Introduction
Ectodermal dysplasia (ED) syndromes are a
heterogeneous group of disorders that are characterized by
congenital defects in two or more ectodermal structures. Among
these structures, at least one must involve the hair, teeth, nails,
or sweat glands (1,2). ED may also impact the skin, eye (lens
or retina), inner ear, development of fingers and toes, nerves and
other body parts (2). At least 150
different types of ED have been observed (2). EDs are classified in subgroups
according to the presence or absence of the four primary defects
observed in ED: Trichodysplasia (hair dysplasia), dental dysplasia,
onychodysplasia (nail dysplasia), dyshidrosis (sweat gland
dysplasia) (2).
A rare type of ED, ED-12 is described by congenital
absent teeth, dystrophic toenails, hypohidrosis, and hair
abnormalities and is linked to mutations in the keratinocyte
differentiation factor 1 (KDF1) gene (3). Additionally, KDF1 is the most
recently identified rare candidate gene for tooth agenesis
(4). This gene is essential for
skin epidermal differentiation and it is known that KDF1
deficiency results in significant abnormalities in skin development
(5). However, the exact role and
importance of KDF1 is still not entirely known (5).
In the present study, a case of a three-generation
family was described where the novel mutation c.812A>C
(p.Lys271Thr or K271T) in KDF1 was identified in four family
members. The family was referred to the Clinical Laboratory
Genetics, Access to Genome P.C. (Athens, Greece), after the birth
of their first child that presented with natal teeth. Following the
detection of the novel KDF1 mutation, further testing was
performed in other family members and it was revealed that the
father and paternal grandfather of the child were also carriers of
the same mutation. Moreover, the second child of the family that
was born 2 years later, was also born with natal teeth and was a
carrier of the same mutation. This is an extremely rare case of the
