus paper
from an international forum on cardiac remodeling. Behalf of an
International Forum on Cardiac Remodeling. J Am Coll Cardiol.
35:569–582. 2000.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Mangold A, Hofbauer TM, Ondracek AS,
Artner T, Scherz T, Speidl WS, Krychtiuk KA, Sadushi-Kolici R,
Jakowitsch J and Lang IM: Neutrophil extracellular traps and
monocyte subsets at the culprit lesion site of myocardial
infarction patients. Sci Rep. 9(16304)2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Jorch SK and Kubes P: An emerging role for
neutrophil extracellular traps in noninfectious disease. Nat Med.
23:279–287. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Li C, Gao P, Zhuang F, Wang T, Wang Z, Wu
G, Zhou Z, Xie H, Xie D, Zhao D, et al: Inhibition of
ALOX12-12-HETE alleviates lung ischemia-reperfusion injury by
reducing endothelial ferroptosis-mediated neutrophil extracellular
trap formation. Research (Wash D C). 7(0473)2024.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Chen J, Wang T, Li X, Gao L, Wang K, Cheng
M, Zeng Z, Chen L, Shen Y and Wen F: DNA of neutrophil
extracellular traps promote NF-κB-dependent autoimmunity via
cGAS/TLR9 in chronic obstructive pulmonary disease. Signal
Transduct Target Ther. 9(163)2024.PubMed/NCBI View Article : Google Scholar
|
|
82
|
He L, Liu R, Yue H, Zhu G, Fu L, Chen H,
Guo Y and Qin C: NETs promote pathogenic cardiac fibrosis and
participate in ventricular aneurysm formation after ischemia injury
through the facilitation of perivascular fibrosis. Biochem Biophys
Res Commun. 583:154–161. 2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Chang CP, Chia RH, Wu TL, Tsao KC, Sun CF
and Wu JT: Elevated cell-free serum DNA detected in patients with
myocardial infarction. Clin Chim Acta. 327:95–101. 2003.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Cao J, Roth S, Zhang S, Kopczak A, Mami S,
Asare Y, Georgakis MK, Messerer D, Horn A, Shemer R, et al:
DNA-sensing inflammasomes cause recurrent atherosclerotic stroke.
Nature. 633:433–441. 2024.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Hally KE, Parker OM, Brunton-O'Sullivan
MM, Harding SA and Larsen PD: Linking neutrophil extracellular
traps and platelet activation: A composite biomarker score for
predicting outcomes after acute myocardial infarction. Thromb
Haemost. 121:1637–1649. 2021.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Yu S, Li M, Li Z, Xu P, Yao Z, Qian S,
Qian F, Gao D and Wang H: Positive correlations between plasma BPI
level and MPO-DNA and S100A8/A9 in myocardial infarction.
Platelets. 33:603–611. 2022.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Nagareddy PR, Sreejit G, Abo-Aly M,
Jaggers RM, Chelvarajan L, Johnson J, Pernes G, Athmanathan B,
Abdel-Latif A and Murphy AJ: NETosis Is Required for
S100A8/A9-induced granulopoiesis after myocardial infarction.
Arterioscler Thromb Vasc Biol. 40:2805–2807. 2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Li YW, Chen SX, Yang Y, Zhang ZH, Zhou WB,
Huang YN, Huang ZQ, He JQ, Chen TF, Wang JF, et al: Colchicine
inhibits NETs and alleviates cardiac remodeling after acute
myocardial infarction. Cardiovasc Drugs Ther. 38:31–41.
2024.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Tan Y, Bao X, Li Y, Song G, Lu H, Sun X,
Gu R, Kang L and Xu B: Colchicine attenuates microvascular
obstruction after myocardial ischemia-reperfusion injury by
inhibiting the proliferation of neutrophil in bone marrow.
Cardiovasc Drugs Ther. 39:259–273. 2025.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Banach M and Penson PE: Colchicine and
cardiovascular outcomes: A critical appraisal of recent studies.
Curr Atheroscler Rep. 23(32)2021.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Tardif JC, Kouz S, Waters DD, Bertrand OF,
Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, et al:
Efficacy and safety of low-dose colchicine after myocardial
infarction. N Engl J Med. 381:2497–2505. 2019.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Zhang W, Liu J, Li X, Bai Z, Sun Y and
Chen X: Lidocaine effects on neutrophil extracellular trapping and
angiogenesis biomarkers in postoperative breast cancer patients
with different anesthesia methods: A prospective, randomized trial.