presence of natal teeth in carriers of a KDF1 mutation,
since only 2 cases have been described thus far (3,6).
Case report
The family was referred to Clinical Laboratory
Genetics, Access to Genome P.C., after a newborn presented with 12
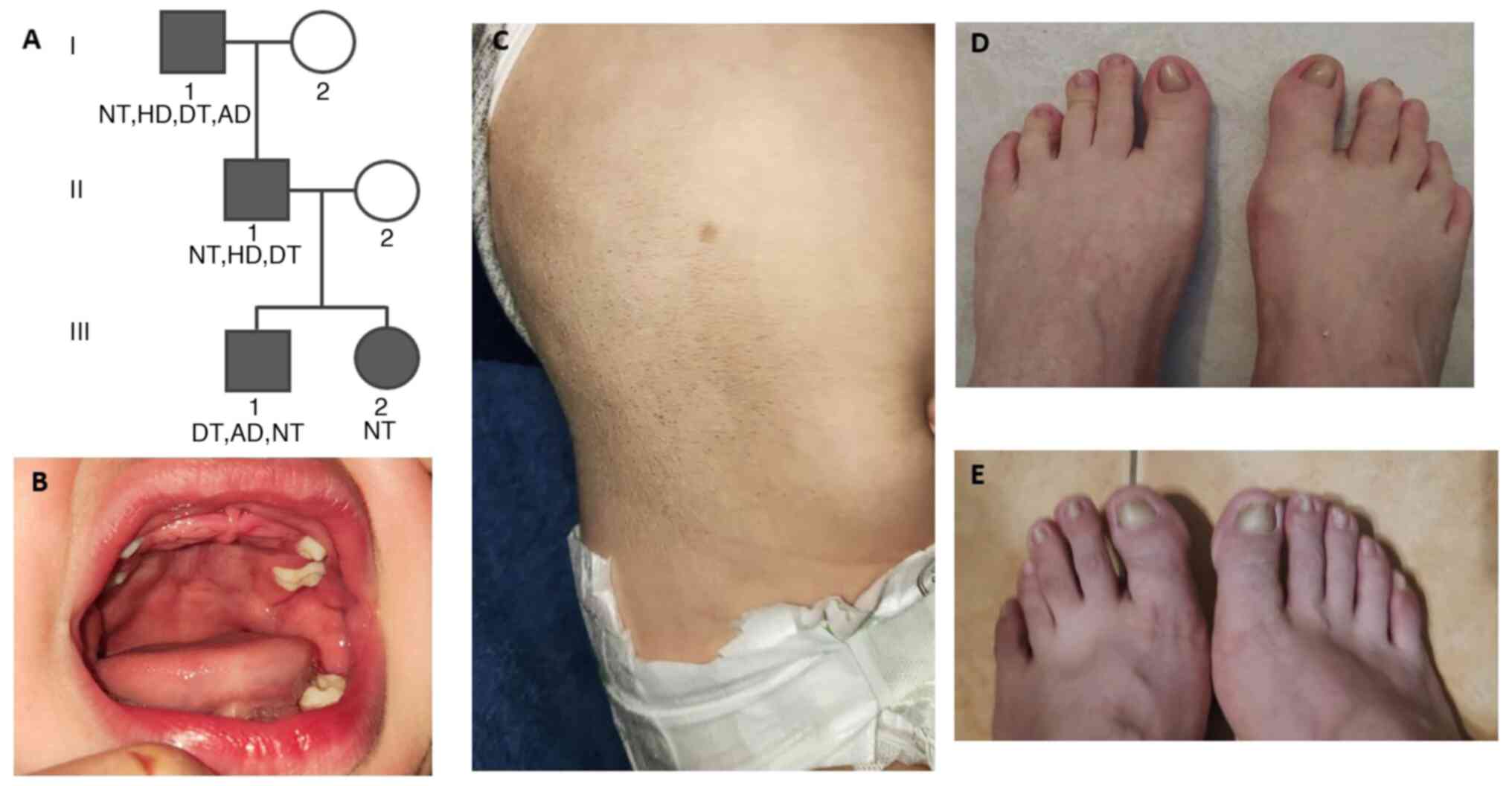
natal teeth at birth (III-1) (Fig.
1). Whole exome sequencing (WES) was performed using next
generation sequencing [accession no. ERR14252058; European
Nucleotide Archive (ENA)]. DNA was isolated from the newborn's
whole blood cells. Exome amplification was performed using Ion
AmpliSeq™ Exome RDY (cat. no. A38264; Thermo Fisher
Scientific, Inc.). Nucleotide Sequencing was performed using Ion
Chef Instrument and Ion GeneStudio™ S5 System (Thermo
Fisher Scientific, Inc.). The analysis involved 4,431 genes that
are associated with known genetic diseases and syndromes.
Alamut® Visual (version 1.7.1; SOPHiA GENETICS SAS) and
Varsome Clinical (V.9.2.2; Saphetor SA) bioinformatic analysis
systems were used for the data analysis. All findings from the
analysis above were evaluated according to the American College of
Medical Genetics and Genomics guidelines as well as the
international scientific literature (7). The reference genome that was used is
UCSC hg19. Bioinformatic analysis revealed a novel mutation in
KDF1, namely c.812A>C (p.Lys271Thr or K271T).
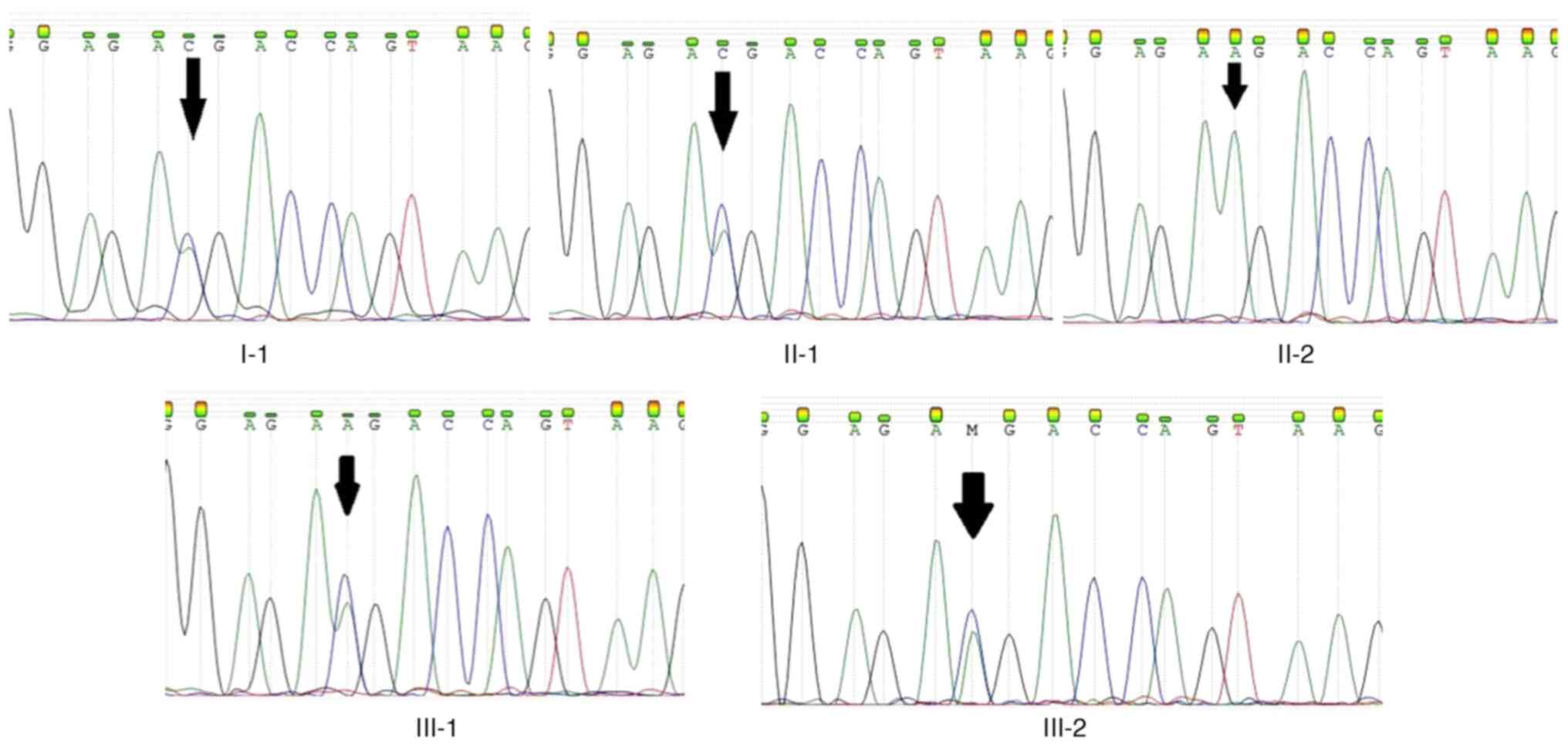
In order to confirm the presence of the mutation,
Sanger sequencing was performed in the patient. Following DNA
amplification by PCR, part of the KDF1 gene was sequenced
and was compared with the control sequence. The sequences of the
primers used were forward, 5'-CCACAGTGACTAAAGACCCCATTCC-3' and
reverse, 5'-TCGTGGCTCCGAGGAGTACTAT-3'. The results confirmed the
presence of the novel c.812A>C mutation in KDF1.
Sanger sequencing of KDF1 was performed on
parental DNA, isolated from whole blood cells, in order to
determine if the mutation was inherited or if it occurred de
novo. Sequencing analysis revealed that the father of the
patient (II-1) was also a carrier of the same mutation, indicating
that the c.812A>C mutation was inherited. The paternal
grandfather (I-1) and the second child (III-2) of the family, born
2 years later, were also tested and the results confirmed that they
are carriers of the c.812A>C mutation in KDF1 (Fig. 2).
Clinical characteristics of the
carrier members
All four family members that carried the KDF1
mutation had characteristics of ED. According to the clinical
history of the family, I-1 was born with natal teeth and now, at
age 74, I-1 has hypodontia. Moreover, I-1 has dystrophic toenails
and atopic dermatitis. Next, II-2 was born with five natal teeth
and now, at age 40, has hypodontia. Moreover, II-2 has dystrophic
toenails. Patients III-1 and III-2 were both born with natal teeth
(III-1 had 12 and III-2 had seven natal teeth), and patient III-1
also has atopic dermatitis (Fig. 1;
Table I).
 | Table ISummary of the clinical
characteristics of the affected family members. |
Table I
Summary of the clinical
characteristics of the affected family members.
| Member | Natal teeth | Hypodontia | Atopic
dermatitis | Dystrophic
toenails |
|---|
| I-1 | + (unknown
number) | + | + | + |
| II-1 | 5 | + | - | + |
| III-1 | 12 | - | + | - |
| III-2 | 7 | - | - | - |
Prenatal ultrasound examination of III-1 and III-2
during pregnancy did not reveal any anomalies. Regarding patient
III-1, the scan at 12 weeks of gestation revealed a nuchal
translucency measurement of 1.22 mm and the risk for common
triploidies was low. The anomaly scan at 21+2 weeks and the third
trimester growth scan at 32+2 weeks of gestation revealed normal
fetal structure and growth. For patient III-2, the first trimester
scan at 12+2 weeks of gestation revealed a nuchal translucency
measurement of 1.98 mm and the risk for Trisomy 21 was 1 in 381.
Both the anomaly scan at 21+2 weeks and the growth scan at 32+2
weeks of gestation showed normal fetal structure and growth.