BMC Anesthesiol. 24(162)2024.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Sapey E, Patel JM, Greenwood H, Walton GM,
Grudzinska F, Parekh D, Mahida RY, Dancer RCA, Lugg ST, Howells PA,
et al: Simvastatin improves neutrophil function and clinical
outcomes in pneumonia. A pilot randomized controlled clinical
trial. Am J Respir Crit Care Med. 200:1282–1293. 2019.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Ebrahimi F, Giaglis S, Hahn S, Blum CA,
Baumgartner C, Kutz A, van Breda SV, Mueller B, Schuetz P,
Christ-Crain M and Hasler P: Markers of neutrophil extracellular
traps predict adverse outcome in community-acquired pneumonia:
Secondary analysis of a randomised controlled trial. Eur Respir J.
51(1701389)2018.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Shin D, Kim J, Lee S and Chae MS: Impact
of perioperative lidocaine on neutrophil extracellular trapping and
serum cytokines in robot-assisted radical prostatectomy: Randomized
controlled study. Medicina (Kaunas). 60(1452)2024.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Hu Z, Hua X, Mo X, Chang Y, Chen X, Xu Z,
Tao M, Hu G and Song J: Inhibition of NETosis via PAD4 alleviated
inflammation in giant cell myocarditis. IScience.
26(107162)2023.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Ai P, Pan H, Chen K, Zheng J, Gao Z and
Jin G: Viral mimetic poly(I:C) induces neutrophil extracellular
traps via PAD4 to promote inflammation and thrombosis. Biochem
Biophys Res Commun. 565:64–71. 2021.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Savchenko AS, Borissoff JI, Martinod K, De
Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y and Wagner DD:
VWF-mediated leukocyte recruitment with chromatin decondensation by
PAD4 increases myocardial ischemia/reperfusion injury in mice.
Blood. 123:141–148. 2014.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang
YD, Ma YQ, Zhao JH and Li YM: Neutrophil extracellular traps in
ischemia-reperfusion injury-induced myocardial no-reflow:
Therapeutic potential of DNase-based reperfusion strategy. Am J
Physiol Heart Circ Physiol. 308:H500–H509. 2015.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Di G, Vázquez-Reyes S, Díaz B,
Peña-Martinez C, García-Culebras A, Cuartero MI, Moraga A, Pradillo
JM, Esposito E, Lo EH, et al: Daytime DNase-I administration
protects mice from ischemic stroke without inducing bleeding or
tPA-induced hemorrhagic transformation, even with aspirin
pretreatment. Stroke. 56:527–532. 2025.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Carminita E, Crescence L, Brouilly N,
Altié A, Panicot-Dubois L and Dubois C: DNAse-dependent,
NET-independent pathway of thrombus formation in vivo. Proc Natl
Acad Sci USA. 118(e2100561118)2021.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Mangold A, Alias S, Scherz T, Hofbauer M,
Jakowitsch J, Panzenböck A, Simon D, Laimer D, Bangert C,
Kammerlander A, et al: Coronary neutrophil extracellular trap
burden and deoxyribonuclease activity in ST-elevation acute
coronary syndrome are predictors of ST-segment resolution and
infarct size. Circ Res. 116:1182–1192. 2015.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Kolaczkowska E, Jenne CN, Surewaard BG,
Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G and Kubes
P: Molecular mechanisms of NET formation and degradation revealed
by intravital imaging in the liver vasculature. Nat Commun.
6(6673)2015.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Englert H, Göbel J, Khong D, Omidi M,
Wolska N, Konrath S, Frye M, Mailer RK, Beerens M, Gerwers JC, et
al: Targeting NETs using dual-active DNase1 variants. Front
Immunol. 14(1181761)2023.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Liu T, Lv X, Xu Q, Qi X, Qiu S, Luan Y,
Shen N, Cheng J, Jin L, Tian T, et al: Stroke-homing peptide-DNase1
alleviates intestinal ischemia reperfusion injury by selectively
degrading neutrophil extracellular traps. Cell Prolif.