Discussion
Mutations in KDF1 are associated with the
manifestation of clinical characteristics of ED-12
(hypohidrotic/hair/tooth/nail type, Phenotype MIM no. 617337;
OMIM®, John Hopkins University, Baltimore, USA)
following autosomal dominant mode of inheritance. In the present
study, a case of a family with a familial, previously undescribed
mutation in KDF1, present in four family members and three
generations is described. All carrier members exhibited clinical
manifestations of ED. The novel mutation is a missense mutation
that has not been reported in the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and Decipher
(https://www.deciphergenomics.org/)
databases and is not described in the international literature.
Bioinformatic analysis suggested that the mutation likely affects
the structure and function of the KDF1 protein and according to the
ACMG/AMP guidelines, it is categorized as a variant of unknown
significance based on the PM2, PP2, PP3 criteria (7).
In 2013, KDF1 was first described by Lee
et al (8). They described a
Kdf1 mouse mutant that had a short snout and short limbs.
This gene was expressed in epidermal cells during epidermal
development. The Kdf1 mouse mutant showed a thickened
epidermis and defective epidermal barrier formation due to
keratinocyte defects. Zeng et al (9) showed that KDF1 is also
expressed in tooth germs during tooth development, indicating that
KDF1 plays an important role during tooth germ development
(9).
To date, only a few cases of KDF1 mutations
have been reported and the phenotype associated with the mutations
varies (Table II). A heterozygous
KDF1 mutation was first reported to cause ED-12 (autosomal
dominant hypohidrotic ED) by Shamseldin et al (3). In their study, they described the case
of a multi-generational family with ED, where the affected members
exhibited tooth agenesis, hypohidrosis, lusterless hair, dystrophic
toenails, lateral thinning of eyebrows and keratosis pilaris
(3). Only one of the patients had
natal teeth. Another case of a newborn with natal teeth was
described by Aljohar et al (6). This was the first case of multiple
teeth present at birth due to a KDF1 mutation (6). ED due to a KDF1 mutation has
also been described by Manaspon et al (10). They identified a novel de
novo mutation in KDF1 in a 5-year-old patient with ED.
Moreover, Kamat et al (11)
described a patient with hypohidrotic ED that had peg-shaped teeth
due to the presence of a novel mutation in KDF1.
| Table IIData regarding the four family members
of this case report and patients from other studies that carry a
KDF1 mutation. |
Table II
Data regarding the four family members
of this case report and patients from other studies that carry a
KDF1 mutation.
| Case | Phenotype | Teeth | Age at diagnosis | Mutation | Inheritance | (Refs.) |
|---|
| I-1 | ED
manifestations | Natal teeth at birth,
presently hypodontia | 71 years | c.812A>C | N/A | Present study |
| II-1 | ED
manifestations | 5 Natal teeth at
birth, presently hypodontia | 37 years | c.812A>C | Paternal | Present study |
| III-1 | ED
manifestations | 12 Natal teeth | 2 weeks | c.812A>C | Paternal | Present study |
| III-2 | ED
manifestations | 7 Natal teeth | 2 weeks | c.812A>C | Paternal | Present study |
| Patient from Zeng
et al | NSTA | Several absent
teeth | 7 years | c.908G>C | Maternal | (9) |
| Patient from Manaspon
et al | ED | Loss of all
teeth | 5 years | c.823A>C | De novo | (10) |
| Patient from
Aljohar et al | ED | Multiple natal
teeth | 1 week | c.753 > A | Maternal | (6) |
| Patient II:8 from
Shamseldin et al | ED | Loss of all
teeth | 16 years | c.753C>A | Paternal | (3) |
| Patient II:2 from
Shamseldin et al | ED | Loss of all
teeth | 30 years | c.753C>A | Paternal | (3) |
| Patient II:6 from
Shamseldin et al | ED | Loss of all
teeth | 21 years | c.753C>A | Paternal | (3) |
| Patient III:2 from
Shamseldin et al | ED | Natal | 1 year | c.753C>A | Maternal | (3) |
| Patient II:7 from
Shamseldin et al | ED | Loss of all
teeth | 17 years | c.753C>A | Paternal | (3) |
| Patient I:1 from
Shamseldin et al | ED | Loss of all
teeth | 52 years | c.753C>A | N/A | (3) |
| Patient II-1 from
Yu et al | NSTA | Several absent
teeth | 21 years | c.920G>C | Paternal | (4) |
| Patient from Kamat
et al | Hypohidrotic
ED | Peg shaped | 3 years | c.449G>A | N/A | (11) |
As aforementioned, KDF1 is the most recently
identified rare candidate gene for tooth agenesis. Zeng et
al (9) described the case of a
7-year-old patient with non-syndromic tooth agenesis (NSTA) that
had invaginated lingual fossa and high susceptibility to dental
caries. The patient was found to be a carrier of a novel
KDF1 mutation. Yu et al (4) have also described a case of NSTA in a
patient carrying a novel KDF1 mutation. In the present case
report, the novel KDF1 mutation led to the presence of natal
teeth in all carriers. Thus far, only 2 cases of KDF1
mutations leading to natal teeth at birth have been reported
(3,6). To the best of our knowledge, this was
the third instance of the presence of natal teeth at birth due to a
KDF1 mutation. The presence of natal teeth in newborns with
ED is highly uncommon (6).
As Zeng et al (9) suggested, the phenotype differences
between patients with KDF1 mutations may be due to the
different mutations or the difference in genetic background of the
patients. In the present case report, all affected family members
that were reported were born with natal teeth. Moreover, both adult
members that are carriers of the mutation now have hypodontia.
However, dystrophic toenails and atopic dermatitis are present in
two out of the four affected members.
In the present study, the case of four family
members across three generations who are carriers of the previously
unreported c.812A>C mutation in KDF1 were described. The
present case report included the clinical manifestations of all
carrier members. Notably, all four affected family members had
natal teeth at birth and to the best of our knowledge, this was the
third report of natal teeth at birth due to a KDF1 mutation.
The findings of the present case report are significant for
establishing genotype-phenotype associations related to KDF1
mutations. Furthermore, they are crucial for genetic counseling,
particularly in cases where this KDF1 mutation is detected
prenatally.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found
in the European Nucleotide Archive (ENA), under accession no.
ERR14252058 or at the following URL: https://www.ebi.ac.uk/ena/browser/view/ERR14252058.
Authors' contributions
CK substantially contributed to the design of the
present study and prepared the manuscript. EM and IP were in charge
of patient management and project supervision, as well as
critically revised the manuscript. IP, EM, ES, CE, and ElP
performed WES and Sanger sequencing and analyzed patient data. EfP,
SS, AC, PA, ME and GN were responsible for assessment of the
patients and advised on patient treatment. EM and IP confirmed the
authenticity of all raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All procedures were conducted according to The
Declaration of Helsinki 1975, as revised in 2008.
Patient consent for publication
Written informed consent was obtained from all adult
family members for the inclusion of their data and images, as well
as the data and images of their children. Any information revealing
the identities of family members was not included.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Majmundar VD and Baxi K: Ectodermal
dysplasia. In StatPearls[Internet]; Treasure Island (FL):
StatPearls Publishing, 2025.
|
|
2
|
Deshmukh S and Prashanth S: Ectodermal
dysplasia: A genetic review. Int J Clin Pediatr Dent. 5:197–202.
2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Shamseldin HE, Khalifa O, Binamer YM,
Almutawa A, Arold ST, Zaidan H and Alkuraya FS: KDF1, encoding
keratinocyte differentiation factor 1, is mutated in a
multigenerational family with ectodermal dysplasia. Hum Genet.
136:99–105. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yu M, Liu H, Liu Y, Zheng J, Wu J, Sun K,
Feng H, Liu H and Han D: KDF1 novel variant causes unique dental
and oral epithelial defects. Int J Mol Sci.
23(12465)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li Y, Tang L, Yue J, Gou X, Lin A,
Weatherbee SD and Wu X: Regulation of epidermal differentiation
through KDF1-mediated deubiquitination of IKKα. EMBO Rep.
21(e48566)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Aljohar A, Alwakeel H and Palma A:
Multiple natal teeth in a one-week-old baby: A case report. Clin
Case Rep. 9:1292–1294. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lee S, Kong Y and Weatherbee SD: Forward
genetics identifies Kdf1/1810019J16Rik as an essential regulator of
the proliferation-differentiation decision in epidermal progenitor
cells. Dev Biol. 383:201–213. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zeng B, Lu H, Xiao X, Yu X, Li S, Zhu L,
Yu D and Zhao W: KDF1 is a novel candidate gene of non-syndromic
tooth agenesis. Arch Oral Biol. 97:131–136. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Manaspon C, Thaweesapphithak S, Osathanon
T, Suphapeetiporn K, Porntaveetus T and Shotelersuk V: A novel de
novo mutation substantiates KDF1 as a gene causing ectodermal
dysplasia. Br J Dermatol. 181:419–420. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kamat D, Mahajan R, Chatterjee D, Yadav J,
Kumar R, Dayal D, De D and Handa S: Clinical and genetic
characteristics of ectodermal dysplasia in four Indian children.
Indian J Dermatol. 67:54–57. 2022.PubMed/NCBI View Article : Google Scholar
|