(e70010)2025.PubMed/NCBI View Article : Google Scholar : (Epub ahead of
print).
|
|
106
|
Yao D, Bao L, Wang S, Tan M, Xu Y, Wu T,
Zhang Z and Gong K: Isoliquiritigenin alleviates myocardial
ischemia-reperfusion injury by regulating the
Nrf2/HO-1/SLC7a11/GPX4 axis in mice. Free Radic Biol Med. 221:1–12.
2024.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Yang K, Gao R, Chen H, Hu J, Zhang P, Wei
X, Shi J, Chen Y, Zhang L, Chen J, et al: Myocardial reperfusion
injury exacerbation due to ALDH2 deficiency is mediated by
neutrophil extracellular traps and prevented by leukotriene C4
inhibition. Eur Heart J. 45:1662–1680. 2024.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Lin K, Fang S, Cai B, Huang X, Zhang X, Lu
Y, Zhang W and Wei E: ERK/Egr-1 signaling pathway is involved in
CysLT2 receptor-mediated IL-8 production in HEK293 cells. Eur J
Cell Biol. 93:278–288. 2014.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Dölling M, Eckstein M, Singh J, Schauer C,
Schoen J, Shan X, Bozec A, Knopf J, Schett G, Muñoz LE and Herrmann
M: Hypoxia promotes neutrophil survival after acute myocardial
infarction. Front Immunol. 13(726153)2022.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Tang X, Wang P, Zhang R, Watanabe I, Chang
E, Vinayachandran V, Nayak L, Lapping S, Liao S, Madera A, et al:
KLF2 regulates neutrophil activation and thrombosis in cardiac
hypertrophy and heart failure progression. J Clin Invest.
132(e147191)2022.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Vorobjeva N, Galkin I, Pletjushkina O,
Golyshev S, Zinovkin R, Prikhodko A, Pinegin V, Kondratenko I,
Pinegin B and Chernyak B: Mitochondrial permeability transition
pore is involved in oxidative burst and NETosis of human
neutrophils. Biochim Biophys Acta Mol Basis Dis.
1866(165664)2020.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Vajen T, Koenen RR, Werner I, Staudt M,
Projahn D, Curaj A, Sönmez TT, Simsekyilmaz S, Schumacher D,
Möllmann J, et al: Blocking CCL5-CXCL4 heteromerization preserves
heart function after myocardial infarction by attenuating leukocyte
recruitment and NETosis. Sci Rep. 8(10647)2018.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Xu K, Cooney KA, Shin EY, Wang L, Deppen
JN, Ginn SC and Levit RD: Adenosine from a biologic source
regulates neutrophil extracellular traps (NETs). J Leukoc Biol.
105:1225–1234. 2019.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Allard B, Longhi MS, Robson SC and Stagg
J: The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor
targets. Immunol Rev. 276:121–144. 2017.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Borg N, Alter C, Görldt N, Jacoby C, Ding
Z, Steckel B, Quast C, Bönner F, Friebe D, Temme S, et al: CD73 on
T cells orchestrates cardiac wound healing after myocardial
infarction by purinergic metabolic reprogramming. Circulation.
136:297–313. 2017.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Sayegh MN, Cooney KA, Han WM, Cicka M,
Strobel F, Wang L, García AJ and Levit RD: Hydrogel delivery of
purinergic enzymes improves cardiac ischemia/reperfusion injury. J
Mol Cell Cardiol. 176:98–109. 2023.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Chilingaryan Z, Deshmukh T, Leung HHL,
Perdomo J, Emerson P, Kurup R, Chong BH and Chong JJH: Erythrocyte
interaction with neutrophil extracellular traps in coronary artery
thrombosis following myocardial infarction. Pathology. 54:87–94.
2022.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Dou H, Kotini A, Liu W, Fidler T,
Endo-Umeda K, Sun X, Olszewska M, Xiao T, Abramowicz S, Yalcinkaya
M, et al: Oxidized phospholipids promote NETosis and arterial
thrombosis in LNK(SH2B3) deficiency. Circulation. 144:1940–1954.
2021.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Fordyce CB, Gersh BJ, Stone GW and Granger
CB: Novel therapeutics in myocardial infarction: Targeting
microvascular dysfunction and reperfusion injury. Trends Pharmacol
Sci. 36:605–616. 2015.PubMed/NCBI View Article : Google Scholar
|